
Translational Attenuation by an Intron Retention in the 5′ UTR of ENAM Causes Amelogenesis Imperfecta

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Subject Enrollment
2.2. DNA Isolation & Whole Exome Sequencing
2.3. Bioinformatics
2.4. Sanger Sequencing
2.5. Cloning and Splicing Assay of the Wild-Type and Mutant ENAM Splicing Vectors
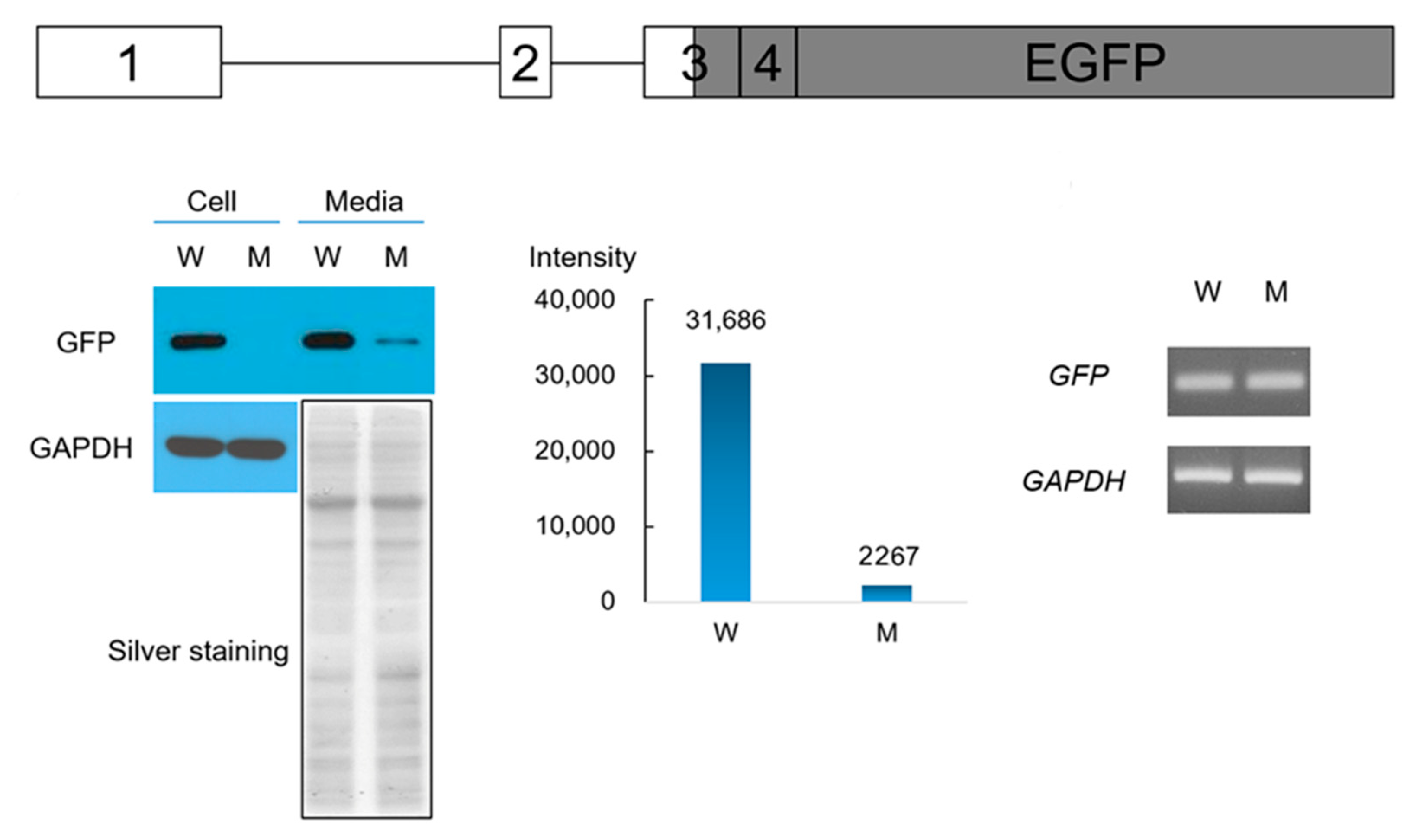
2.6. Western Blot and RT-PCR of the Wild-Type and Mutant ENAM
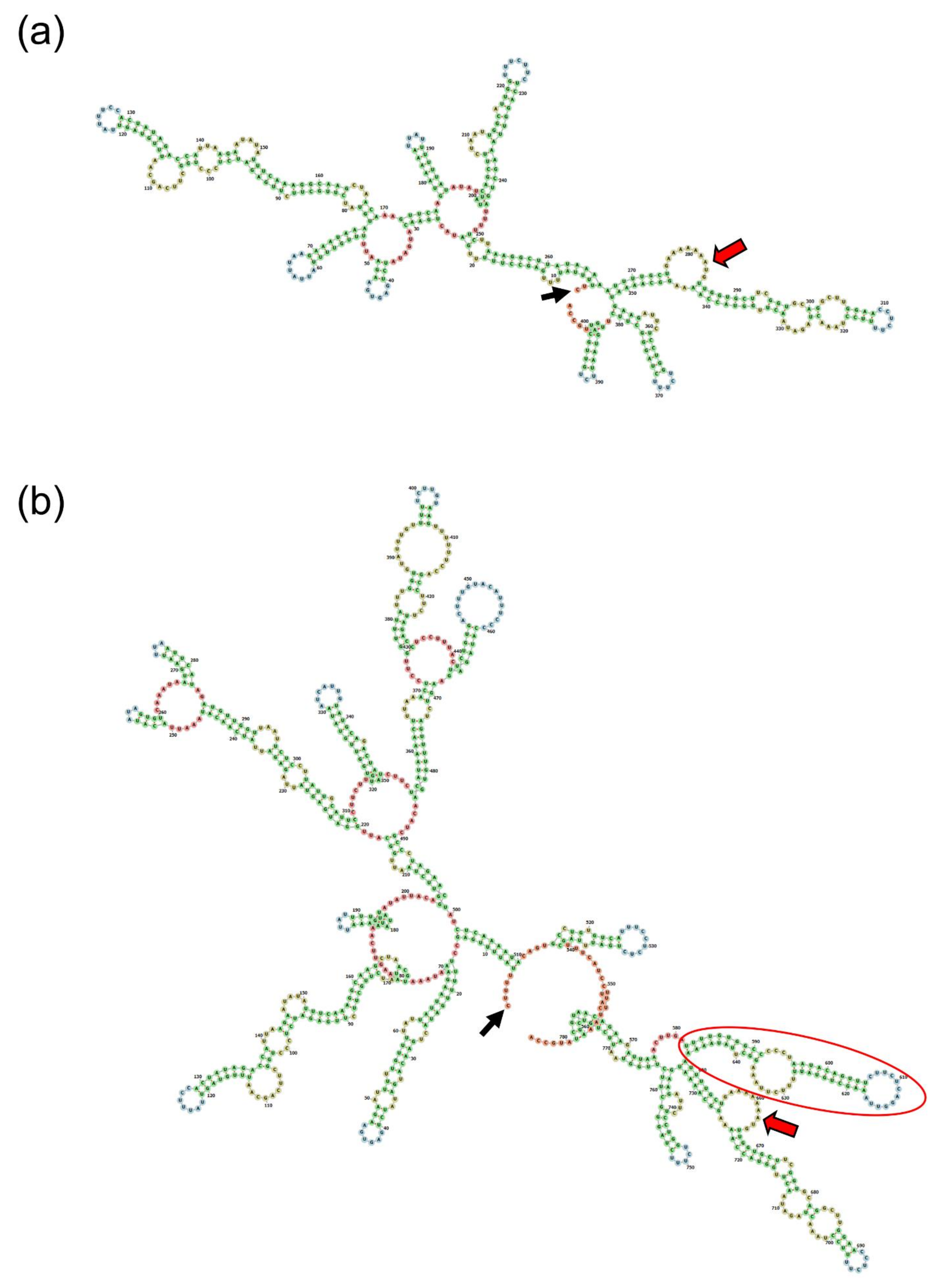
2.7. Prediction of Secondary Structures and Translation of the Wild-Type and Mutant ENAM mRNA
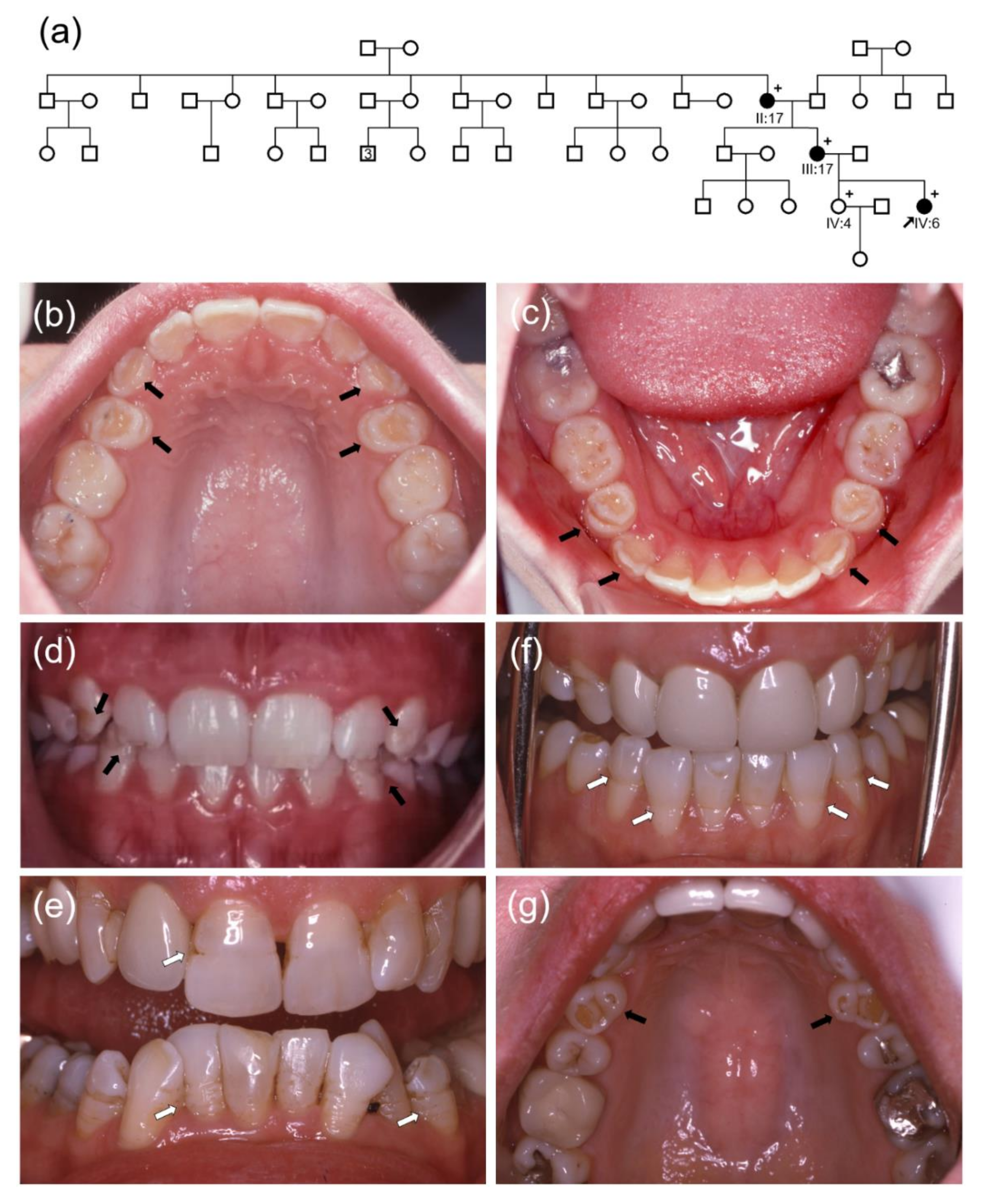
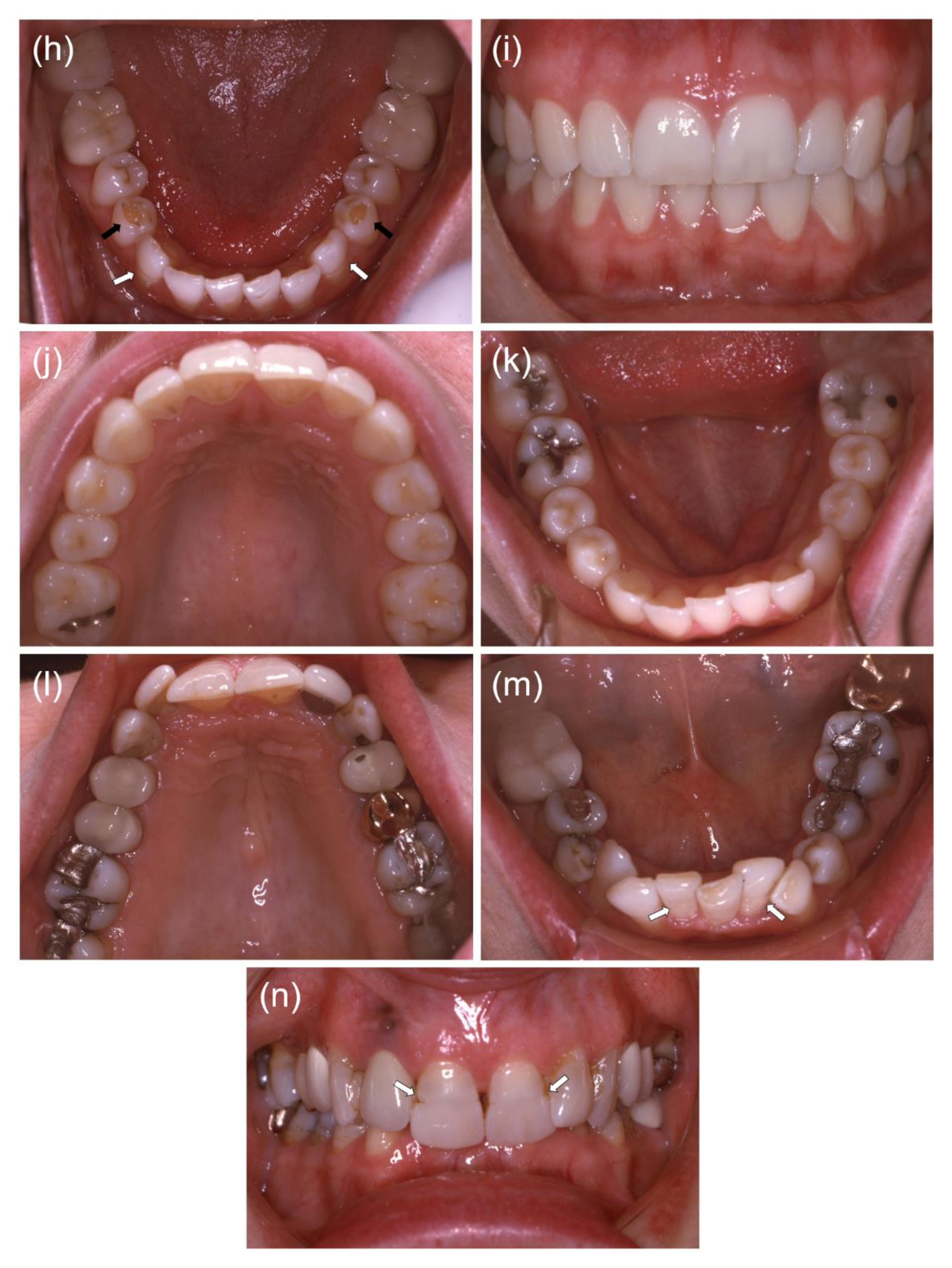
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Simmer, J.P.; Fincham, A.G. Molecular mechanisms of dental enamel formation. Crit. Rev. Oral Biol. Med. 1995, 6, 84–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, M.K.; Laouina, S.; El Alloussi, M.; Dollfus, H.; Bloch-Zupan, A. Amelogenesis Imperfecta: 1 Family, 2 Phenotypes, and 2 Mutated Genes. J. Dent. Res. 2016, 95, 1457–1463. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Kang, J.; Seymen, F.; Koruyucu, M.; Zhang, H.; Kasimoglu, Y.; Bayram, M.; Tuna-Ince, E.B.; Bayrak, S.; Tuloglu, N.; et al. Alteration of Exon Definition Causes Amelogenesis Imperfecta. J. Dent. Res. 2020, 99, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Lagerstrom, M.; Dahl, N.; Nakahori, Y.; Nakagome, Y.; Backman, B.; Landegren, U.; Pettersson, U. A deletion in the amelogenin gene (AMG) causes X-linked amelogenesis imperfecta (AIH1). Genomics 1991, 10, 971–975. [Google Scholar] [CrossRef]
- Rajpar, M.H.; Harley, K.; Laing, C.; Davies, R.M.; Dixon, M.J. Mutation of the gene encoding the enamel-specific protein, enamelin, causes autosomal-dominant amelogenesis imperfecta. Hum. Mol. Genet. 2001, 10, 1673–1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulter, J.A.; El-Sayed, W.; Shore, R.C.; Kirkham, J.; Inglehearn, C.F.; Mighell, A.J. Whole-exome sequencing, without prior linkage, identifies a mutation in LAMB3 as a cause of dominant hypoplastic amelogenesis imperfecta. Eur. J. Hum. Genet. 2014, 22, 132–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulter, J.A.; Murillo, G.; Brookes, S.J.; Smith, C.E.; Parry, D.A.; Silva, S.; Kirkham, J.; Inglehearn, C.F.; Mighell, A.J. Deletion of ameloblastin exon 6 is associated with amelogenesis imperfecta. Hum. Mol. Genet. 2014, 23, 5317–5324. [Google Scholar] [CrossRef] [Green Version]
- Seymen, F.; Kim, Y.J.; Lee, Y.J.; Kang, J.; Kim, T.H.; Choi, H.; Koruyucu, M.; Kasimoglu, Y.; Tuna, E.B.; Gencay, K.; et al. Recessive Mutations in ACPT, Encoding Testicular Acid Phosphatase, Cause Hypoplastic Amelogenesis Imperfecta. Am. J. Hum. Genet. 2016, 99, 1199–1205. [Google Scholar] [CrossRef] [Green Version]
- Sundell, S.; Valentin, J. Hereditary aspects and classification of hereditary amelogenesis imperfecta. Community Dent. Oral Epidemiol. 1986, 14, 211–216. [Google Scholar] [CrossRef]
- Lee, K.E.; Ko, J.; Le, C.G.; Shin, T.J.; Hyun, H.K.; Lee, S.H.; Kim, J.W. Novel LAMB3 mutations cause non-syndromic amelogenesis imperfecta with variable expressivity. Clin. Genet. 2015, 87, 90–92. [Google Scholar] [CrossRef]
- Koruyucu, M.; Kang, J.; Kim, Y.J.; Seymen, F.; Kasimoglu, Y.; Lee, Z.H.; Shin, T.J.; Hyun, H.K.; Kim, Y.J.; Lee, S.H.; et al. Hypoplastic AI with Highly Variable Expressivity Caused by ENAM Mutations. J. Dent. Res. 2018, 97, 1064–1069. [Google Scholar] [CrossRef]
- Kida, M.; Ariga, T.; Shirakawa, T.; Oguchi, H.; Sakiyama, Y. Autosomal-dominant hypoplastic form of amelogenesis imperfecta caused by an enamelin gene mutation at the exon-intron boundary. J. Dent. Res. 2002, 81, 738–742. [Google Scholar] [CrossRef]
- Kim, J.W.; Seymen, F.; Lin, B.P.; Kiziltan, B.; Gencay, K.; Simmer, J.P.; Hu, J.C. ENAM mutations in autosomal-dominant amelogenesis imperfecta. J. Dent. Res. 2005, 84, 278–282. [Google Scholar] [CrossRef]
- Seymen, F.; Lee, K.E.; Koruyucu, M.; Gencay, K.; Bayram, M.; Tuna, E.B.; Lee, Z.H.; Kim, J.W. ENAM Mutations with Incomplete Penetrance. J. Dent. Res. 2014, 93, 988–992. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.W.; Zhang, H.; Seymen, F.; Koruyucu, M.; Hu, Y.; Kang, J.; Kim, Y.J.; Ikeda, A.; Kasimoglu, Y.; Bayram, M.; et al. Mutations in RELT cause autosomal recessive amelogenesis imperfecta. Clin. Genet. 2019, 95, 375–383. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 3. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Hu, J.C.; Chun, Y.H.; Al Hazzazzi, T.; Simmer, J.P. Enamel formation and amelogenesis imperfecta. Cells Tissues Organs 2007, 186, 78–85. [Google Scholar] [CrossRef]
- Hu, J.C.; Yamakoshi, Y. Enamelin and autosomal-dominant amelogenesis imperfecta. Crit. Rev. Oral. Biol. Med. 2003, 14, 387–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawasaki, K.; Weiss, K.M. Mineralized tissue and vertebrate evolution: The secretory calcium-binding phosphoprotein gene cluster. Proc. Natl. Acad. Sci. USA 2003, 100, 4060–4065. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.C.; Hart, T.C.; Dupont, B.R.; Chen, J.J.; Sun, X.; Qian, Q.; Zhang, C.H.; Jiang, H.; Mattern, V.L.; Wright, J.T.; et al. Cloning human enamelin cDNA, chromosomal localization, and analysis of expression during tooth development. J. Dent. Res. 2000, 79, 912–919. [Google Scholar] [CrossRef]
- Hu, C.-C.; Fukae, M.; Uchida, T.; Qian, Q.; Zhang, C.H.; Ryu, O.H.; Tanabe, T.; Yamakoshi, Y.; Murakami, C.; Dohi, N.; et al. Cloning and characterization of porcine enamelin mRNAs. J. Dent. Res. 1997, 76, 1720–1729. [Google Scholar] [CrossRef]
- Hu, C.-C.; Simmer, J.P.; Bartlett, J.D.; Qian, Q.; Zhang, C.; Ryu, O.H.; Xue, J.; Fukae, M.; Uchida, T.; MacDougall, M.J. Murine enamelin: cDNA and derived protein sequences. Connect. Tissue Res. 1998, 39, 351–365. [Google Scholar] [CrossRef]
- Hu, J.C.-C.; Zhang, C.H.; Ying, Y.; Simmer, J.P. Cloning and Characterization of the Mouse and Human Enamelin Genes. J. Dent. Res. 2001, 80, 898–902. [Google Scholar] [CrossRef]
- Leppek, K.; Das, R.; Barna, M. Functional 5′ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat. Rev. Mol. Cell Biol. 2018, 19, 158–174. [Google Scholar] [CrossRef]
- Jacob, A.G.; Smith, C.W.J. Intron retention as a component of regulated gene expression programs. Hum. Genet. 2017, 136, 1043–1057. [Google Scholar] [CrossRef] [Green Version]
- Hinnebusch, A.G.; Ivanov, I.P.; Sonenberg, N. Translational control by 5′-untranslated regions of eukaryotic mRNAs. Science 2016, 352, 1413–1416. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.J.; Lee, Y.; Zhang, H.; Wright, J.T.; Simmer, J.P.; Hu, J.C.-C.; Kim, J.-W. Translational Attenuation by an Intron Retention in the 5′ UTR of ENAM Causes Amelogenesis Imperfecta. Biomedicines 2021, 9, 456. https://doi.org/10.3390/biomedicines9050456
Kim YJ, Lee Y, Zhang H, Wright JT, Simmer JP, Hu JC-C, Kim J-W. Translational Attenuation by an Intron Retention in the 5′ UTR of ENAM Causes Amelogenesis Imperfecta. Biomedicines. 2021; 9(5):456. https://doi.org/10.3390/biomedicines9050456
Chicago/Turabian StyleKim, Youn Jung, Yejin Lee, Hong Zhang, John Timothy Wright, James P. Simmer, Jan C.-C. Hu, and Jung-Wook Kim. 2021. "Translational Attenuation by an Intron Retention in the 5′ UTR of ENAM Causes Amelogenesis Imperfecta" Biomedicines 9, no. 5: 456. https://doi.org/10.3390/biomedicines9050456
APA StyleKim, Y. J., Lee, Y., Zhang, H., Wright, J. T., Simmer, J. P., Hu, J. C.-C., & Kim, J.-W. (2021). Translational Attenuation by an Intron Retention in the 5′ UTR of ENAM Causes Amelogenesis Imperfecta. Biomedicines, 9(5), 456. https://doi.org/10.3390/biomedicines9050456