
Combination of Mesenchymal Stem Cell-Delivered Oncolytic Virus with Prodrug Activation Increases Efficacy and Safety of Colorectal Cancer Therapy

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Flow Cytometry
2.3. In Vitro Migration Assay
2.4. Adenovirus Infection
2.5. Combination Cytotoxicity Assay
2.6. Genomic DNA Extraction
2.7. PCR
2.8. Quantitative Real-Time PCR
2.9. Human Xenograft HT29 Colorectal Cancer Model
2.10. Immunohistochemistry
2.11. UPLC/MS-QTOF
2.11.1. Sample Preparation
2.11.2. Chromatographic System
2.11.3. Mobile Phase Solutions
2.12. Pharmacokinetics of CRAdNTR in Venous Blood
2.13. Statistical Analysis
3. Results
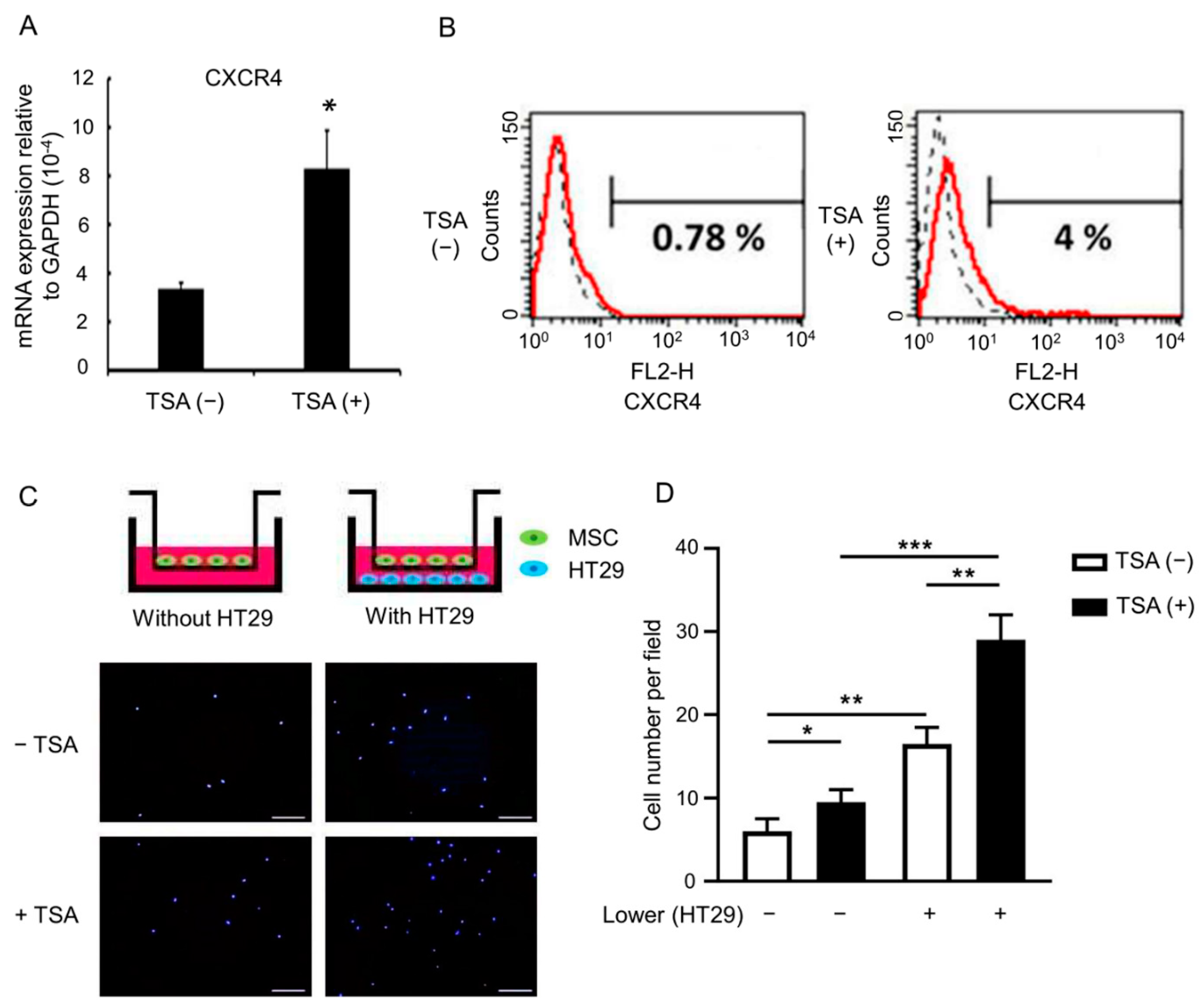
3.1. Trichostatin A (TSA)-Primed MSCs Have a Normal MSC Surface Phenotype
3.2. TSA Enhances MSC Tumor Tropism
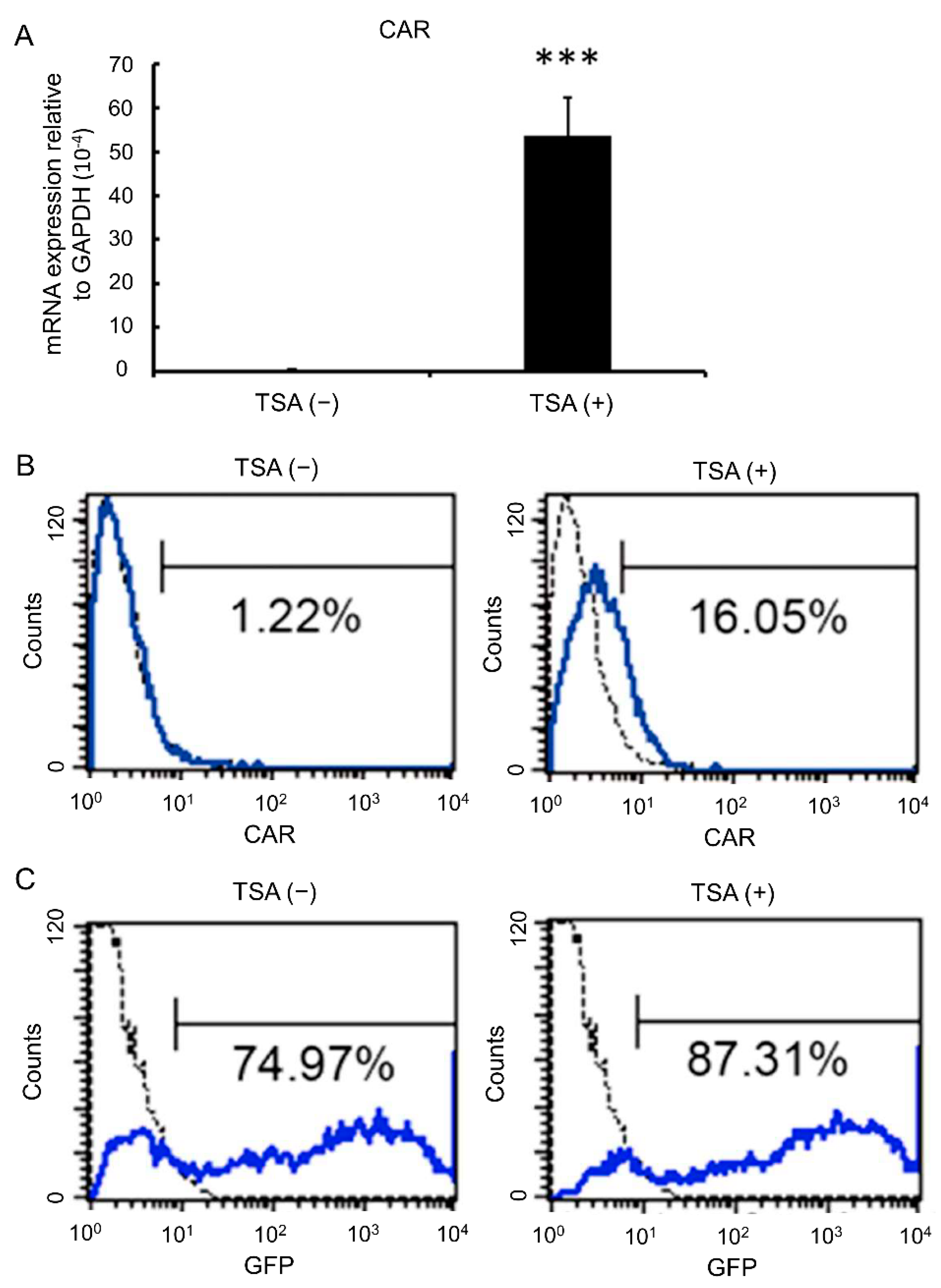
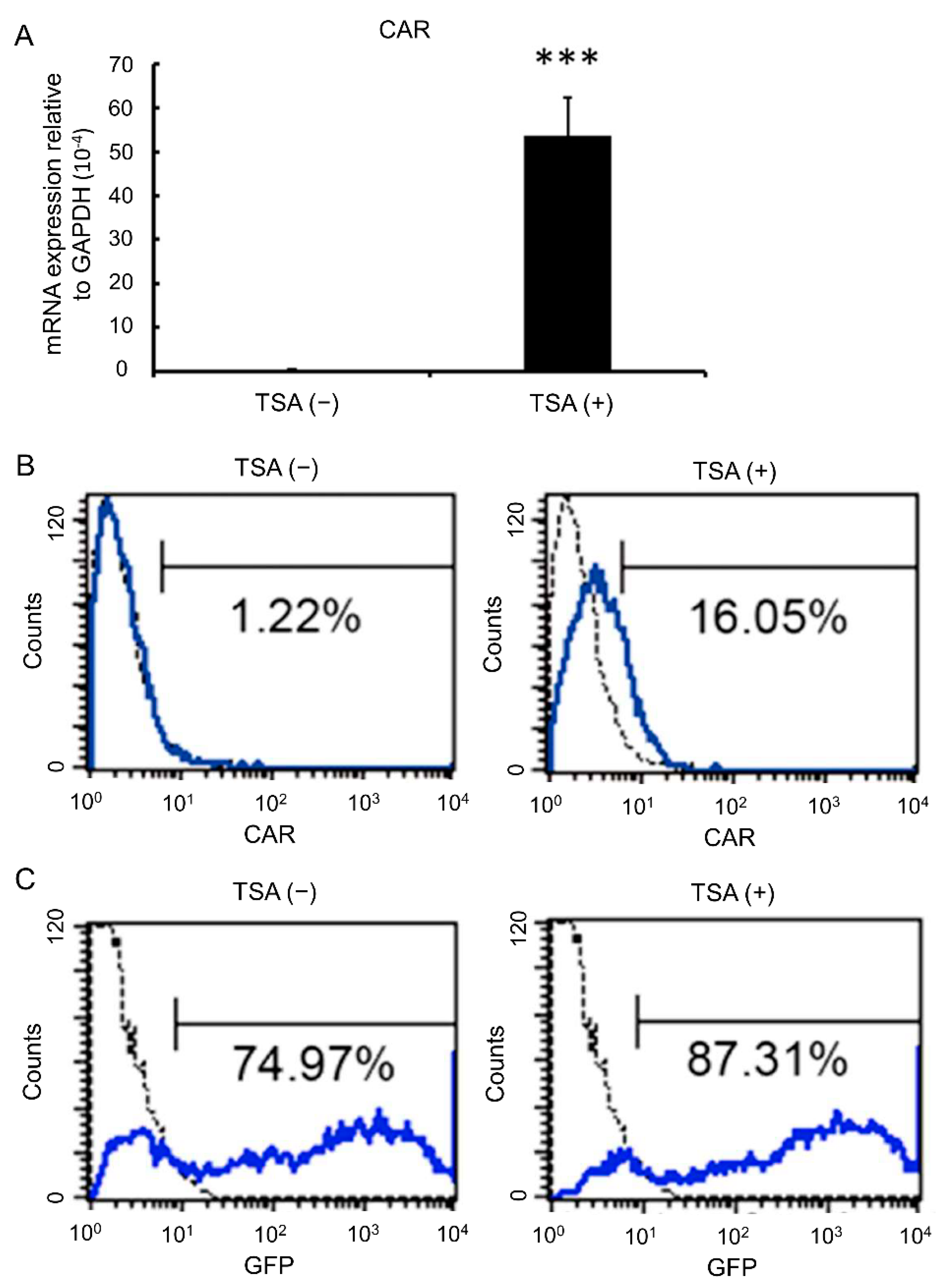
3.3. TSA Enhances CAR Expression and Adenovirus Infection Rates
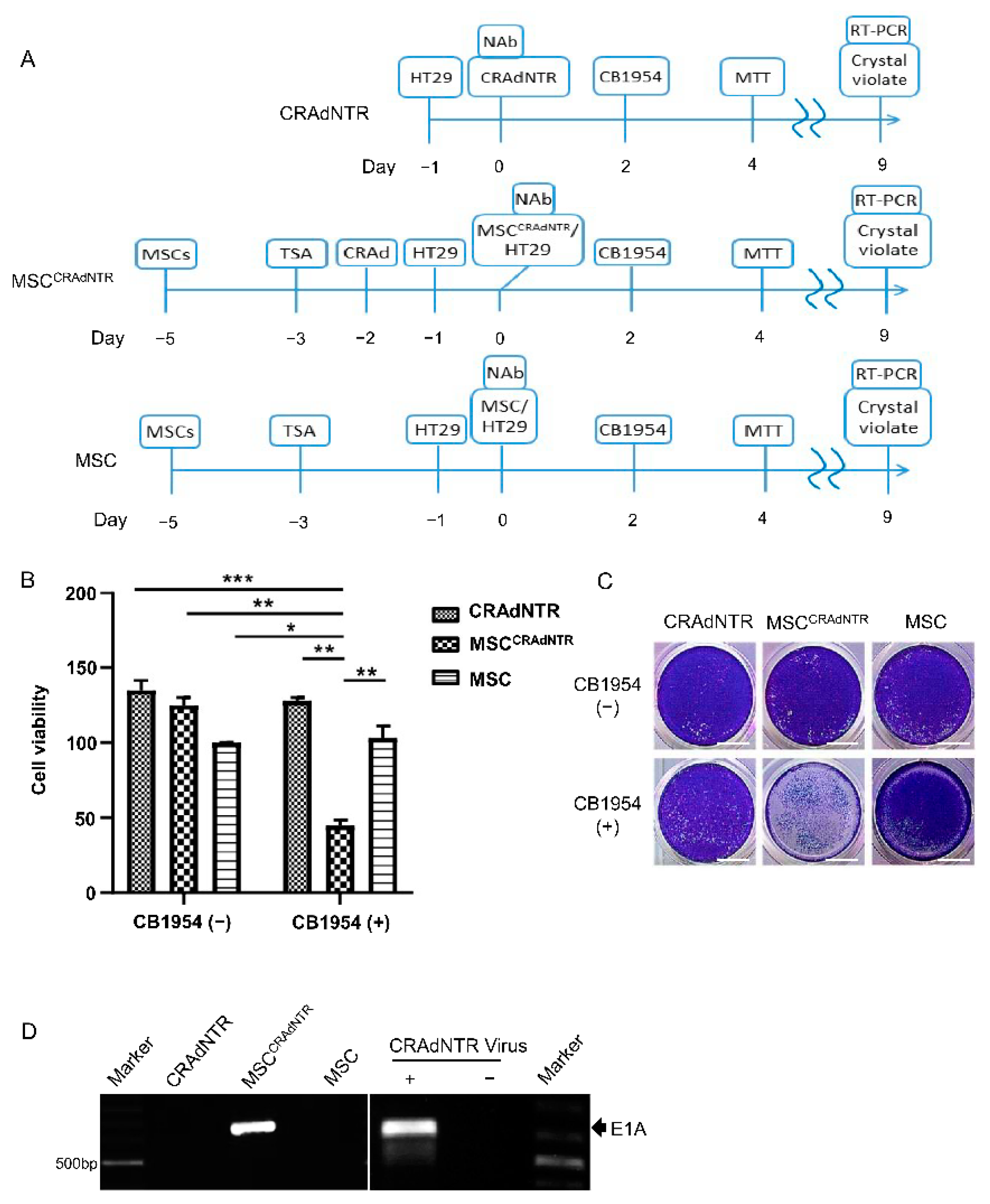
3.4. Combination of MSC-Delivered CRAdNTR with a Prodrug Induces Cytotoxicity in Colorectal Cancer Cells in the Presence of NAb In Vitro
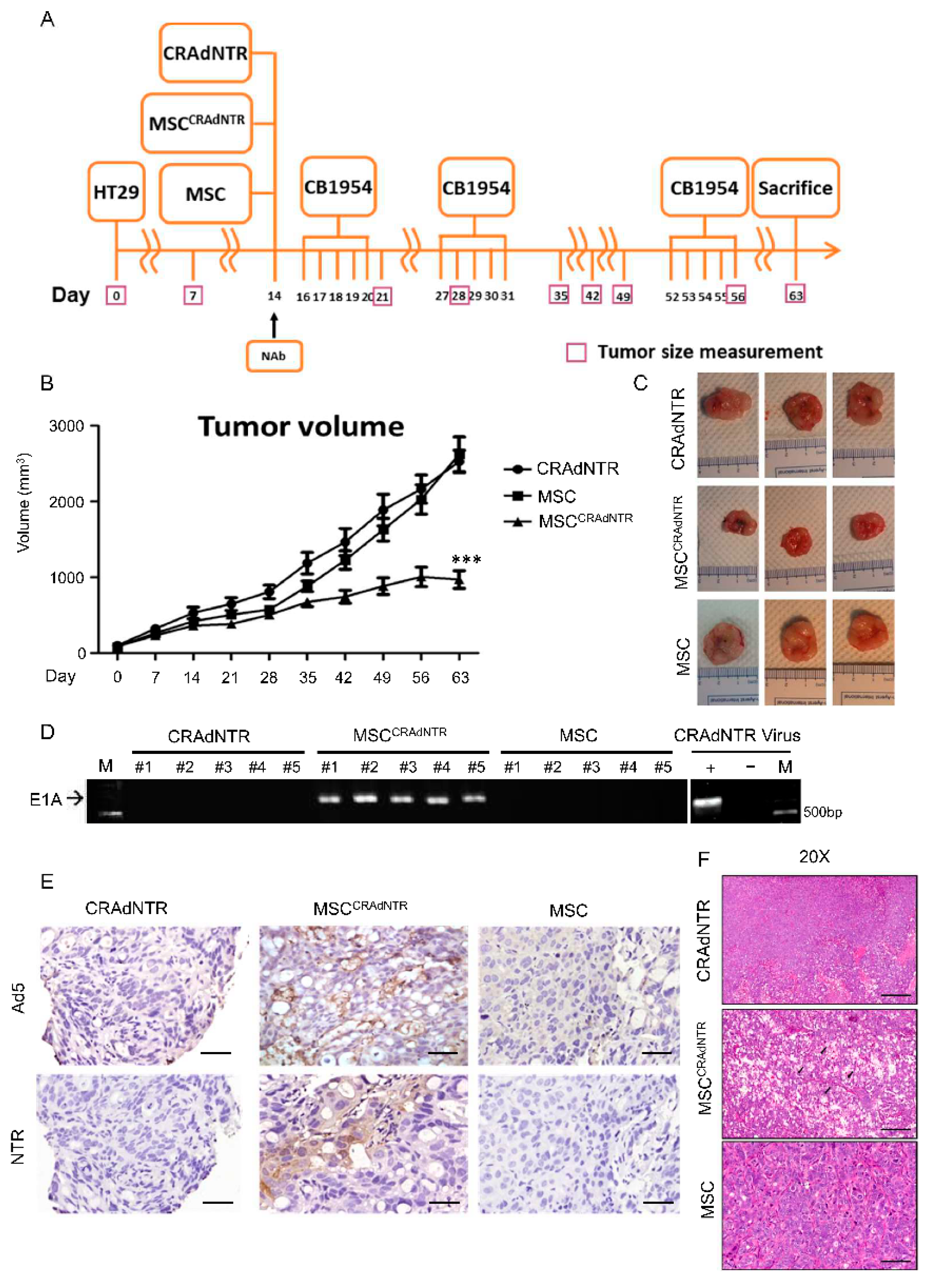
3.5. MSC-Delivered CRAdNTR Combined with a Prodrug Reduces Colorectal Cancer Growth in the Presence of NAb In Vivo
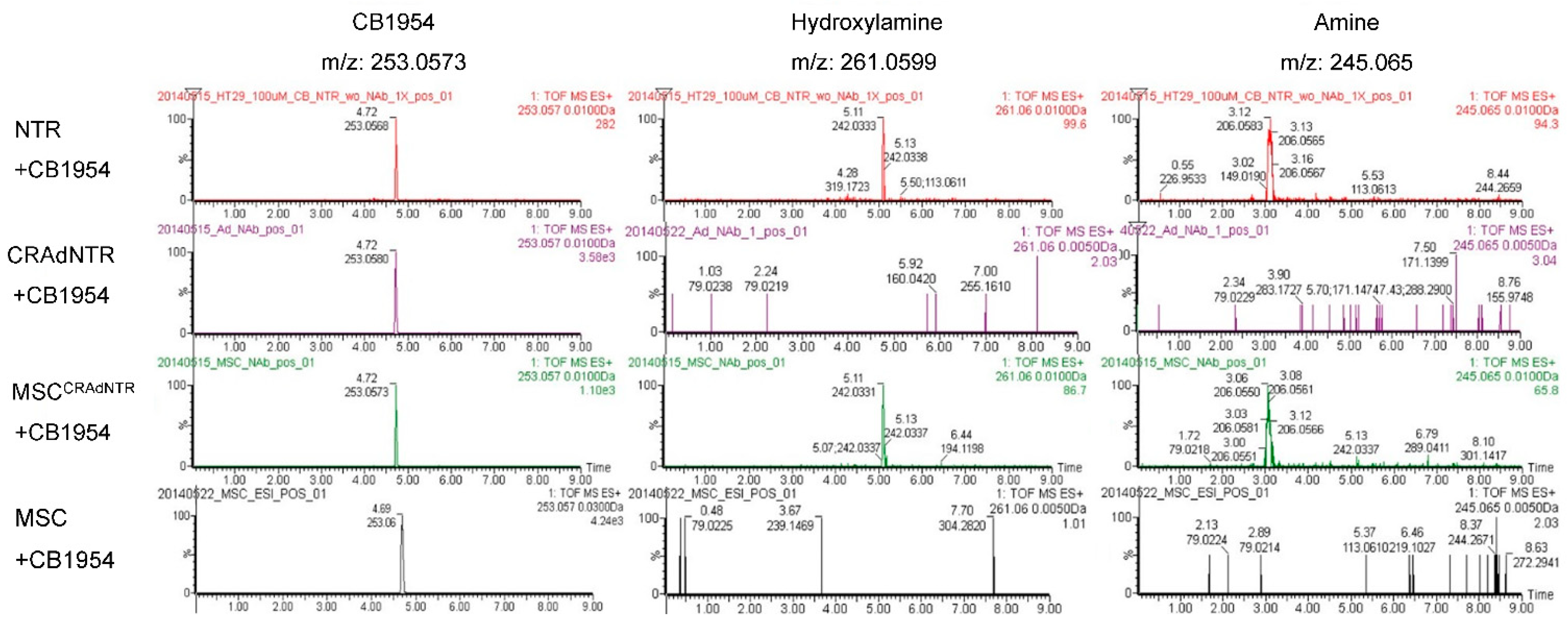
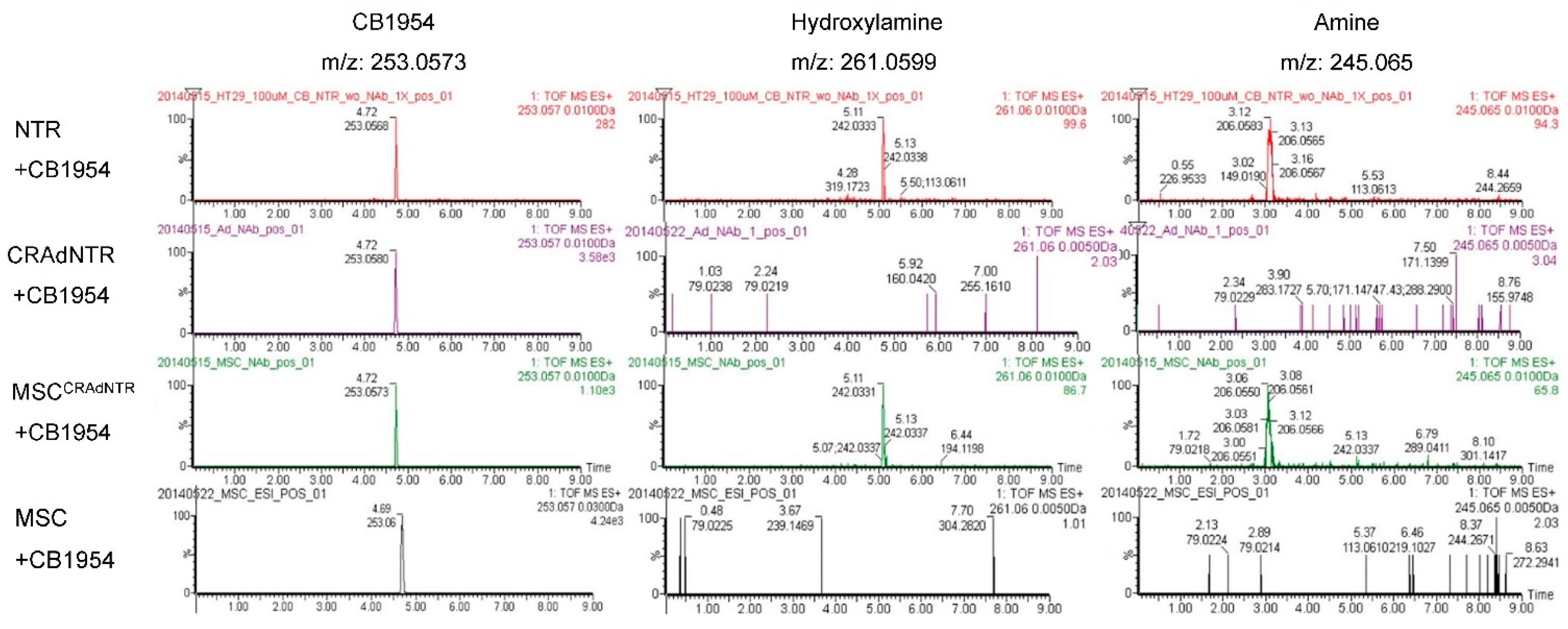
3.6. MSC-Delivered CRAdNTR Combined with a Prodrug Induces Cytotoxicity in Colorectal Cancer Cells In Vitro via the Generation of Cytotoxic Metabolites
3.7. Cytotoxic Metabolites from CB1954 Specifically Exist in the Tumor Area Rather than in Other Vital Organs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Ad-5 | Adenovirus type 5 |
| CRAd | Conditionally replicative adenovirus |
| FBS | Fetal bovine serum |
| GDEPT | Gene-directed enzyme-prodrug therapy |
| i.p. | Intraperitoneal |
| MSC | Mesenchymal stem cell |
| NAb | Neutralized antibody |
| NTR | Nitroreductase |
| s.c. | Subcutaneous |
| SPE | Solid-phase extraction |
| TSA | Trichostatin A |
| qTOF | Quadrupole-time-of-flight |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, E.; Nemunaitis, J. Oncolytic viral therapies. Cancer Gene Ther. 2004, 11, 643–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; Gomez-Manzano, C.; Lang, F.F.; Alemany, R.; Fueyo, J. Oncolytic adenovirus: Preclinical and clinical studies in patients with human malignant gliomas. Curr. Gene Ther. 2009, 9, 422–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergelson, J.M.; Cunningham, J.A.; Droguett, G.; Kurt-Jones, E.A.; Krithivas, A.; Hong, J.S.; Horwitz, M.S.; Crowell, R.L.; Finberg, R.W. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science 1997, 275, 1320–1323. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.C.; Lu, C.Y.; Shyue, S.K.; Liu, H.C.; Ho, L.L. Lineage differentiation-associated loss of adenoviral susceptibility and Coxsackie-adenovirus receptor expression in human mesenchymal stem cells. Stem Cells 2004, 22, 1321–1329. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Dmitriev, I.; O’Malley, J.P.; Wang, M.; Saddekni, S.; You, Z.; Preuss, M.A.; Harris, R.D.; Aurigemma, R.; Siegal, G.P.; et al. A phase I clinical trial of Ad5.SSTR/TK.RGD, a novel infectivity-enhanced bicistronic adenovirus, in patients with recurrent gynecologic cancer. Clin. Cancer Res. 2012, 18, 3440–3451. [Google Scholar] [CrossRef] [Green Version]
- Kirn, D. Clinical research results with dl1520 (Onyx-015), a replication-selective adenovirus for the treatment of cancer: What have we learned? Gene Ther. 2001, 8, 89–98. [Google Scholar] [CrossRef] [Green Version]
- Ries, S.J.; Brandts, C.H.; Chung, A.S.; Biederer, C.H.; Hann, B.C.; Lipner, E.M.; McCormick, F.; Korn, W.M. Loss of p14ARF in tumor cells facilitates replication of the adenovirus mutant dl1520 (ONYX-015). Nat. Med. 2000, 6, 1128–1133. [Google Scholar] [CrossRef]
- Bischoff, J.R.; Kirn, D.H.; Williams, A.; Heise, C.; Horn, S.; Muna, M.; Ng, L.; Nye, J.A.; Sampson-Johannes, A.; Fattaey, A.; et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science 1996, 274, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Huebner, R.J.; Rowe, W.P.; Schatten, W.E.; Smith, R.R.; Thomas, L.B. Studies on the use of viruses in the treatment of carcinoma of the cervix. Cancer 1956, 9, 1211–1218. [Google Scholar] [PubMed]
- Wong, H.H.; Lemoine, N.R.; Wang, Y. Oncolytic Viruses for Cancer Therapy: Overcoming the Obstacles. Viruses 2010, 2, 78–106. [Google Scholar] [CrossRef]
- Hung, S.-C.; Deng, W.-P.; Yang, W.K.; Liu, R.-S.; Lee, C.-C.; Su, T.-C.; Lin, R.-J.; Yang, D.-M.; Chang, C.-W.; Chen, W.-H.; et al. Mesenchymal stem cell targeting of microscopic tumors and tumor stroma development monitored by noninvasive in vivo positron emission tomography imaging. Clin. Cancer Res. 2005, 11, 7749–7756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klauzinska, M.; Castro, N.P.; Rangel, M.C.; Spike, B.T.; Gray, P.C.; Bertolette, D.; Cuttitta, F.; Salomon, D. The multifaceted role of the embryonic gene Cripto-1 in cancer, stem cells and epithelial-mesenchymal transition. Semin. Cancer Biol. 2014, 29, 51–58. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Zhang, M.; Fu, L.; Lin, J.; Zhou, X.; Zhou, P.; Huang, P.; Hu, H.; Han, Y. Liver-targeted delivery of TSG-6 by calcium phosphate nanoparticles for the management of liver fibrosis. Theranostics 2020, 10, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.F.; Chen, M.J.; Wu, M.H.; Hung, S.C. The use of hypoxic cultured mesenchymal stem cell for oncolytic virus therapy. Cancer Gene Ther. 2013, 20, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Wynn, R.F.; Hart, C.A.; Corradi-Perini, C.; O’Neill, L.; Evans, C.A.; Wraith, J.E.; Fairbairn, L.J.; Bellantuono, I. A small proportion of mesenchymal stem cells strongly expresses functionally active CXCR4 receptor capable of promoting migration to bone marrow. Blood 2004, 104, 2643–2645. [Google Scholar] [CrossRef]
- Hung, S.-C.; Pochampally, R.R.; Hsu, S.-C.; Sanchez, C.; Chen, S.-C.; Spees, J.; Prockop, D.J. Short-term exposure of multipotent stromal cells to low oxygen increases their expression of CX3CR1 and CXCR4 and their engraftment in vivo. PLoS ONE 2007, 2, e416. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-C.; Chen, Y.-J.; Yew, T.-L.; Chen, L.-L.; Wang, J.-Y.; Chiu, C.-H.; Hung, S.-C. Hypoxia inhibits senescence and maintains mesenchymal stem cell properties through down-regulation of E2A-p21 by HIF-TWIST. Blood 2011, 117, 459–469. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.-H.; Chen, H.-L.; Yew, T.-L.; Lin, M.-W.; Hung, S.-C. Hypoxic mesenchymal stem cells engraft and ameliorate limb ischaemia in allogeneic recipients. Cardiovasc. Res. 2014, 101, 266–276. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; McLeod, H.L. Strategies for enzyme/prodrug cancer therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2001, 7, 3314–3324. [Google Scholar]
- Chung-Faye, G.; Palmer, D.; Anderson, D.; Clark, J.; Downes, M.; Baddeley, J.; Hussain, S.; Murray, P.I.; Searle, P.; Seymour, L.; et al. Virus-directed, enzyme prodrug therapy with nitroimidazole reductase: A phase I and pharmacokinetic study of its prodrug, CB1954. Clin. Cancer Res. 2001, 7, 2662–2668. [Google Scholar] [PubMed]
- Tietze, L.F.; Schmuck, K. Prodrugs for targeted tumor therapies: Recent developments in ADEPT, GDEPT and PMT. Curr. Pharm. Des. 2011, 17, 3527–3547. [Google Scholar] [CrossRef]
- Wu, P.; Wang, J.; Chen, C.; Chao, K.; Chang, M.; Chen, W.-M.; Hung, S. Early Passage Mesenchymal Stem Cells Display Decreased Radiosensitivity and Increased DNA Repair Activity. Stem Cells Transl. Med. 2017, 6, 1504–1514. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, N.R.; Rowan, A.; Smith, M.E.; Kerr, I.B.; Bodmer, W.F.; Gannon, J.V.; Lane, D.P. p53 mutations in colorectal cancer. Proc. Natl. Acad. Sci. USA 1990, 87, 7555–7559. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Gul, H.; Marquez-Curtis, L.A.; Lo, J.; Jahroudi, N.; Turner, A.R.; Larratt, L.M.; Janowska-Wieczorek, A. The Potent Deacetylase Inhibitor Trichostatin a (TSA) Increases CXCR4 Expression in Hematopoietic Stem/Progenitor Cells by Chromatin Remodelling. ASH Annu. Meet. Abstr. 2008, 112, 3487. [Google Scholar] [CrossRef]
- Sachs, M.D.; Ramamurthy, M.; van der Poel, H.; Wickham, T.J.; Lamfers, M.; Gerritsen, W.; Chowdhury, W.; Li, Y.; Schoenberg, M.P.; Rodriguez, R. Histone deacetylase inhibitors upregulate expression of the coxsackie adenovirus receptor (CAR) preferentially in bladder cancer cells. Cancer Gene Ther. 2004, 11, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Deak, E.; Seifried, E.; Henschler, R. Homing pathways of mesenchymal stromal cells (MSCs) and their role in clinical applications. Int. Rev. Immunol. 2010, 29, 514–529. [Google Scholar] [CrossRef]
- Kang, S.K.; Shin, I.S.; Ko, M.S.; Jo, J.Y.; Ra, J.C. Journey of mesenchymal stem cells for homing: Strategies to enhance efficacy and safety of stem cell therapy. Stem Cells Int. 2012, 2012, 342968. [Google Scholar] [CrossRef] [Green Version]
- Lourenco, S.; Teixeira, V.H.; Kalber, T.; Jose, R.J.; Floto, R.A.; Janes, S.M. Macrophage migration inhibitory factor-CXCR4 is the dominant chemotactic axis in human mesenchymal stem cell recruitment to tumors. J. Immunol. 2015, 194, 3463–3474. [Google Scholar] [CrossRef] [Green Version]
- Gul, H.; Marquez-Curtis, L.A.; Jahroudi, N.; Lo, J.; Turner, A.R.; Janowska-Wieczorek, A. Valproic acid increases CXCR4 expression in hematopoietic stem/progenitor cells by chromatin remodeling. Stem Cells Dev. 2009, 18, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, D.; Eide, P.W.; Eilertsen, I.A.; Danielsen, S.A.; Eknaes, M.; Hektoen, M.; Lind, G.E.; Lothe, R.A. Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis 2013, 2, e71. [Google Scholar] [CrossRef] [PubMed]
- Grove, J.I.; Searle, P.F.; Weedon, S.J.; Green, N.K.; McNeish, I.A.; Kerr, D.J. Virus-directed enzyme prodrug therapy using CB1954. Anticancer Drug Des. 1999, 14, 461–472. [Google Scholar]
- Buijs, P.R.; Verhagen, J.H.; van Eijck, C.H.; van den Hoogen, B.G. Oncolytic viruses: From bench to bedside with a focus on safety. Hum. Vaccin. Immunother. 2015, 11, 1573–1584. [Google Scholar] [CrossRef] [Green Version]
- Dominici, M.; le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Gurgul, A.; Opiela, J.; Pawlina, K.; Szmatola, T.; Bochenek, M.; Bugno-Poniewierska, M. The effect of histone deacetylase inhibitor trichostatin A on porcine mesenchymal stem cell transcriptome. Biochimie 2017, 139, 56–73. [Google Scholar] [CrossRef] [PubMed]
- Samiec, M.; Opiela, J.; Lipiński, D.; Romanek, J. Trichostatin A-mediated epigenetic transformation of adult bone marrow-derived mesenchymal stem cells biases the in vitro developmental capability, quality, and pluripotency extent of porcine cloned embryos. BioMed Res. Int. 2015, 2015, 814686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalimuthu, S.; Oh, J.M.; Gangadaran, P.; Zhu, L.; Lee, H.W.; Rajendran, R.L.; Baek, S.H.; Jeon, Y.H.; Jeong, S.Y.; Lee, S.-W.; et al. In Vivo Tracking of Chemokine Receptor CXCR4-Engineered Mesenchymal Stem Cell Migration by Optical Molecular Imaging. Stem Cells Int. 2017, 2017, 8085637. [Google Scholar] [CrossRef]
- Jeun, S.-S.; Park, S.A.; Ryu, C.H.; Kim, S.M.; Lim, J.Y.; Park, S.I.; Jeong, C.H.; Jun, J.A.; Oh, J.H.; Park, S.H.; et al. CXCR4-transfected human umbilical cord blood-derived mesenchymal stem cells exhibit enhanced migratory capacity toward gliomas. Int. J. Oncol. 2011, 38, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Stoff-Khalili, M.A.; Rivera, A.A.; Mathis, J.M.; Banerjee, N.S.; Moon, A.S.; Hess, A.; Rocconi, R.P.; Numnum, T.M.; Everts, M.; Chow, L.T.; et al. Mesenchymal stem cells as a vehicle for targeted delivery of CRAds to lung metastases of breast carcinoma. Breast Cancer Res. Treat. 2007, 105, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Nakao, S.; Arai, Y.; Tasaki, M.; Yamashita, M.; Murakami, R.; Kawase, T.; Amino, N.; Nakatake, M.; Kurosaki, H.; Mori, M.; et al. Intratumoral expression of IL-7 and IL-12 using an oncolytic virus increases systemic sensitivity to immune checkpoint blockade. Sci. Transl. Med. 2020, 12, eaax7992. [Google Scholar] [CrossRef] [PubMed]
- Saha, D.; Martuza, R.L.; Rabkin, S.D. Macrophage Polarization Contributes to Glioblastoma Eradication by Combination Immunovirotherapy and Immune Checkpoint Blockade. Cancer Cell 2017, 32, 253–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ho, C.-T.; Wu, M.-H.; Chen, M.-J.; Lin, S.-P.; Yen, Y.-T.; Hung, S.-C. Combination of Mesenchymal Stem Cell-Delivered Oncolytic Virus with Prodrug Activation Increases Efficacy and Safety of Colorectal Cancer Therapy. Biomedicines 2021, 9, 548. https://doi.org/10.3390/biomedicines9050548
Ho C-T, Wu M-H, Chen M-J, Lin S-P, Yen Y-T, Hung S-C. Combination of Mesenchymal Stem Cell-Delivered Oncolytic Virus with Prodrug Activation Increases Efficacy and Safety of Colorectal Cancer Therapy. Biomedicines. 2021; 9(5):548. https://doi.org/10.3390/biomedicines9050548
Chicago/Turabian StyleHo, Chun-Te, Mei-Hsuan Wu, Ming-Jen Chen, Shih-Pei Lin, Yu-Ting Yen, and Shih-Chieh Hung. 2021. "Combination of Mesenchymal Stem Cell-Delivered Oncolytic Virus with Prodrug Activation Increases Efficacy and Safety of Colorectal Cancer Therapy" Biomedicines 9, no. 5: 548. https://doi.org/10.3390/biomedicines9050548
APA StyleHo, C.-T., Wu, M.-H., Chen, M.-J., Lin, S.-P., Yen, Y.-T., & Hung, S.-C. (2021). Combination of Mesenchymal Stem Cell-Delivered Oncolytic Virus with Prodrug Activation Increases Efficacy and Safety of Colorectal Cancer Therapy. Biomedicines, 9(5), 548. https://doi.org/10.3390/biomedicines9050548