
Novel Pharmaceutical Strategy for Selective Abrogation of TSP1-Induced Vascular Dysfunction by Decoy Recombinant CD47 Soluble Receptor in Prophylaxis and Treatment Models


 and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. In Silico Modeling and Analysis
2.1.1. Simulations
2.1.2. Energy Minimization and Calculating the Active Site Sequence
2.1.3. Building of the rh-CD47p Model and Protein–Protein Docking Experiments
2.2. Materials and Reagents
2.3. Methods
2.3.1. Measurement of Mouse Thoracic Aorta Relaxation
2.3.2. Protocol for Pretreatment with Recombinant Human CD47 Peptide (rh-CD47p)
2.3.3. Protocol for Post-Treatment with rh-CD47p
2.3.4. Immunoblot
2.3.5. Modified CD47 ELISA
2.4. Data Analysis
3. Results
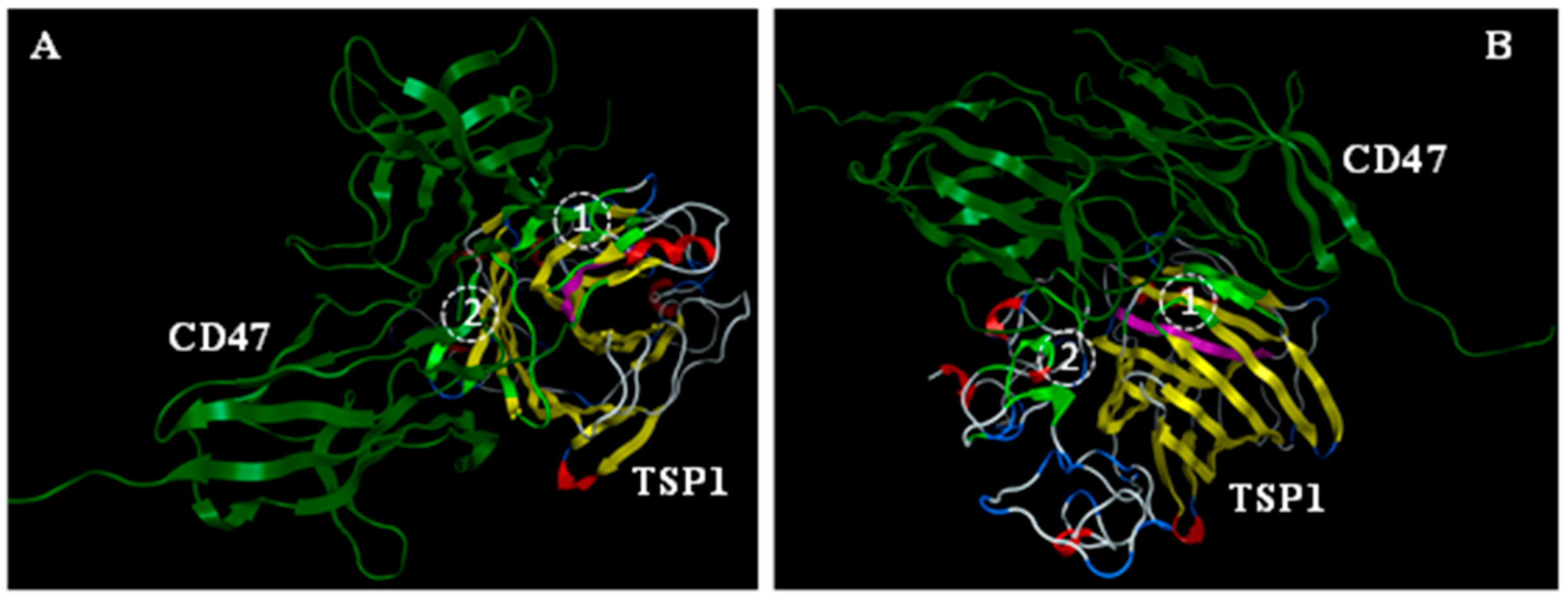
3.1. In Silico Modeling and Visualization of Putative Extracellular CD47:TSP1 Interactions
3.2. In Vitro Confirmation of Qualitative and Quantitative Association between mTSP1 and Soluble rh-CD47p
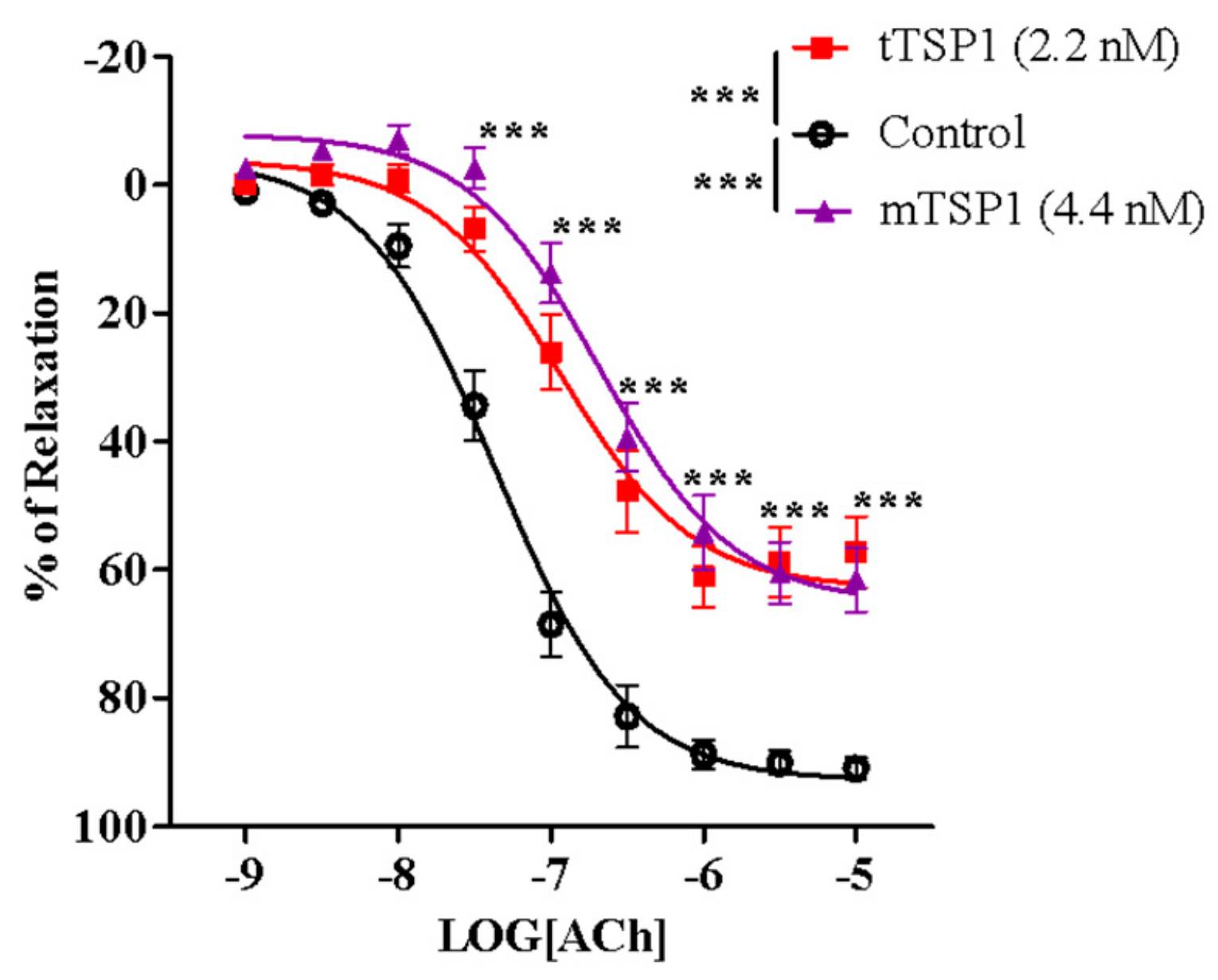
3.3. Ex Vivo Inhibition of Endothelium-Dependent Vasodilation Due to Trimeric and Monomeric Forms of TSP1
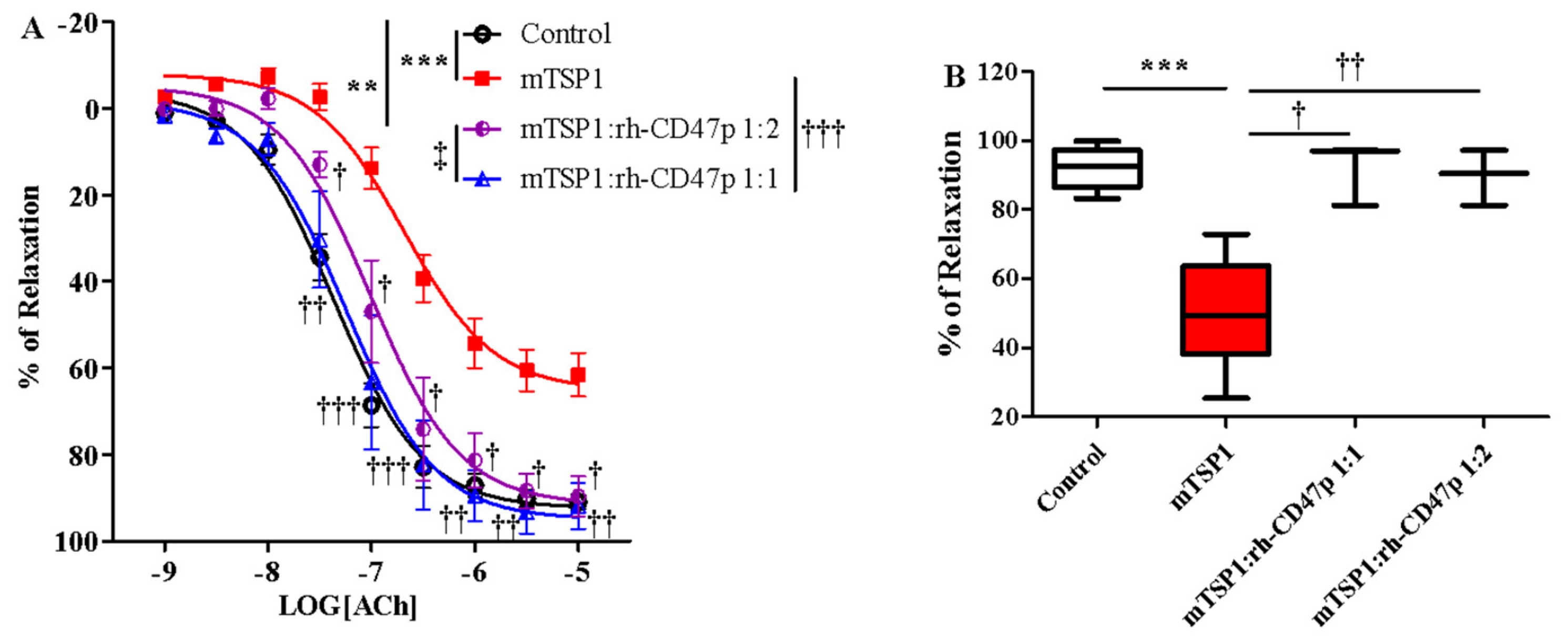
3.4. Pretreatment with rh-CD47p Prevents TSP1-Blunted Vasodilation
3.5. Post-Treatment with rh-CD47p Completely Restored Vasodilation Following mTSP1-Mediated Injury to Endothelial Vasoreactivity
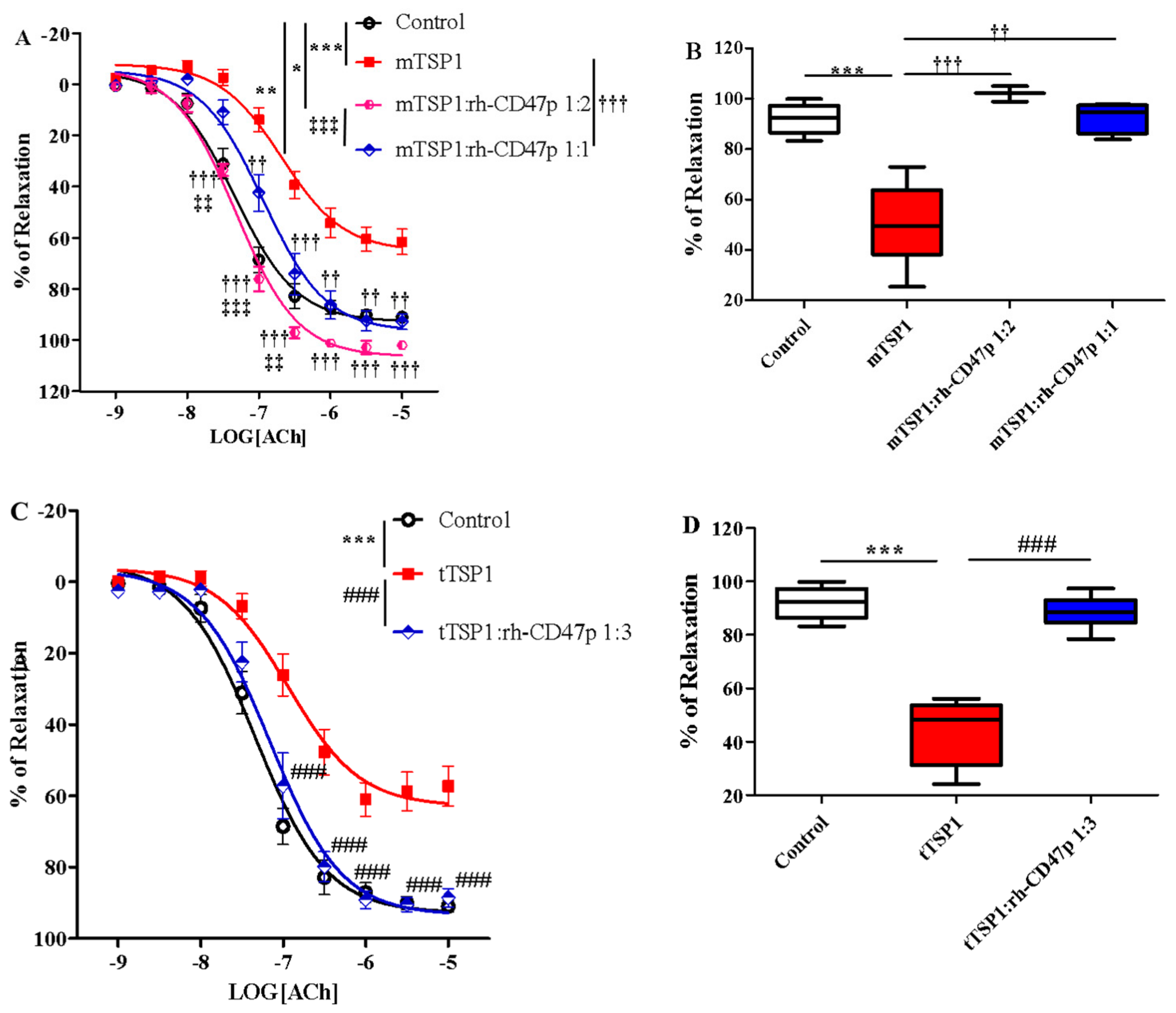
3.6. Post-Treatment With Increasing Molar Ratios of rh-CD47p:tTSP1 to Achieve Complete Mitigation of tTSP1-Blunted Vasodilation
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, H.; Herndon, M.E.; Lawler, J. The cell biology of thrombospondin-1. Matrix Biol. 2000, 19, 597–614. [Google Scholar] [CrossRef]
- Choi, K.Y.; Kim, D.B.; Kim, M.J.; Kwon, B.J.; Chang, S.Y.; Jang, S.W.; Cho, E.J.; Rho, T.H.; Kim, J.H. Higher plasma thrombospondin-1 levels in patients with coronary artery disease and diabetes mellitus. Korean Circ. J. 2012, 42, 100–106. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.M.; Hu, Y.; Miller, G.G.; Mitchell, R.N.; Libby, P. Association of thrombospondin-1 and cardiac allograft vasculopathy in human cardiac allografts. Circulation 2001, 103, 525–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.L.; Jong, Y.S.; Wu, Y.W.; Wang, W.J.; Hsieh, A.R.; Chao, C.L.; Chen, W.J.; Yang, W.S. Association of Plasma Thrombospondin-1 Level with Cardiovascular Disease and Mortality in Hemodialysis Patients. Acta Cardiol. Sin. 2015, 31, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, R.; Khanal, S.; Mathias, A.; Gupta, S.; Lallo, J.; Sahu, S.; Ohanyan, V.; Patel, A.; Storm, K.; Datta, S.; et al. TSP-1 (Thrombospondin-1) Deficiency Protects ApoE−/− Mice Against Leptin-Induced Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2021, 41, e112–e127. [Google Scholar] [CrossRef] [PubMed]
- Favier, J.; Germain, S.; Emmerich, J.; Corvol, P.; Gasc, J.M. Critical overexpression of thrombospondin 1 in chronic leg ischaemia. J. Pathol. 2005, 207, 358–366. [Google Scholar] [CrossRef]
- Bauer, P.M.; Bauer, E.M.; Rogers, N.M.; Yao, M.; Feijoo-Cuaresma, M.; Pilewski, J.M.; Champion, H.C.; Zuckerbraun, B.S.; Calzada, M.J.; Isenberg, J.S. Activated CD47 promotes pulmonary arterial hypertension through targeting caveolin-1. Cardiovasc. Res. 2012, 93, 682–693. [Google Scholar] [CrossRef] [Green Version]
- Rogers, N.M.; Sharifi-Sanjani, M.; Yao, M.; Ghimire, K.; Bienes-Martinez, R.; Mutchler, S.M.; Knupp, H.E.; Baust, J.; Novelli, E.M.; Ross, M.; et al. TSP1-CD47 signaling is upregulated in clinical pulmonary hypertension and contributes to pulmonary arterial vasculopathy and dysfunction. Cardiovasc. Res. 2017, 113, 15–29. [Google Scholar] [CrossRef]
- Resovi, A.; Pinessi, D.; Chiorino, G.; Taraboletti, G. Current understanding of the thrombospondin-1 interactome. Matrix Biol. 2014, 37, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.W.; Isenberg, J.S.; Roberts, D.D. Thrombospondin-1 is an inhibitor of pharmacological activation of soluble guanylate cyclase. Br. J. Pharmacol. 2010, 159, 1542–1547. [Google Scholar] [CrossRef] [Green Version]
- Bauer, E.M.; Qin, Y.; Miller, T.W.; Bandle, R.W.; Csanyi, G.; Pagano, P.J.; Bauer, P.M.; Schnermann, J.; Roberts, D.D.; Isenberg, J.S. Thrombospondin-1 supports blood pressure by limiting eNOS activation and endothelial-dependent vasorelaxation. Cardiovasc. Res. 2010, 88, 471–481. [Google Scholar] [CrossRef] [Green Version]
- Isenberg, J.S.; Wink, D.A.; Roberts, D.D. Thrombospondin-1 antagonizes nitric oxide-stimulated vascular smooth muscle cell responses. Cardiovasc. Res. 2006, 71, 785–793. [Google Scholar] [CrossRef]
- Csanyi, G.; Yao, M.; Rodriguez, A.I.; Al Ghouleh, I.; Sharifi-Sanjani, M.; Frazziano, G.; Huang, X.; Kelley, E.E.; Isenberg, J.S.; Pagano, P.J. Thrombospondin-1 regulates blood flow via CD47 receptor-mediated activation of NADPH oxidase 1. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2966–2973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharifi-Sanjani, M.; Shoushtari, A.H.; Quiroz, M.; Baust, J.; Sestito, S.F.; Mosher, M.; Ross, M.; McTiernan, C.F.; St Croix, C.M.; Bilonick, R.A.; et al. Cardiac CD47 drives left ventricular heart failure through Ca2+-CaMKII-regulated induction of HDAC3. J. Am. Heart Assoc. 2014, 3, e000670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isenberg, J.S.; Romeo, M.J.; Yu, C.; Yu, C.K.; Nghiem, K.; Monsale, J.; Rick, M.E.; Wink, D.A.; Frazier, W.A.; Roberts, D.D. Thrombospondin-1 stimulates platelet aggregation by blocking the antithrombotic activity of nitric oxide/cGMP signaling. Blood 2008, 111, 613–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, L.Y.; Ramakrishnan, D.P.; Silverstein, R.L. Thrombospondin-1 modulates VEGF signaling via CD36 by recruiting SHP-1 to VEGFR2 complex in microvascular endothelial cells. Blood 2013, 122, 1822–1832. [Google Scholar] [CrossRef] [Green Version]
- Kaur, S.; Martin-Manso, G.; Pendrak, M.L.; Garfield, S.H.; Isenberg, J.S.; Roberts, D.D. Thrombospondin-1 inhibits VEGF receptor-2 signaling by disrupting its association with CD47. J. Biol. Chem. 2010, 285, 38923–38932. [Google Scholar] [CrossRef] [Green Version]
- Oldenborg, P.A.; Zheleznyak, A.; Fang, Y.F.; Lagenaur, C.F.; Gresham, H.D.; Lindberg, F.P. Role of CD47 as a marker of self on red blood cells. Science 2000, 288, 2051–2054. [Google Scholar] [CrossRef]
- Okazawa, H.; Motegi, S.; Ohyama, N.; Ohnishi, H.; Tomizawa, T.; Kaneko, Y.; Oldenborg, P.A.; Ishikawa, O.; Matozaki, T. Negative regulation of phagocytosis in macrophages by the CD47-SHPS-1 system. J. Immunol. 2005, 174, 2004–2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeanne, A.; Schneider, C.; Martiny, L.; Dedieu, S. Original insights on thrombospondin-1-related antireceptor strategies in cancer. Front. Pharmacol. 2015, 6, 252. [Google Scholar] [CrossRef] [Green Version]
- Sikic, B.I.; Lakhani, N.; Patnaik, A.; Shah, S.A.; Chandana, S.R.; Rasco, D.; Colevas, A.D.; O’Rourke, T.; Narayanan, S.; Papadopoulos, K.; et al. First-in-Human, First-in-Class Phase I Trial of the Anti-CD47 Antibody Hu5F9-G4 in Patients With Advanced Cancers. J. Clin. Oncol. 2019, 37, 946–953. [Google Scholar] [CrossRef]
- Jeanne, A.; Sick, E.; Devy, J.; Floquet, N.; Belloy, N.; Theret, L.; Boulagnon-Rombi, C.; Diebold, M.D.; Dauchez, M.; Martiny, L.; et al. Identification of TAX2 peptide as a new unpredicted anti-cancer agent. Oncotarget 2015, 6, 17981–18000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Bonecchi, R.; Locati, M. Tuning inflammation and immunity by chemokine sequestration: Decoys and more. Nat. Rev. Immunol. 2006, 6, 907–918. [Google Scholar] [CrossRef] [PubMed]
- Borroni, E.M.; Bonecchi, R.; Buracchi, C.; Savino, B.; Mantovani, A.; Locati, M. Chemokine decoy receptors: New players in reproductive immunology. Immunol. Investig. 2008, 37, 483–497. [Google Scholar] [CrossRef]
- Pontejo, S.M.; Alejo, A.; Alcami, A. Comparative Biochemical and Functional Analysis of Viral and Human Secreted Tumor Necrosis Factor (TNF) Decoy Receptors. J. Biol. Chem. 2015, 290, 15973–15984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schieferdecker, A.; Voigt, M.; Riecken, K.; Braig, F.; Schinke, T.; Loges, S.; Bokemeyer, C.; Fehse, B.; Binder, M. Denosumab mimics the natural decoy receptor osteoprotegerin by interacting with its major binding site on RANKL. Oncotarget 2014, 5, 6647–6653. [Google Scholar] [CrossRef] [Green Version]
- Floquet, N.; Dedieu, S.; Martiny, L.; Dauchez, M.; Perahia, D. Human thrombospondin’s (TSP-1) C-terminal domain opens to interact with the CD-47 receptor: A molecular modeling study. Arch. Biochem. Biophys. 2008, 478, 103–109. [Google Scholar] [CrossRef]
- Isenberg, J.S.; Annis, D.S.; Pendrak, M.L.; Ptaszynska, M.; Frazier, W.A.; Mosher, D.F.; Roberts, D.D. Differential interactions of thrombospondin-1, -2, and -4 with CD47 and effects on cGMP signaling and ischemic injury responses. J. Biol. Chem. 2009, 284, 1116–1125. [Google Scholar] [CrossRef] [Green Version]
- Gao, A.G.; Lindberg, F.P.; Finn, M.B.; Blystone, S.D.; Brown, E.J.; Frazier, W.A. Integrin-associated protein is a receptor for the C-terminal domain of thrombospondin. J. Biol. Chem. 1996, 271, 21–24. [Google Scholar] [CrossRef] [Green Version]
- Kvansakul, M.; Adams, J.C.; Hohenester, E. Structure of a thrombospondin C-terminal fragment reveals a novel calcium core in the type 3 repeats. EMBO J. 2004, 23, 1223–1233. [Google Scholar] [CrossRef]
- Jiang, W.; Phillips, J.C.; Huang, L.; Fajer, M.; Meng, Y.; Gumbart, J.C.; Luo, Y.; Schulten, K.; Roux, B. Generalized Scalable Multiple Copy Algorithms for Molecular Dynamics Simulations in NAMD. Comput. Phys. Commun. 2014, 185, 908–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, S.; Cheng, X.; Lee, J.; Kim, S.; Park, S.J.; Patel, D.S.; Beaven, A.H.; Lee, K.I.; Rui, H.; Park, S.; et al. CHARMM-GUI 10 years for biomolecular modeling and simulation. J. Comput. Chem. 2017, 38, 1114–1124. [Google Scholar] [CrossRef] [PubMed]
- Mallajosyula, S.S.; Jo, S.; Im, W.; MacKerell, A.D., Jr. Molecular dynamics simulations of glycoproteins using CHARMM. Methods Mol. Biol. 2015, 1273, 407–429. [Google Scholar] [CrossRef] [Green Version]
- Pedretti, A.; Villa, L.; Vistoli, G. VEGA—An open platform to develop chemo-bio-informatics applications, using plug-in architecture and script programming. J. Comput. Aided Mol. Des. 2004, 18, 167–173. [Google Scholar] [CrossRef]
- Laurie, A.T.; Jackson, R.M. Q-SiteFinder: An energy-based method for the prediction of protein-ligand binding sites. Bioinformatics 2005, 21, 1908–1916. [Google Scholar] [CrossRef]
- Cereto-Massague, A.; Ojeda, M.J.; Valls, C.; Mulero, M.; Pujadas, G.; Garcia-Vallve, S. Tools for in silico target fishing. Methods 2015, 71, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Isenberg, J.S.; Martin-Manso, G.; Maxhimer, J.B.; Roberts, D.D. Regulation of nitric oxide signalling by thrombospondin 1: Implications for anti-angiogenic therapies. Nat. Rev. Cancer 2009, 9, 182–194. [Google Scholar] [CrossRef] [Green Version]
- Leclair, P.; Lim, C.J. CD47-independent effects mediated by the TSP-derived 4N1K peptide. PLoS ONE 2014, 9, e98358. [Google Scholar] [CrossRef]
- Kaiser, R.; Frantz, C.; Bals, R.; Wilkens, H. The role of circulating thrombospondin-1 in patients with precapillary pulmonary hypertension. Respir. Res. 2016, 17, 96. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.B.; Tang, W.D.; Wang, H.X.; Xu, Y. Predictive value of thrombospondin-1 for outcomes in patients with acute ischemic stroke. Clin. Chim. Acta 2015, 450, 176–180. [Google Scholar] [CrossRef]
- Penn, A.M.; Bibok, M.B.; Saly, V.K.; Coutts, S.B.; Lesperance, M.L.; Balshaw, R.F.; Votova, K.; Croteau, N.S.; Trivedi, A.; Jackson, A.M.; et al. Verification of a proteomic biomarker panel to diagnose minor stroke and transient ischaemic attack: Phase 1 of SpecTRA, a large scale translational study. Biomarkers 2018, 23, 392–405. [Google Scholar] [CrossRef] [Green Version]
- Procter, N.E.; Ball, J.; Ngo, D.T.; Chirkov, Y.Y.; Isenberg, J.S.; Hylek, E.M.; Stewart, S.; Horowitz, J.D. Platelet hyperaggregability in patients with atrial fibrillation. Evidence of a background proinflammatory milieu. Herz 2016, 41, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Procter, N.E.; Ball, J.; Liu, S.; Hurst, N.; Nooney, V.B.; Goh, V.; Stafford, I.; Heresztyn, T.; Carrington, M.; Ngo, D.T.; et al. Impaired platelet nitric oxide response in patients with new onset atrial fibrillation. Int. J. Cardiol. 2015, 179, 160–165. [Google Scholar] [CrossRef]
- Isenberg, J.S.; Frazier, W.A.; Roberts, D.D. Thrombospondin-1: A physiological regulator of nitric oxide signaling. Cell Mol. Life Sci. 2008, 65, 728–742. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Wu, H.; Cui, L.; Gao, Y.; Chen, L.; Li, X.; Liang, T.; Yang, X.; Cheng, J.; Luo, J. CD47-retargeted oncolytic adenovirus armed with melanoma differentiation-associated gene-7/interleukin-24 suppresses in vivo leukemia cell growth. Oncotarget 2015, 6, 43496–43507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayat, S.M.G.; Jaafari, M.R.; Hatamipour, M.; Penson, P.E.; Sahebkar, A. Liposome Circulation Time is Prolonged by CD47 Coating. Protein Pept. Lett. 2020, 27, 1029–1037. [Google Scholar] [CrossRef]
- Kumar, R.; Mickael, C.; Kassa, B.; Gebreab, L.; Robinson, J.C.; Koyanagi, D.E.; Sanders, L.; Barthel, L.; Meadows, C.; Fox, D.; et al. TGF-beta activation by bone marrow-derived thrombospondin-1 causes Schistosoma- and hypoxia-induced pulmonary hypertension. Nat. Commun. 2017, 8, 15494. [Google Scholar] [CrossRef] [PubMed]
- Labrousse-Arias, D.; Castillo-Gonzalez, R.; Rogers, N.M.; Torres-Capelli, M.; Barreira, B.; Aragones, J.; Cogolludo, A.; Isenberg, J.S.; Calzada, M.J. HIF-2alpha-mediated induction of pulmonary thrombospondin-1 contributes to hypoxia-driven vascular remodelling and vasoconstriction. Cardiovasc. Res. 2016, 109, 115–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cohort | EC50 | Fold of Shift vs. Control | Fold of Shift vs. TSP1 Alone |
|---|---|---|---|
| Control | 43.5 ± 6.3 | ||
| tTSP1 alone | 213.8 ± 81.5 * | 4.9 | |
| mTSP1 alone | 238.6 ± 76.7 † | 5.5 | |
| Pre-treatment | |||
| tTSP1:CD47 1:3 | 59.7 ± 9.1 * | 1.4 | −3.6 |
| mTSP1:CD47 1:1 | 130.2 ± 31.1 † | 3.0 | −1.8 |
| mTSP1:CD47 1:2 | 47.8 ± 3.2 † | 1.1 | −5.0 |
| Post-treatment | |||
| tTSP1:CD47 1:3 | 147.9 ± 48.7 | 3.4 | −1.4 |
| tTSP1:CD47 1:4 | 79.8 ± 17.9 ## | 1.8 | −2.7 |
| tTSP1:CD47 1:5 | 45.3 ± 17.0 ## | 1.0 | −4.7 |
| tTSP1:CD47 1:6 | 44.4 ± 8.0 ## | 1.0 | −4.8 |
| mTSP1:CD47 1:1 | 74.8 ± 32.1 † | 1.9 | −3.2 |
| mTSP1:CD47 1:2 | 172.1 ± 68.5 † | 4.0 | −1.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, M.; Sturdivant, J.; Ebrahimi, A.; Ganguly, S.; Elbayoumi, T. Novel Pharmaceutical Strategy for Selective Abrogation of TSP1-Induced Vascular Dysfunction by Decoy Recombinant CD47 Soluble Receptor in Prophylaxis and Treatment Models. Biomedicines 2021, 9, 642. https://doi.org/10.3390/biomedicines9060642
Yao M, Sturdivant J, Ebrahimi A, Ganguly S, Elbayoumi T. Novel Pharmaceutical Strategy for Selective Abrogation of TSP1-Induced Vascular Dysfunction by Decoy Recombinant CD47 Soluble Receptor in Prophylaxis and Treatment Models. Biomedicines. 2021; 9(6):642. https://doi.org/10.3390/biomedicines9060642
Chicago/Turabian StyleYao, Molly, Jalicia Sturdivant, Aren Ebrahimi, Samayita Ganguly, and Tamer Elbayoumi. 2021. "Novel Pharmaceutical Strategy for Selective Abrogation of TSP1-Induced Vascular Dysfunction by Decoy Recombinant CD47 Soluble Receptor in Prophylaxis and Treatment Models" Biomedicines 9, no. 6: 642. https://doi.org/10.3390/biomedicines9060642
APA StyleYao, M., Sturdivant, J., Ebrahimi, A., Ganguly, S., & Elbayoumi, T. (2021). Novel Pharmaceutical Strategy for Selective Abrogation of TSP1-Induced Vascular Dysfunction by Decoy Recombinant CD47 Soluble Receptor in Prophylaxis and Treatment Models. Biomedicines, 9(6), 642. https://doi.org/10.3390/biomedicines9060642