
Functional Characterization of the Dopaminergic Psychostimulant Sydnocarb as an Allosteric Modulator of the Human Dopamine Transporter

 , , and
, , and 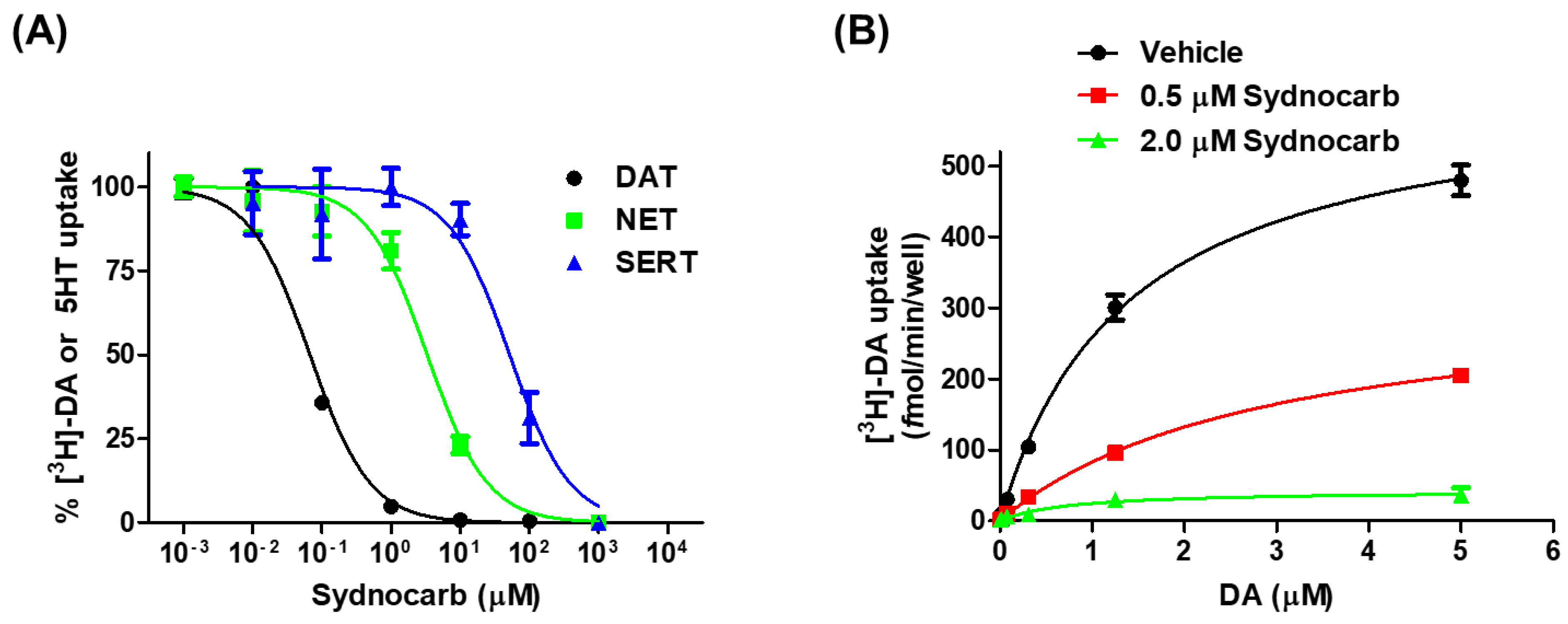{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Drugs
2.2. Structural Models of WT DAT in the OF and IF States
2.3. Force Field Parameters for Sydnocarb and KM822
2.4. Docking Simulations
2.5. MD Simulations
2.6. Site-Directed Mutagenesis, Cell Culture, and Transfections
2.7. Transport Kinetic Assays Using COS-7 Cells
2.8. Transport Inhibition Assays Using COS-7 Cells
2.9. Biotinylation
2.10. Data Analysis
3. Results and Discussion
3.1. Pharmacology of Sydnocarb Reveals the High Potency and Selectivity of Sydnocarb as a DAT Noncompetitive Inhibitor
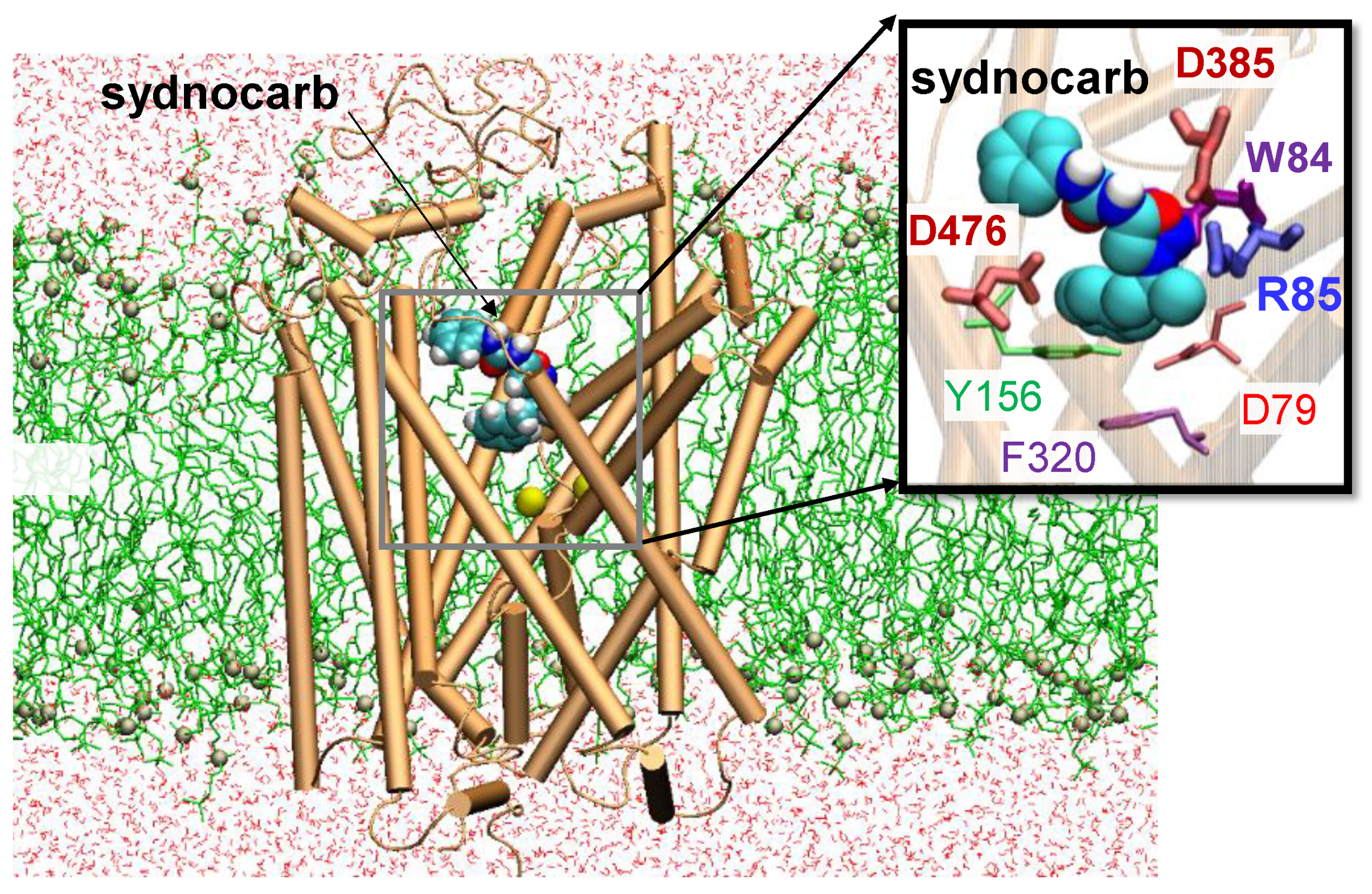
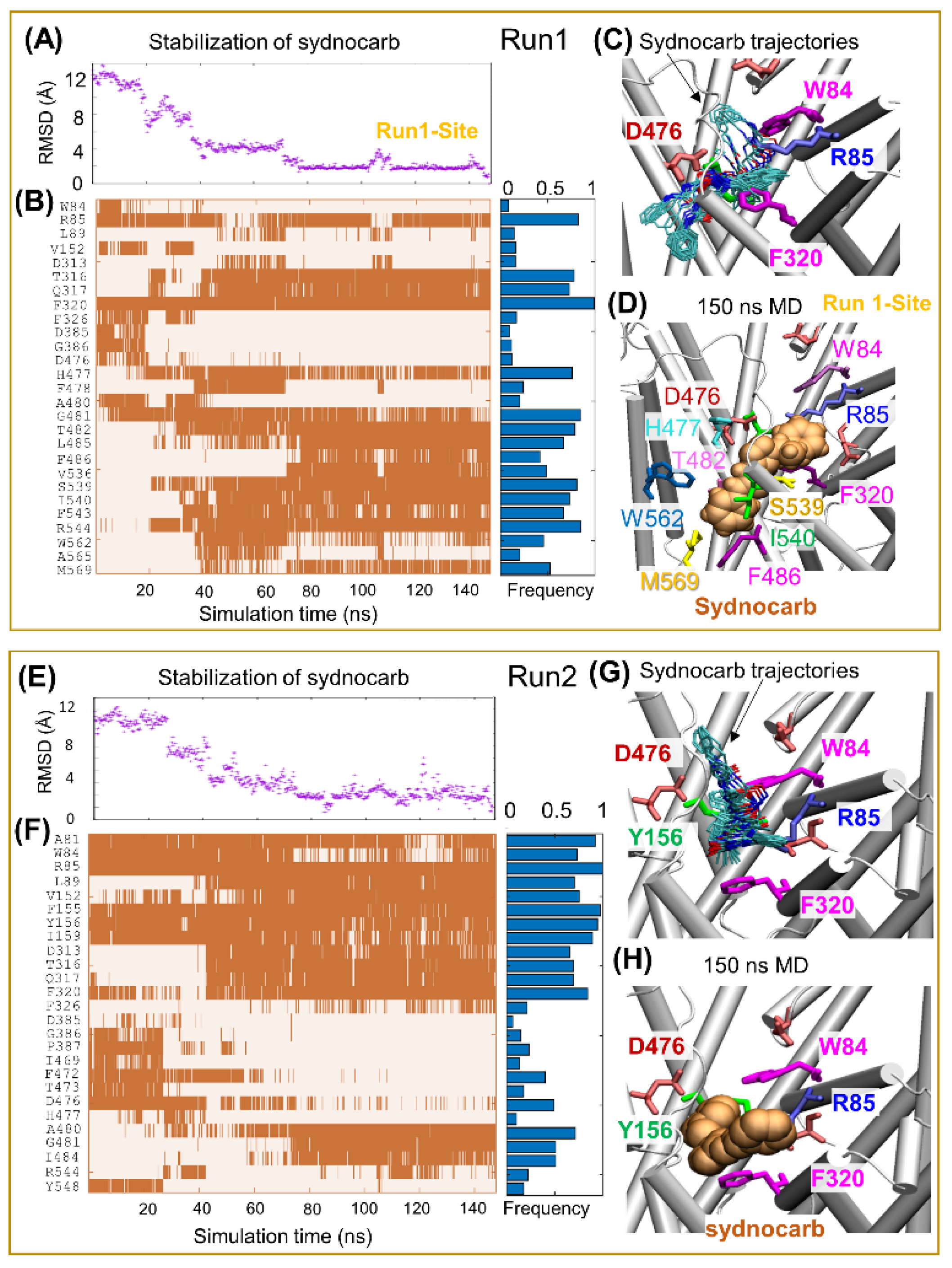
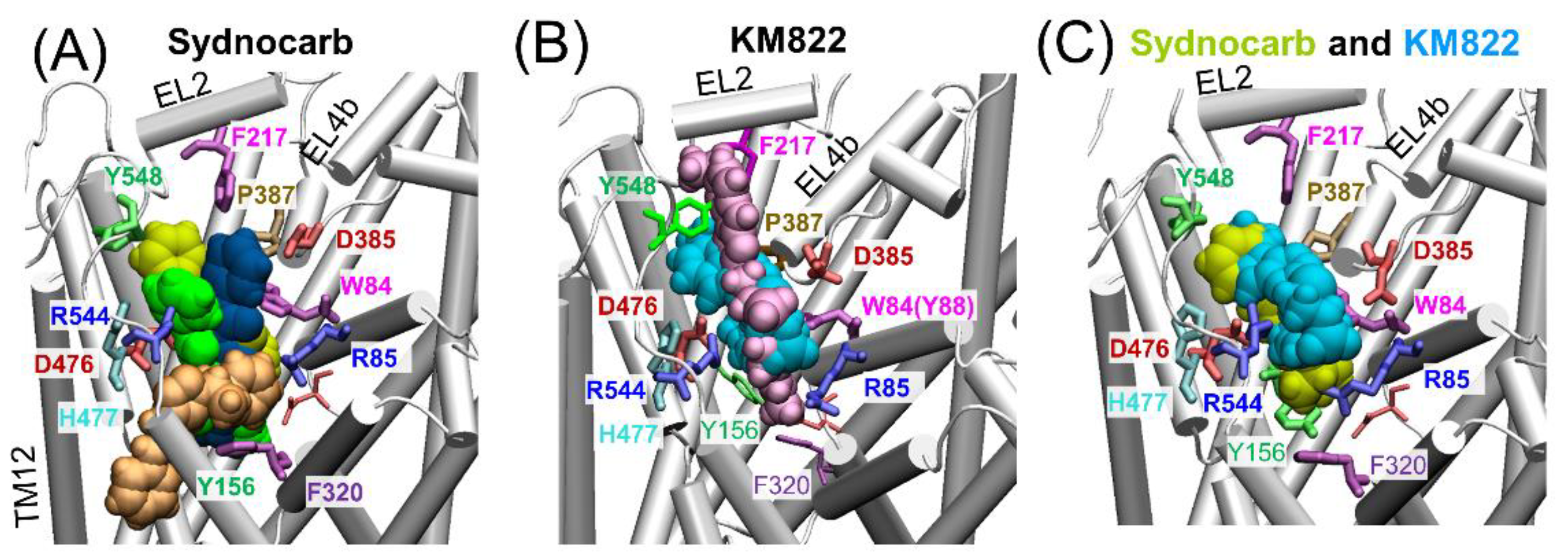
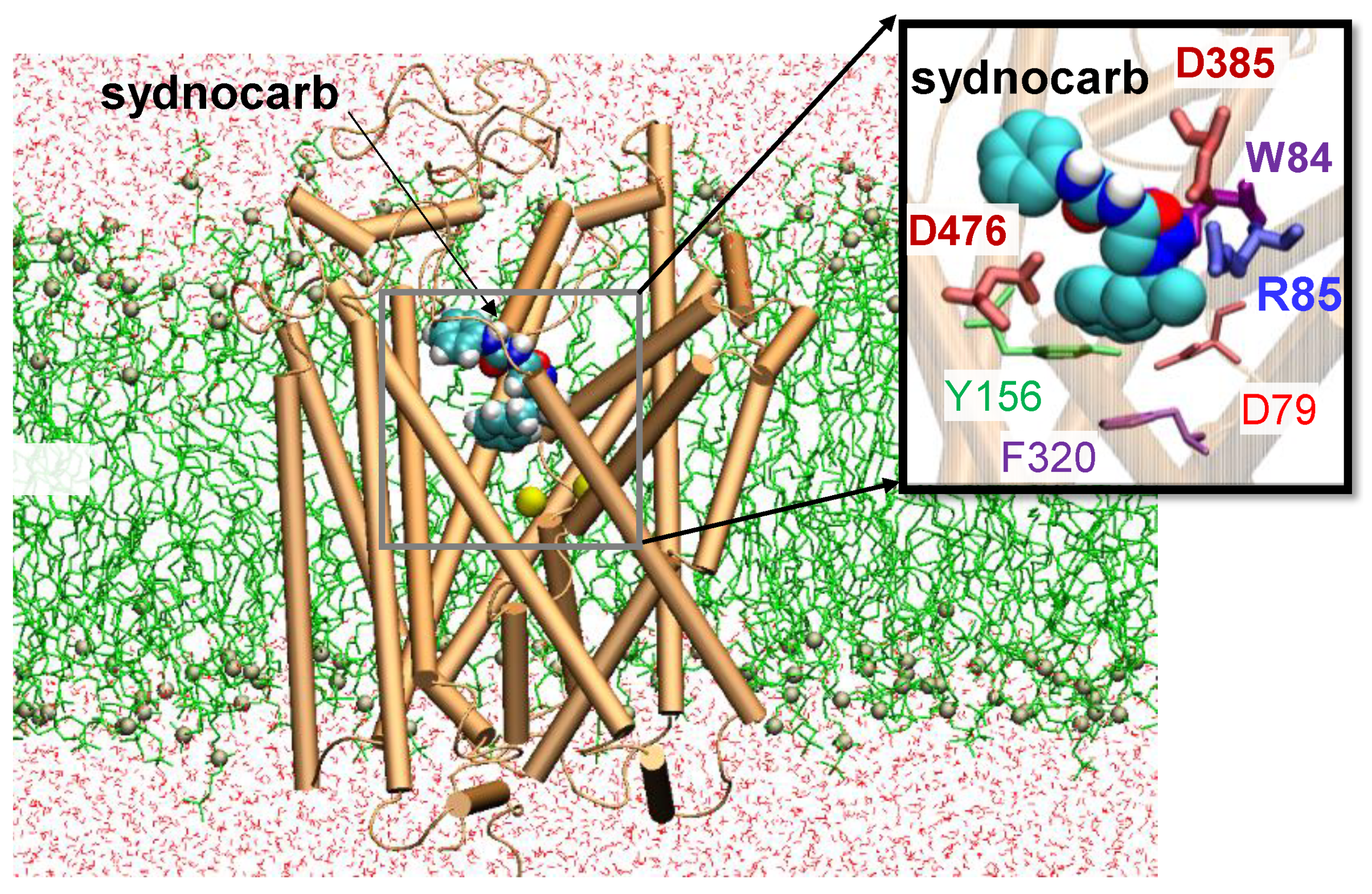
3.2. Docking and MD Simulations Reveal Allosteric Binding Sites for Sydnocarb
3.3. EC Gate Closure Leading to the Transition to an Occluded state Does Not Take Place in the Presence of Sydnocarb
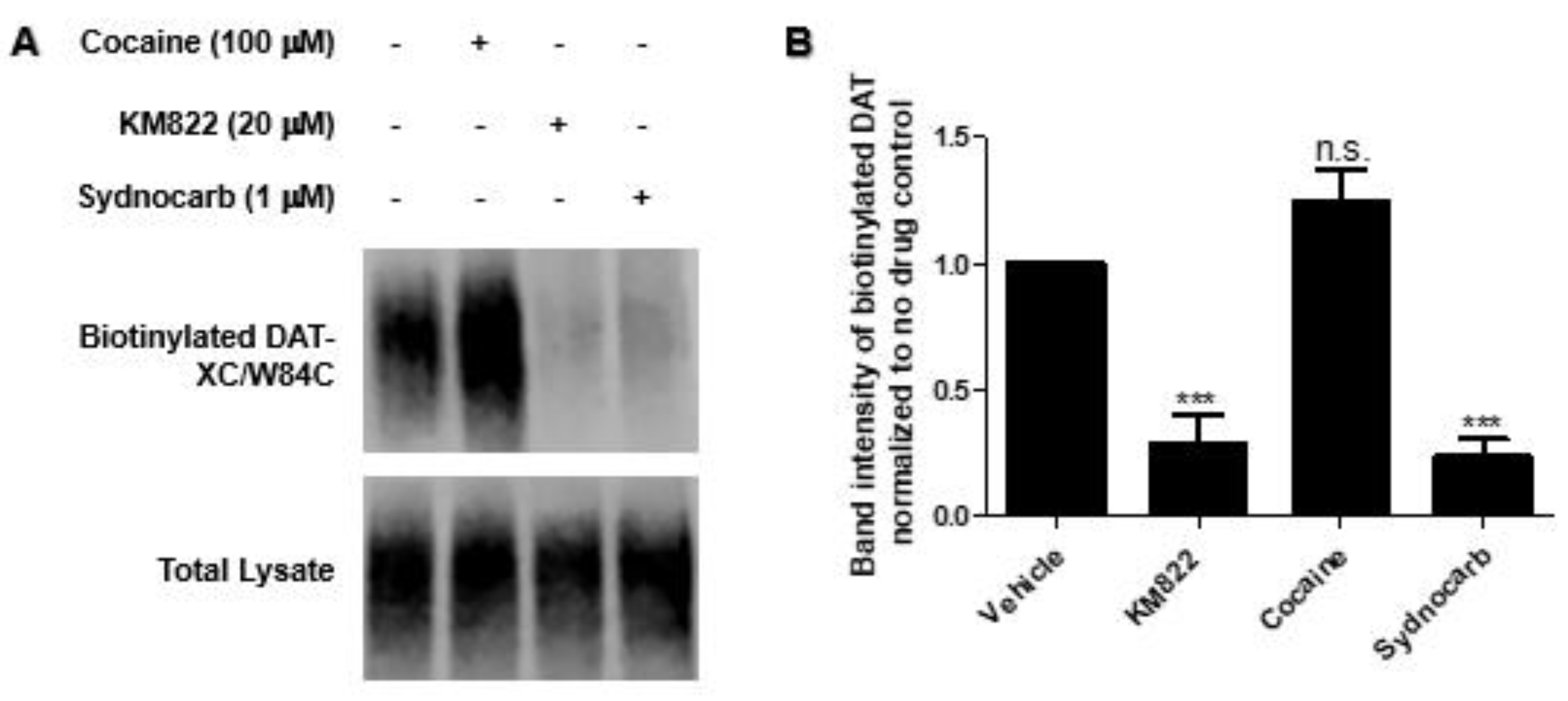
3.4. Binding Site Characterization of Sydnocarb Confirms the Location of the Allosteric Site as Predicted by Docking and MD Simulations
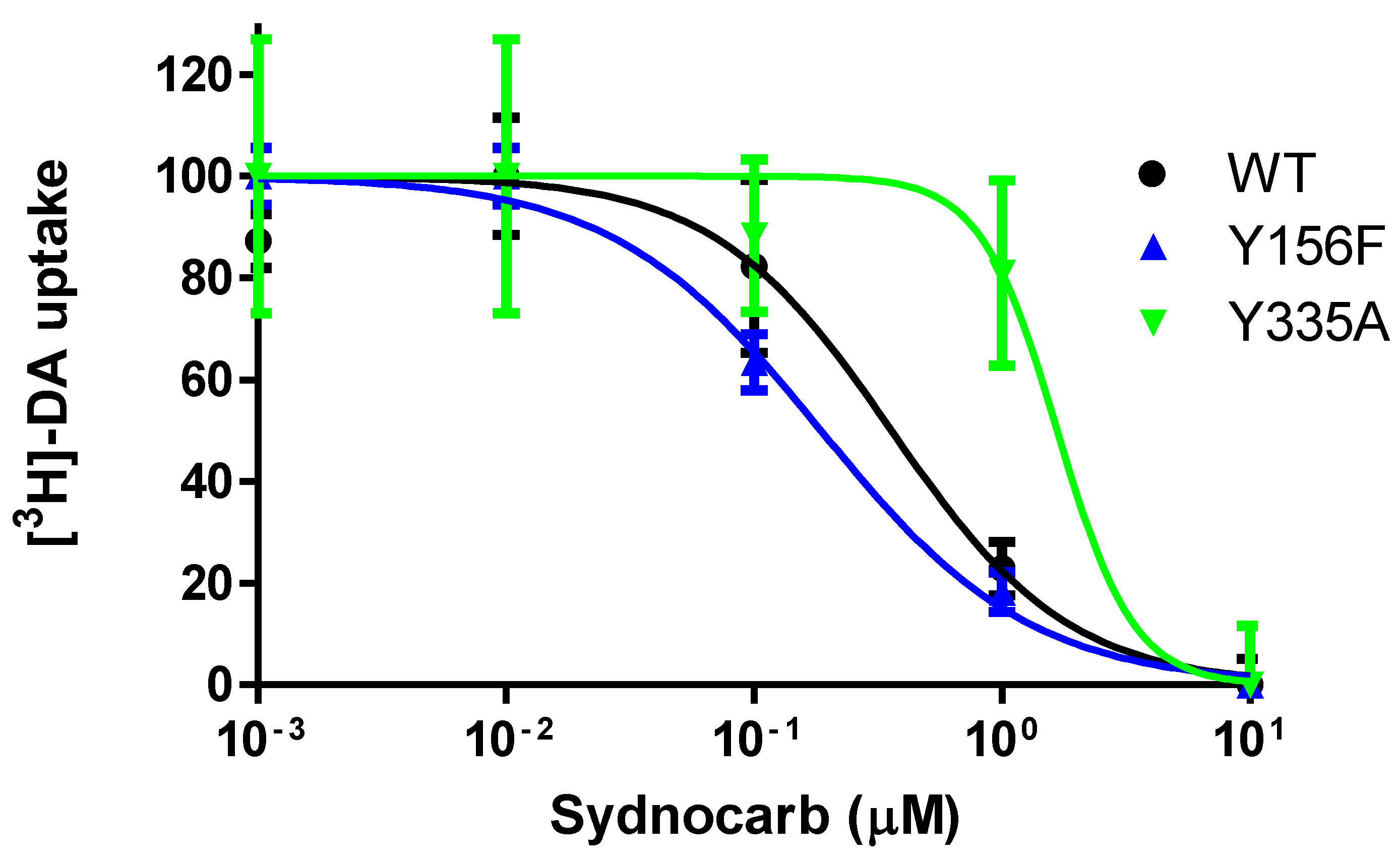
3.5. Interaction of Sydnocarb with Inward (Y335A) versus Outward (Y156F) Equilibrium Shifting DAT Mutations
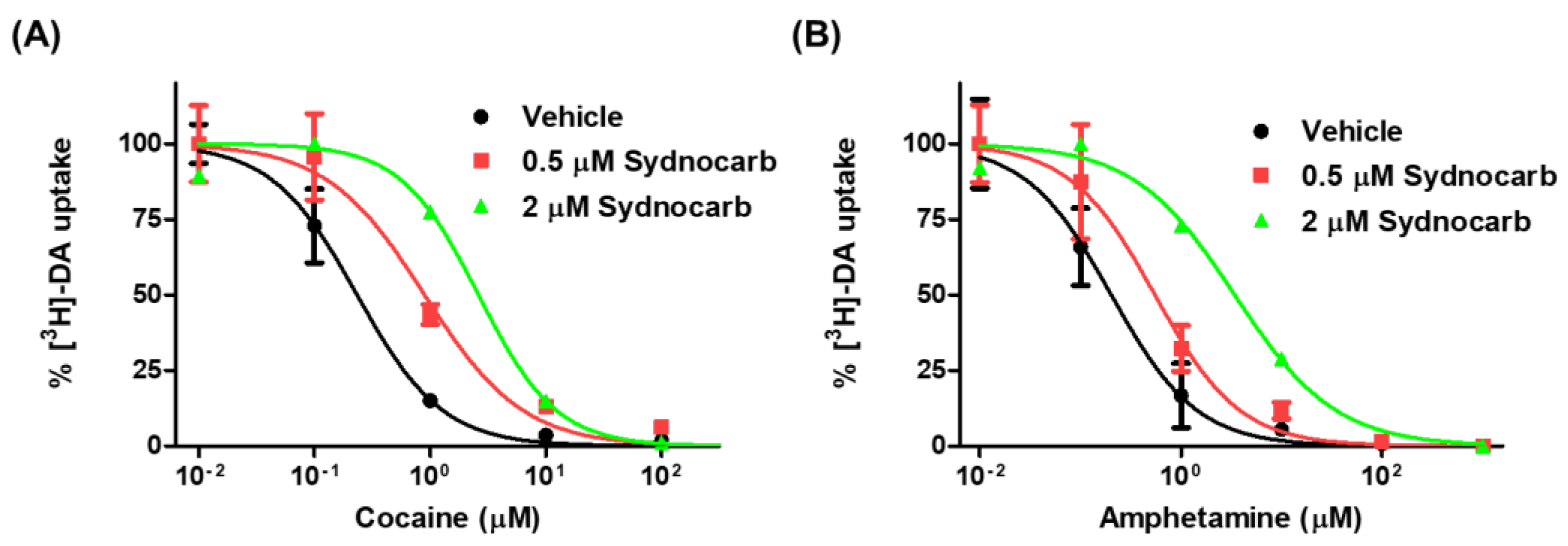
3.6. Sydnocarb Affects Psychostimulant Activity in In Vitro Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kristensen, A.S.; Andersen, J.; Jorgensen, T.N.; Sorensen, L.; Eriksen, J.; Loland, C.J.; Stromgaard, K.; Gether, U. SLC6 neurotransmitter transporters: Structure, function, and regulation. Pharmacol. Rev. 2011, 63, 585–640. [Google Scholar] [CrossRef]
- Lin, Z.; Canales, J.J.; Bjorgvinsson, T.; Thomsen, M.; Qu, H.; Liu, Q.R.; Torres, G.E.; Caine, S.B. Monoamine transporters: Vulnerable and vital doorkeepers. Prog. Mol. Biol. Transl. Sci. 2011, 98, 1–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, C. Developing Treatments for Stimulant Abuse: A Brief Overview. East Asian Arch. Psychiatry 2016, 26, 52–59. [Google Scholar] [PubMed]
- Aggarwal, S.; Mortensen, O.V. Overview of Monoamine Transporters. Curr. Protoc. Pharmacol. 2017, 79, 12.16.1–12.16.17. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, K.C.; Reith, M.E. The atypical stimulant and nootropic modafinil interacts with the dopamine transporter in a different manner than classical cocaine-like inhibitors. PLoS ONE 2011, 6, e25790. [Google Scholar] [CrossRef] [Green Version]
- Desai, R.I.; Kopajtic, T.A.; Koffarnus, M.; Newman, A.H.; Katz, J.L. Identification of a dopamine transporter ligand that blocks the stimulant effects of cocaine. J. Neurosci. 2005, 25, 1889–1893. [Google Scholar] [CrossRef]
- Li, S.M.; Kopajtic, T.A.; O’Callaghan, M.J.; Agoston, G.E.; Cao, J.; Newman, A.H.; Katz, J.L. N-substituted benztropine analogs: Selective dopamine transporter ligands with a fast onset of action and minimal cocaine-like behavioral effects. J. Pharmacol. Exp. Ther. 2011, 336, 575–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loland, C.J.; Desai, R.I.; Zou, M.F.; Cao, J.; Grundt, P.; Gerstbrein, K.; Sitte, H.H.; Newman, A.H.; Katz, J.L.; Gether, U. Relationship between conformational changes in the dopamine transporter and cocaine-like subjective effects of uptake inhibitors. Mol. Pharmacol. 2008, 73, 813–823. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, K.C.; Rothman, R.B.; Reith, M.E. Nonclassical pharmacology of the dopamine transporter: Atypical inhibitors, allosteric modulators, and partial substrates. J. Pharmacol. Exp. Ther. 2013, 346, 2–10. [Google Scholar] [CrossRef] [Green Version]
- Cheng, M.H.; Bahar, I. Monoamine transporters: Structure, intrinsic dynamics and allosteric regulation. Nature Struct. Mol. Biol. 2019, 26, 545–556. [Google Scholar] [CrossRef]
- Yamashita, A.; Singh, S.K.; Kawate, T.; Jin, Y.; Gouaux, E. Crystal structure of a bacterial homologue of Na+/Cl--dependent neurotransmitter transporters. Nature 2005, 437, 215–223. [Google Scholar] [CrossRef]
- Penmatsa, A.; Wang, K.H.; Gouaux, E. X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature 2013, 503, 85–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, J.A.; Green, E.M.; Gouaux, E. X-ray structures and mechanism of the human serotonin transporter. Nature 2016, 532, 334–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Zhen, J.; Karpowich, N.K.; Goetz, R.M.; Law, C.J.; Reith, M.E.; Wang, D.N. LeuT-desipramine structure reveals how antidepressants block neurotransmitter reuptake. Science 2007, 317, 1390–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.K.; Yamashita, A.; Gouaux, E. Antidepressant binding site in a bacterial homologue of neurotransmitter transporters. Nature 2007, 448, 952–956. [Google Scholar] [CrossRef]
- Penmatsa, A.; Wang, K.H.; Gouaux, E. X-ray structures of Drosophila dopamine transporter in complex with nisoxetine and reboxetine. Nat. Struct. Mol. Biol. 2015, 22, 506–508. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.H.; Penmatsa, A.; Gouaux, E. Neurotransmitter and psychostimulant recognition by the dopamine transporter. Nature 2015, 521, 322–327. [Google Scholar] [CrossRef] [Green Version]
- Coleman, J.A.; Yang, D.; Zhao, Z.; Wen, P.C.; Yoshioka, C.; Tajkhorshid, E.; Gouaux, E. Serotonin transporter-ibogaine complexes illuminate mechanisms of inhibition and transport. Nature 2019, 569, 141–145. [Google Scholar] [CrossRef]
- Coleman, J.A.; Gouaux, E. Structural basis for recognition of diverse antidepressants by the human serotonin transporter. Nat. Struct. Mol. Biol. 2018, 25, 170–175. [Google Scholar] [CrossRef]
- Cheng, M.H.; Bahar, I. Molecular Mechanism of Dopamine Transport by Human Dopamine Transporter. Structure 2015, 23, 2171–2181. [Google Scholar] [CrossRef] [Green Version]
- Quick, M.; Abramyan, A.M.; Wiriyasermkul, P.; Weinstein, H.; Shi, L.; Javitch, J.A. The LeuT-fold neurotransmitter:sodium symporter MhsT has two substrate sites. Proc. Natl. Acad. Sci. USA 2018, 115, E792–E7931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.; Quick, M.; Zhao, Y.; Weinstein, H.; Javitch, J.A. The mechanism of a neurotransmitter:sodium symporter--inward release of Na+ and substrate is triggered by substrate in a second binding site. Mol. Cell 2008, 30, 667–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, M.H.; Bahar, I. Complete mapping of substrate translocation highlights the role of LeuT N-terminal segment in regulating transport cycle. PLoS Comput. Biol. 2014, 10, e1003879. [Google Scholar] [CrossRef] [Green Version]
- Quick, M.; Winther, A.M.; Shi, L.; Nissen, P.; Weinstein, H.; Javitch, J.A. Binding of an octylglucoside detergent molecule in the second substrate (S2) site of LeuT establishes an inhibitor-bound conformation. Proc. Natl. Acad. Sci. USA 2009, 106, 5563–5568. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.K.; Piscitelli, C.L.; Yamashita, A.; Gouaux, E. A competitive inhibitor traps LeuT in an open-to-out conformation. Science 2008, 322, 1655–1661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kortagere, S.; Fontana, A.C.; Rose, D.R.; Mortensen, O.V. Identification of an allosteric modulator of the serotonin transporter with novel mechanism of action. Neuropharmacology 2013, 72, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Plenge, P.; Abramyan, A.M.; Sorensen, G.; Mork, A.; Weikop, P.; Gether, U.; Bang-Andersen, B.; Shi, L.; Loland, C.J. The mechanism of a high-affinity allosteric inhibitor of the serotonin transporter. Nat. Commun. 2020, 11, 1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostyn, S.N.; Wilson, K.A.; Schumann-Gillett, A.; Frangos, Z.J.; Shimmon, S.; Rawling, T.; Ryan, R.M.; O’Mara, M.L.; Vandenberg, R.J. Identification of an allosteric binding site on the human glycine transporter, GlyT2, for bioactive lipid analgesics. Elife 2019, 8. [Google Scholar] [CrossRef]
- Larsen, M.B.; Fontana, A.C.; Magalhaes, L.G.; Rodrigues, V.; Mortensen, O.V. A catecholamine transporter from the human parasite Schistosoma mansoni with low affinity for psychostimulants. Mol. Biochem. Parasitol. 2011, 177, 35–41. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, S.; Liu, X.; Rice, C.; Menell, P.; Clark, P.J.; Paparoidamis, N.; Xiao, Y.C.; Salvino, J.M.; Fontana, A.C.K.; Espana, R.A.; et al. Identification of a Novel Allosteric Modulator of the Human Dopamine Transporter. ACS Chem. Neurosci. 2019, 10, 3718–3730. [Google Scholar] [CrossRef]
- Erdo, S.L.; Kiss, B.; Rosdy, B. Inhibition of dopamine uptake by a new psychostimulant mesocarb (Sydnocarb). Pol. J. Pharmacol. Pharm. 1981, 33, 141–147. [Google Scholar]
- Witkin, J.M.; Savtchenko, N.; Mashkovsky, M.; Beekman, M.; Munzar, P.; Gasior, M.; Goldberg, S.R.; Ungard, J.T.; Kim, J.; Shippenberg, T.; et al. Behavioral, toxic, and neurochemical effects of sydnocarb, a novel psychomotor stimulant: Comparisons with methamphetamine. J. Pharmacol. Exp. Ther. 1999, 288, 1298–1310. [Google Scholar]
- Gainetdinov, R.R.; Sotnikova, T.D.; Grekhova, T.V.; Rayevsky, K.S. Effects of a psychostimulant drug sydnocarb on rat brain dopaminergic transmission in vivo. Eur. J. Pharmacol. 1997, 340, 53–58. [Google Scholar] [CrossRef]
- Bashkatova, V.; Mathieu-Kia, A.M.; Durand, C.; Penit-Soria, J. Neurochemical changes and neurotoxic effects of an acute treatment with sydnocarb, a novel psychostimulant: Comparison with D-amphetamine. Ann. N. Y. Acad. Sci. 2002, 965, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Rudenko, G.M.; Altshuler, R.A. Peculiarities of clinical activity and pharmacokinetics of sydnocarb (sydnocarbum), an original psychostimulant. Agressologie 1979, 20, 265–270. [Google Scholar] [PubMed]
- Afanas’ev, I.I.; Anderzhanova, E.A.; Kudrin, V.S.; Rayevsky, K.S. Effects of amphetamine and sydnocarb on dopamine release and free radical generation in rat striatum. Pharmacol. Biochem. Behav. 2001, 69, 653–658. [Google Scholar] [CrossRef]
- Gruner, J.A.; Mathiasen, J.R.; Flood, D.G.; Gasior, M. Characterization of pharmacological and wake-promoting properties of the dopaminergic stimulant sydnocarb in rats. J. Pharmacol. Exp. Ther. 2011, 337, 380–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, M.H.; Block, E.; Hu, F.; Cobanoglu, M.C.; Sorkin, A.; Bahar, I. Insights into the Modulation of Dopamine Transporter Function by Amphetamine, Orphenadrine, and Cocaine Binding. Front. Neurol. 2015, 6, 134. [Google Scholar] [CrossRef] [Green Version]
- Fiser, A.; Šali, A. Modeller: Generation and refinement of homology-based protein structure models. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2003; Volume 374, pp. 461–491. [Google Scholar]
- Sterling, T.; Irwin, J.J. ZINC 15--Ligand Discovery for Everyone. J. Chem. Inf. Model 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Raman, E.P.; MacKerell Jr, A.D. Automation of the CHARMM General Force Field (CGenFF) II: Assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model 2012, 52, 3155–3168. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Wu, E.L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Davila-Contreras, E.M.; Qi, Y.; Lee, J.; Monje-Galvan, V.; Venable, R.M.; et al. CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. J. Comput. Chem. 2014, 35, 1997–2004. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, M.H.; Kaya, C.; Bahar, I. Quantitative assessment of the energetics of dopamine translocation by human dopamine transporter. J. Phys. Chem. B 2018, 122, 5336–5346. [Google Scholar] [CrossRef]
- Plenge, P.; Shi, L.; Beuming, T.; Te, J.; Newman, A.H.; Weinstein, H.; Gether, U.; Loland, C.J. Steric hindrance mutagenesis in the conserved extracellular vestibule impedes allosteric binding of antidepressants to the serotonin transporter. J. Biol. Chem. 2012, 287, 39316–39326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramyan, A.M.; Stolzenberg, S.; Li, Z.; Loland, C.J.; Noe, F.; Shi, L. The Isomeric Preference of an Atypical Dopamine Transporter Inhibitor Contributes to Its Selection of the Transporter Conformation. ACS Chem. Neurosci. 2017, 8, 1735–1746. [Google Scholar] [CrossRef] [PubMed]
- Loland, C.J.; Mereu, M.; Okunola, O.M.; Cao, J.; Prisinzano, T.E.; Mazier, S.; Kopajtic, T.; Shi, L.; Katz, J.L.; Tanda, G.; et al. R-modafinil (armodafinil): A unique dopamine uptake inhibitor and potential medication for psychostimulant abuse. Biol. Psychiatry 2012, 72, 405–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, W.C.; Wasko, M.J.; Wilkinson, D.S.; Hiranita, T.; Li, L.; Hayashi, S.; Snell, D.B.; Madura, J.D.; Surratt, C.K.; Katz, J.L. Dopamine Transporter Dynamics of N-Substituted Benztropine Analogs with Atypical Behavioral Effects. J. Pharmacol. Exp. Ther. 2018, 366, 527–540. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aggarwal, S.; Cheng, M.H.; Salvino, J.M.; Bahar, I.; Mortensen, O.V. Functional Characterization of the Dopaminergic Psychostimulant Sydnocarb as an Allosteric Modulator of the Human Dopamine Transporter. Biomedicines 2021, 9, 634. https://doi.org/10.3390/biomedicines9060634
Aggarwal S, Cheng MH, Salvino JM, Bahar I, Mortensen OV. Functional Characterization of the Dopaminergic Psychostimulant Sydnocarb as an Allosteric Modulator of the Human Dopamine Transporter. Biomedicines. 2021; 9(6):634. https://doi.org/10.3390/biomedicines9060634
Chicago/Turabian StyleAggarwal, Shaili, Mary Hongying Cheng, Joseph M. Salvino, Ivet Bahar, and Ole Valente Mortensen. 2021. "Functional Characterization of the Dopaminergic Psychostimulant Sydnocarb as an Allosteric Modulator of the Human Dopamine Transporter" Biomedicines 9, no. 6: 634. https://doi.org/10.3390/biomedicines9060634
APA StyleAggarwal, S., Cheng, M. H., Salvino, J. M., Bahar, I., & Mortensen, O. V. (2021). Functional Characterization of the Dopaminergic Psychostimulant Sydnocarb as an Allosteric Modulator of the Human Dopamine Transporter. Biomedicines, 9(6), 634. https://doi.org/10.3390/biomedicines9060634