
Animal Models of Autosomal Recessive Parkinsonism


 , , ,
, , ,
Abstract
:1. Introduction
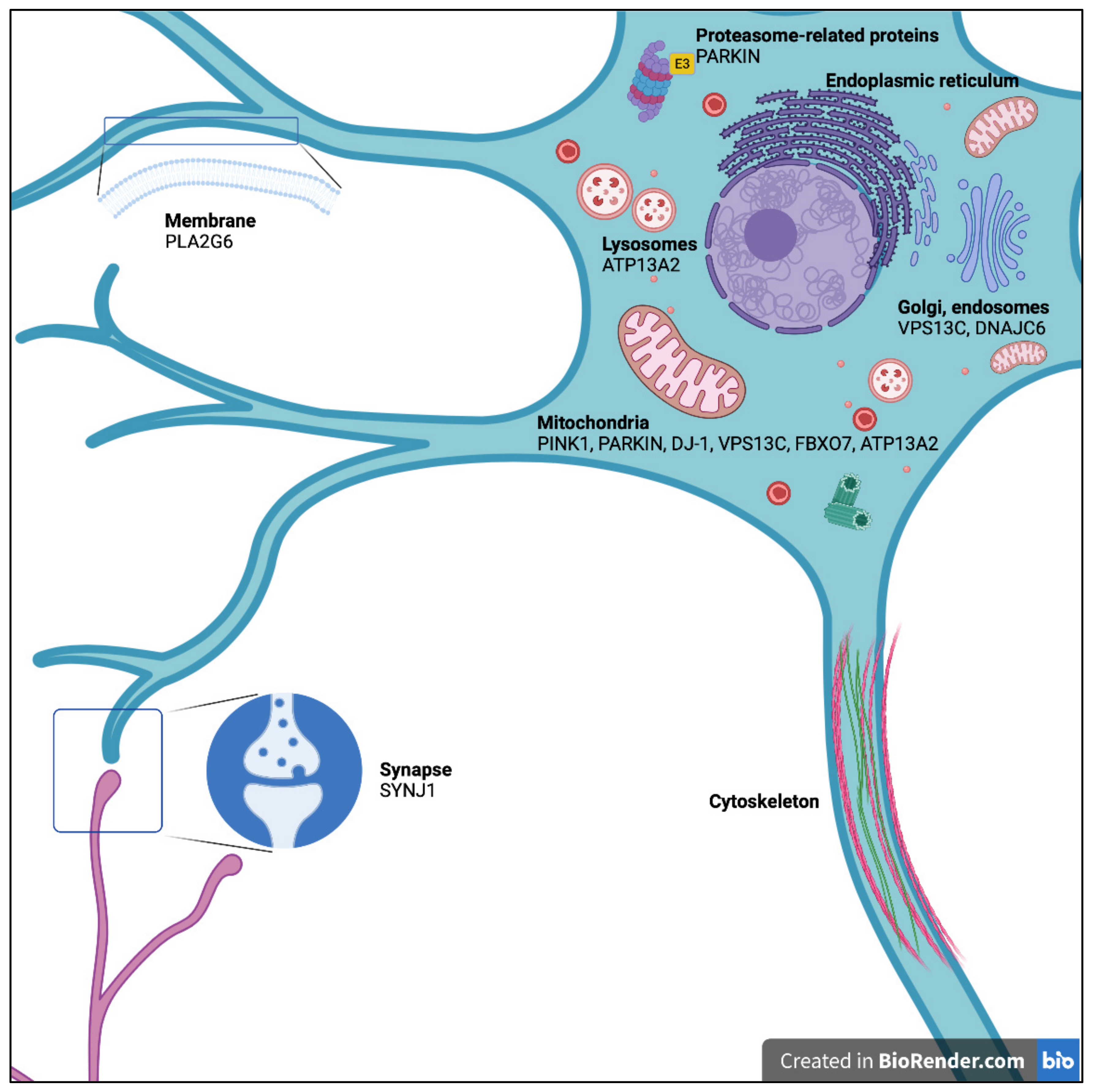
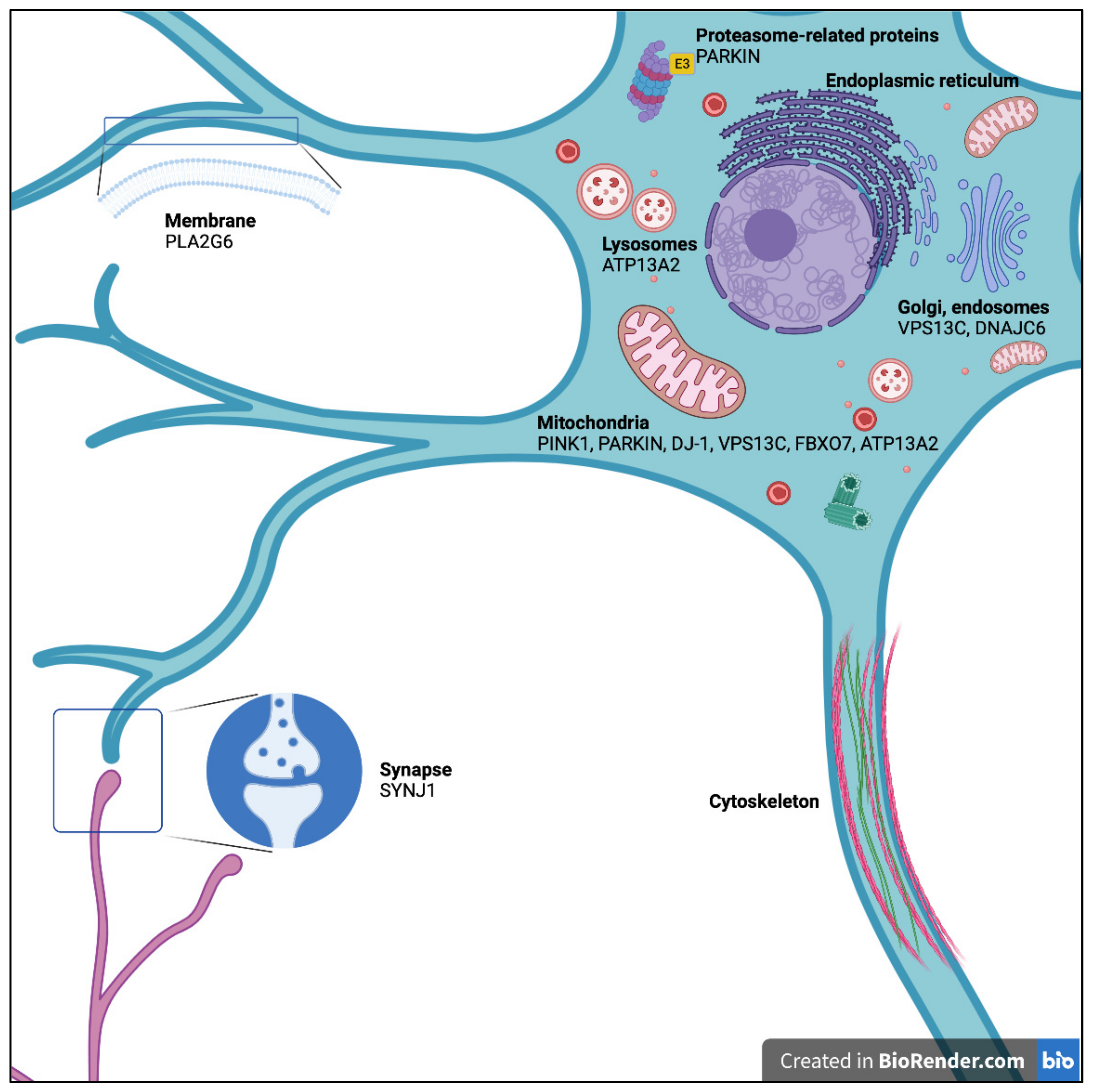
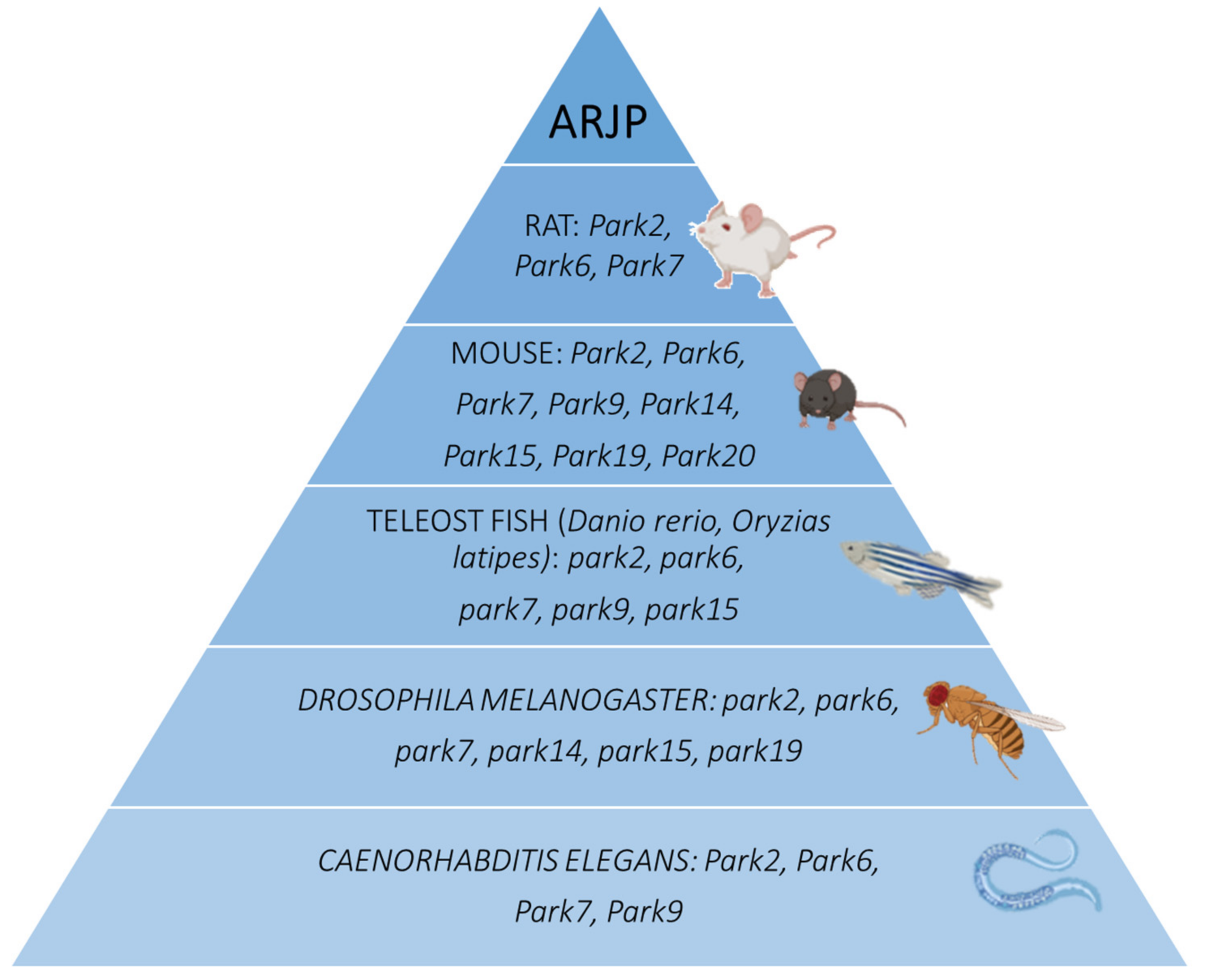
2. PARK2: The Parkin RBR E3 Ubiquitin Protein Ligase Gene (PARKIN)
2.1. Mouse Model
2.2. Rat Model
2.3. Zebrafish (Danio rerio)
2.4. Drosophila melanogaster
2.5. Caenorhabditis elegans
3. PARK6: PTEN-Induced Putative Kinase 1 (PINK1)
3.1. Mouse Model
3.2. Rat Model
3.3. Zebrafish (Danio rerio)
3.4. Medaka Fish (Oryzias latipes)
3.5. Drosophila melanogaster
3.6. Caenorhabditis elegans
4. PARK7: The Parkinsonism-Associated Deglycase Gene (DJ-1)
4.1. Mouse Model
4.2. Rat Model
4.3. Zebrafish (Danio rerio)
4.4. Drosophila melanogaster
4.5. Caenorhabditis elegans
5. PARK9: The ATPase 13A2 Gene (ATP13A2)
5.1. Mouse Model
5.2. Zebrafish (Danio rerio)
5.3. Medaka Fish (Oryzias latipes)
5.4. Caenorhabditis elegans
6. PARK14: The Phospholipase A2 Group VI Gene (PLA2G6)
6.1. Mouse Model
6.2. Drosophila melanogaster
7. PARK15: The F-Box Protein 7 Gene (FBXO7)
7.1. Mouse Model
7.2. Zebrafish (Danio rerio)
7.3. Drosophila melanogaster
8. PARK19: The DnaJ Heat Shock Protein Family (Hsp40) Member C6 Gene (DNAJC6)
8.1. Mouse Model
8.2. Drosophila melanogaster
9. PARK20: The Synaptojanin 1 Gene (SYNJ1)
Mouse Model
10. PARK23: The Vacuolar Protein-Sorting 13 Homolog C Gene (VPS13C)
11. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| (pS129) α-syn | phospho-Ser129 α-synuclein |
| 5-HT | serotonin |
| 6-OHDA | 6-hydroxydopamine |
| ADE | anterior deirids neurons |
| AM | anteromedial |
| ANAD | atypical neuroaxonal dystrophy |
| AR EOPD | autosomal recessive early-onset Parkinson’s Disease |
| AR | autosomal-recessive |
| ARJP | autosomal recessive Juvenile Parkinsonism |
| ARPD | autosomal recessive Parkinson’s disease |
| ATP | adenosine triphosphate |
| BAC | bacterial artificial chromosome |
| BMP | lysosomal lipid Bis(monoacylglycero)phosphate |
| CAN | canal-associated neurons |
| CATD | cathepsin D |
| CEP | chepalic sensilla neurons |
| ChAc | chorea-acanthocytosis syndrome |
| CME | clathrin-mediated endocytosis |
| CNS | central nervous system |
| DA | dopamine |
| DAT | dopamine transporter |
| DCCD | dicyclohexylcarbodiimide |
| DHA | docosahexaenoic acid |
| DKO | double-KO |
| DL | dorsolateral |
| DM | dorsomedial |
| DOPAC | 3,4-dihydroxyphenylacetic acid |
| Dpf | days postfertilization |
| DRP1 | dynamin related protein-1 |
| ENU | N-ethyl-N-nitrosourea |
| ETC | electron transport chain |
| FBP | F-box-containing protein |
| FBXO7 | F-box protein 7 |
| FCCP | carbonyl cyanide-p-trifluoromethoxyphenylhydrazone |
| GAK | cyclin G-associated kinase protein |
| GFAP | glia fibrillary acidic protein |
| GFP | green fluorescent protein |
| GSK-3β | glycogen synthase kinase 3 β |
| GWAS | genome-wide association study |
| H2O2 | hydrogen peroxide |
| HAD | haloacid dehalogenase domain |
| HIF | hypoxia-inducible factor |
| Hpf | hours post-fertilization |
| HPLC | high performance liquid chromatography |
| HVA | homovanillic acid |
| IMS | mitochondrial intermembrane space |
| INAD | infantile neuroaxonal dystrophy |
| KD | knock-down |
| KI | knock-in |
| KO | knock-out |
| LAMP | lysosome-associated membrane glycoprotein |
| LC | locus coeruleus |
| LC-MS/MS | liquid-chromatography tandem mass spectrometry |
| LGR | L-glutathione reduced |
| LTD | long-term depression |
| LTP | long-term potentiation |
| MAO | monoamine oxidase |
| M-CATD | mature form of pH dependent lysosomal enzyme aspartyl protease ASP-3/CATD |
| MEFs | mouse embryonic fibroblasts |
| MFN2 | mitofusin-2 |
| Mn | manganese |
| MO | morpholino |
| MPP | mitochondrial processing peptidase |
| MPP+ | 1-methyl-4-phenylpyridinium |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrapyridine |
| mPTP | mitochondrial permeability transition pore |
| mtDNA | mitochondrial DNA |
| MTS | mitochondrial targeting sequence |
| NAC | N-acetyl cysteine |
| NBIA | neurodegeneration with brain iron accumulation |
| NEO | neomycin |
| NEX | neuronal helix-loop-helix protein-1 |
| OCR | oxygen consumption rate |
| P | postnatal day |
| PAG | periaqueductal gray |
| PARL | presenilin-associated rhomboid-like protease |
| P-CATD | premature form of pH dependent lysosomal enzyme aspartyl protease ASP-3/CATD |
| PD | Parkinson’s disease |
| PDE | posterior deirids neurons |
| PI | phosphatidylinositol |
| PINK1 | (PTEN)-induced kinase 1 |
| PNS | peripheral nervous system |
| PPM1/2 | paired posterior medial 1 and 2 DA neuron clusters |
| PPM3 | paired posterior medial 3 DA neuron cluster |
| PPS | Parkinsonian-pyramidal syndrome |
| rAAV2/8 | recombinant type 2 adeno-associated viral vector pseudotyped with type 8 capsid |
| RNAi | RNA interference |
| RONS | reactive oxygen and nitrogen species |
| ROS | reactive oxygen species |
| SNc | substantia nigra pars compacta |
| β-PEA | β-phenylenthylamine |
| TGF-β | transforming growth factor-β |
| TH | tyrosine hydroxylase |
| TILLING | targeting induced local lesions in genomes |
| TIM23 | translocase of the inner mitochondrial membrane |
| TMD | transmembrane domain |
| TOM | translocase of the outer mitochondrial membrane |
| TOM20 | mitochondrial outer membrane protein |
| TUNEL | terminal deoxynucleotidyl transferase dUTP nick end labeling |
| USVs | ultrasound vocalizations |
| UVC | ultraviolet C |
| VEGF | vascular endothelial growth factor |
| VMAT2 | vesicular monoamine transporter type-2 |
| VPS | vacuolar protein sorting |
| WES | whole-exome sequencing |
| WT | wild type |
| ZFN | zinc finger nuclease |
| α-syn | α-synuclein |
References
- GBD 2016 Parkinson’s Disease Collaborators. Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef] [Green Version]
- Shulman, J.M.; De Jager, P.L.; Feany, M.B. Parkinson’s disease: Genetics and pathogenesis. Annu. Rev. Pathol. 2011, 6, 193–222. [Google Scholar] [CrossRef] [Green Version]
- Obeso, J.A.; Stamelou, M.; Goetz, C.G.; Poewe, W.; Lang, A.E.; Weintraub, D.; Burn, D.; Halliday, G.M.; Bezard, E.; Przedborski, S.; et al. Past, present, and future of Parkinson’s disease: A special essay on the 200th Anniversary of the Shaking Palsy. Mov. Disord. 2017, 32, 1264–1310. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Wang, P.; Jankovic, J. The genetics of Parkinson disease. Ageing Res. Rev. 2018, 42, 72–85. [Google Scholar] [CrossRef]
- Vingill, S.; Connor-Robson, N.; Wade-Martins, R. Are rodent models of Parkinson’s disease behaving as they should? Behav. Brain Res. 2018, 352, 133–141. [Google Scholar] [CrossRef]
- Chia, S.J.; Tan, E.K.; Chao, Y.X. Historical Perspective: Models of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 2464. [Google Scholar] [CrossRef] [Green Version]
- Vaz, R.L.; Outeiro, T.F.; Ferreira, J.J. Zebrafish as an animal model for drug discovery in Parkinson’s disease and other movement disorders: A systematic review. Front. Neurol. 2018, 9, 347. [Google Scholar] [CrossRef]
- Xi, Y.; Ryan, J.; Noble, S.; Yu, M.; Yilbas, A.E.; Ekker, M. Impaired dopaminergic neuron development and locomotor function in zebrafish with loss of pink1 function. Eur. J. Neurosci. 2010, 31, 623–633. [Google Scholar] [CrossRef]
- Blandini, F.; Armentero, M.T. Animal models of Parkinson’s disease. FEBS J. 2012, 279, 1156–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, H.; Uemura, N.; Yamakado, H.; Takeda, S.; Takahashi, R. Exploring the pathogenetic mechanisms underlying Parkinson’s disease in medaka fish. J. Parkinsons Dis. 2014, 4, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Yu, J. Modeling Parkinson’s disease in drosophila: What have we learned for dominant traits? Front. Neurol. 2018, 9, 228. [Google Scholar] [CrossRef] [Green Version]
- Cooper, J.F.; Van Raamsdonk, J.M. Modeling Parkinson’s disease in C. elegans. J. Parkinsons Dis. 2018, 8, 17–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Hattori, N.; Matsumine, H.; Asakawa, S.; Kitada, T.; Yoshino, H.; Elibol, B.; Brookes, A.J.; Yamamura, Y.; Kobayashi, T.; Wang, M.; et al. Point mutations (Thr240Arg and Gln311Stop) [correction of Thr240Arg and Ala311Stop] in the Parkin gene. Biochem. Biophys. Res. Commun. 1998, 249, 754–758. [Google Scholar] [CrossRef]
- Klein, C.; Schlossmacher, M.G. The genetics of Parkinson disease: Implications for neurological care. Nat. Clin. Pract. Neurol. 2006, 2, 136–146. [Google Scholar] [CrossRef]
- Grunewald, A.; Kasten, M.; Ziegler, A.; Klein, C. Next-generation phenotyping using the parkin example: Time to catch up with genetics. JAMA Neurol. 2013, 70, 1186–1191. [Google Scholar] [CrossRef]
- Mitsuyama, S.; Ohtsubo, M.; Minoshima, S.; Shimizu, N. The KM-parkin-DB: A sub-set mutationview database specialized for PARK2 (PARKIN) variants. Hum. Mutat. 2015, 36, E2430–E2440. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.W.; Hang, L.; Yao, T.P.; Lim, K.L. Parkin regulation and neurodegenerative disorders. Front. Aging Neurosci. 2015, 7, 248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cookson, M.R. Parkin’s substrates and the pathways leading to neuronal damage. Neuromol. Med. 2003, 3, 1–13. [Google Scholar] [CrossRef]
- Hattori, N.; Mizuno, Y. Pathogenetic mechanisms of parkin in Parkinson’s disease. Lancet 2004, 364, 722–724. [Google Scholar] [CrossRef]
- Mata, I.F.; Lockhart, P.J.; Farrer, M.J. Parkin genetics: One model for Parkinson’s disease. Hum. Mol. Genet. 2004, 13, R127–R133. [Google Scholar] [CrossRef] [Green Version]
- Itier, J.M.; Ibanez, P.; Mena, M.A.; Abbas, N.; Cohen-Salmon, C.; Bohme, G.A.; Laville, M.; Pratt, J.; Corti, O.; Pradier, L.; et al. Parkin gene inactivation alters behaviour and dopamine neurotransmission in the mouse. Hum. Mol. Genet. 2003, 12, 2277–2291. [Google Scholar] [CrossRef]
- Goldberg, M.S.; Fleming, S.M.; Palacino, J.J.; Cepeda, C.; Lam, H.A.; Bhatnagar, A.; Meloni, E.G.; Wu, N.; Ackerson, L.C.; Klapstein, G.J.; et al. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J. Biol. Chem. 2003, 278, 43628–43635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palacino, J.J.; Sagi, D.; Goldberg, M.S.; Krauss, S.; Motz, C.; Wacker, M.; Klose, J.; Shen, J. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J. Biol. Chem. 2004, 279, 18614–18622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stichel, C.C.; Zhu, X.R.; Bader, V.; Linnartz, B.; Schmidt, S.; Lubbert, H. Mono- and double-mutant mouse models of Parkinson’s disease display severe mitochondrial damage. Hum. Mol. Genet. 2007, 16, 2377–2393. [Google Scholar] [CrossRef] [Green Version]
- Perez, F.A.; Palmiter, R.D. Parkin-deficient mice are not a robust model of parkinsonism. Proc. Natl Acad. Sci. USA 2005, 102, 2174–2179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, S.; Chiba, T.; Nishiyama, S.; Kakiuchi, T.; Tsukada, H.; Hatano, T.; Fukuda, T.; Yasoshima, Y.; Kai, N.; Kobayashi, K.; et al. Decline of striatal dopamine release in parkin-deficient mice shown by ex vivo autoradiography. J. Neurosci. Res. 2006, 84, 1350–1357. [Google Scholar] [CrossRef] [PubMed]
- von Coelln, R.; Thomas, B.; Savitt, J.M.; Lim, K.-L.; Sasaki, M.; Hess, E.J.; Dawson, V.; Dawson, T.M. Loss of locus coeruleus neurons and reduced startle in parkin null mice. Proc. Natl. Acad. Sci. USA 2004, 101, 10744–10749. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.-H.; Ko, H.S.; Kang, H.; Lee, Y.; Lee, Y.-I.; Pletinkova, O.; Troconso, J.C.; Dawson, V.; Dawson, T.M. PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson’s disease. Cell 2011, 144, 689–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevens, D.A.; Lee, Y.; Kang, H.C.; Lee, B.D.; Lee, Y.I.; Bower, A.; Jiang, H.; Kang, S.; Andrabi, S.A.; Dawson, V.L.; et al. Parkin loss leads to PARIS-dependent declines in mitochondrial mass and respiration. Proc. Natl. Acad. Sci. USA 2015, 112, 11696–11701. [Google Scholar] [CrossRef] [Green Version]
- Lorenzetti, D.; Antalffy, B.; Vogel, H.; Noveroske, J.; Armstrong, D.; Justice, M. The neurological mutant quaking(viable) is Parkin deficient. Mamm. Genome 2004, 15, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Ebersole, T.A.; Chen, Q.; Justice, M.J.; Artzt, K. The quaking gene product necessary in embryogenesis and myelination combines features of RNA binding and signal transduction proteins. Nat. Genet. 1996, 12, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.-H.; Fleming, S.M.; Meurers, B.; Ackerson, L.C.; Mortazavi, F.; Lo, V.; Hernandez, D.; Sulzer, D.; Jackson, G.R.; Maidment, N.T.; et al. Bacterial artificial chromosome transgenic mice expressing a truncated mutant parkin exhibit age-dependent hypokinetic motor deficits, dopaminergic neuron degeneration, and accumulation of proteinase K-resistant alpha-synuclein. J. Neurosci. 2009, 29, 1962–1976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dave, K.D.; De Silva, S.; Sheth, N.P.; Ramboz, S.; Beck, M.J.; Quang, C.; Switzer, R.C.; Ahmad, S.O.; Sunkin, S.M.; Walker, D.; et al. Phenotypic characterization of recessive gene knockout rat models of Parkinson’s disease. Neurobiol. Dis. 2014, 70, 190–203. [Google Scholar] [CrossRef] [Green Version]
- Gemechu, J.M.; Sharma, A.; Yu, D.; Xie, Y.; Merkel, O.M.; Moszczynska, A. Characterization of dopaminergic system in the striatum of young adult Park2-/- knockout rats. Sci. Rep. 2018, 8, 1517. [Google Scholar] [CrossRef] [Green Version]
- Stauch, K.L.; Villeneuve, L.M.; Purnell, P.R.; Pandey, S.; Guda, C.; Fox, H.S. SWATH-MS proteome profiling data comparison of DJ-1, Parkin, and PINK1 knockout rat striatal mitochondria. Data Brief. 2016, 9, 589–593. [Google Scholar] [CrossRef] [Green Version]
- Van Rompuy, A.S.; Lobbestael, E.; Van der Perren, A.; Van den Haute, C.; Baekelandt, V. Long-term overexpression of human wild-type and T240R mutant Parkin in rat substantia nigra induces progressive dopaminergic neurodegeneration. J. Neuropathol. Exp. Neurol. 2014, 73, 159–174. [Google Scholar] [CrossRef] [Green Version]
- Flinn, L.; Mortiboys, H.; Volkmann, K.; Koster, R.W.; Ingham, P.W.; Bandmann, O. Complex I deficiency and dopaminergic neuronal cell loss in parkin-deficient zebrafish (Danio rerio). Brain 2009, 132, 1613–1623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greene, J.C.; Whitworth, A.J.; Kuo, I.; Andrews, L.A.; Feany, M.B.; Pallanck, L.J. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc. Natl. Acad. Sci. USA 2003, 100, 4078–4083. [Google Scholar] [CrossRef] [Green Version]
- Whitworth, A.J.; Theodore, D.A.; Greene, J.C.; Benes, H.; Wes, P.D.; Pallanck, L.J. Increased glutathione S-transferase activity rescues dopaminergic neuron loss in a Drosophila model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 8024–8029. [Google Scholar] [CrossRef] [Green Version]
- Pesah, Y.; Pham, T.; Burgess, H.; Middlebrooks, B.; Verstreken, P.; Zhou, Y.; Mark Harding, H.B. Drosophila parkin mutants have decreased mass and cell size and increased sensitivity to oxygen radical stress. Development 2004, 131, 2183–2194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Lu, R.; Ouyang, X.; Ho, M.W.L.; Chia, W.; Yu, F.; Lim, K.-L. Drosophila overexpressing parkin R275W mutant exhibits dopaminergic neuron degeneration and mitochondrial abnormalities. J. Neurosci. 2007, 27, 8563–8570. [Google Scholar] [CrossRef] [Green Version]
- Cha, G.-H.; Kim, S.; Park, J.; Lee, E.; Kim, M.; Lee, S.B.; Kim, J.M.; Chung, J.; Cho, K.S. Parkin negatively regulates JNK pathway in the dopaminergic neurons of Drosophila. Proc. Natl. Acad. Sci. USA 2005, 102, 10345–10350. [Google Scholar] [CrossRef] [Green Version]
- Sang, T.K.; Chang, H.Y.; Lawless, G.M.; Ratnaparkhi, A.; Mee, L.; Ackerson, L.C.; Maidment, N.T.; Krantz, D.E.; Jackson, G.R. A Drosophila model of mutant human parkin-induced toxicity demonstrates selective loss of dopaminergic neurons and dependence on cellular dopamine. J. Neurosci. 2007, 27, 981–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ved, R.; Saha, S.; Westlund, B.; Perier, C.; Burnam, L.; Sluder, A.; Hoener, M.; Rodrigues, C.M.P.; Alfonso, A.; Steer, C.; et al. Similar patterns of mitochondrial vulnerability and rescue induced by genetic modification of alpha-synuclein, parkin, and DJ-1 in Caenorhabditis elegans. J. Biol. Chem. 2005, 280, 42655–42668. [Google Scholar] [CrossRef] [Green Version]
- Bornhorst, J.; Chakraborty, S.; Meyer, S.; Lohren, H.; Brinkhaus, S.G.; Knight, A.L.; Caldwell, K.; Caldwell, G.; Karst, U.; Schwerdtle, T.; et al. The effects of pdr1, djr1.1 and pink1 loss in manganese-induced toxicity and the role of alpha-synuclein in C. elegans. Metallomics 2014, 6, 476–490. [Google Scholar] [CrossRef] [Green Version]
- Poulopoulos, M.; Levy, O.A.; Alcalay, R.N. The neuropathology of genetic Parkinson’s disease. Mov. Disord. 2012, 27, 831–842. [Google Scholar] [CrossRef] [PubMed]
- El-Brolosy, M.A.; Stainier, D.Y.R. Genetic compensation: A phenomenon in search of mechanisms. PLoS Genet. 2017, 13, e1006780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascual, A.; Hidalgo-Figueroa, M.; Piruat, J.I.; Pintado, C.O.; Gomez-Diaz, R.; Lopez-Barneo, J. Absolute requirement of GDNF for adult catecholaminergic neuron survival. Nat. Neurosci. 2008, 11, 755–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsey, C.P.; Giasson, B.I. Identification and characterization of a novel endogenous murine parkin mutation. J. Neurochem. 2010, 113, 402–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitada, T.; Asakawa, S.; Minoshima, S.; Mizuno, Y.; Shimizu, N. Molecular cloning, gene expression, and identification of a splicing variant of the mouse parkin gene. Mamm. Genome 2000, 11, 417–421. [Google Scholar] [CrossRef]
- Regoni, M.; Cattaneo, S.; Mercatelli, D.; Novello, S.; Passoni, A.; Bagnati, R.; Davoli, E.; Croci, L.; Consalez, G.G.; Albanese, F.; et al. Pharmacological antagonism of kainate receptor rescues dysfunction and loss of dopamine neurons in a mouse model of human parkin-induced toxicity. Cell Death Dis. 2020, 11, 963. [Google Scholar] [CrossRef]
- Creed, R.B.; Goldberg, M.S. New developments in genetic rat models of Parkinson’s disease. Mov. Disord. 2018, 33, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Cookson, M.R.; Lockhart, P.J.; McLendon, C.; O’Farrell, C.; Schlossmacher, M.; Farrer, M.J. RING finger 1 mutations in Parkin produce altered localization of the protein. Hum. Mol. Genet. 2003, 12, 2957–2965. [Google Scholar] [CrossRef] [Green Version]
- Bandmann, O.; Burton, E.A. Genetic zebrafish models of neurodegenerative diseases. Neurobiol. Dis. 2010, 40, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, Y.; Takeda, S.; Furutani-Seiki, M.; Kamei, Y.; Todo, T.; Sasado, T.; Deguchi, T.; Kondoh, H.; Mudde, J.; Yamazoe, M.; et al. Generation of medaka gene knockout models by target-selected mutagenesis. Genome Biol. 2006, 7, R116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [Green Version]
- Voigt, A.; Berlemann, L.A.; Winklhofer, K.F. The mitochondrial kinase PINK1: Functions beyond mitophagy. J. Neurochem. 2016, 139, 232–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silvestri, L.; Caputo, V.; Bellacchio, E.; Atorino, L.; Dallapiccola, B.; Valente, E.M.; Casari, G. Mitochondrial import and enzymatic activity of PINK1 mutants associated to recessive parkinsonism. Hum. Mol. Genet. 2005, 14, 3477–3492. [Google Scholar] [CrossRef] [Green Version]
- Meissner, C.; Lorenz, H.; Weihofen, A.; Selkoe, D.J.; Lemberg, M.K. The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J. Neurochem. 2011, 117, 856–867. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [Green Version]
- Sim, C.H.; Gabriel, K.; Mills, R.D.; Culvenor, J.G.; Cheng, H.C. Analysis of the regulatory and catalytic domains of PTEN-induced kinase-1 (PINK1). Hum. Mutat. 2012, 33, 1408–1422. [Google Scholar] [CrossRef]
- Zhou, C.; Huang, Y.; Shao, Y.; May, J.; Prou, D.; Perier, C.; Dauer, W.; Schon, E.A.; Przedborski, S. The kinase domain of mitochondrial PINK1 faces the cytoplasm. Proc. Natl. Acad. Sci. USA 2008, 105, 12022–12027. [Google Scholar] [CrossRef] [Green Version]
- Springer, W.; Kahle, P.J. Regulation of PINK1-Parkin-mediated mitophagy. Autophagy 2011, 7, 266–278. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, S.; Wood-Kaczmar, A.; Yao, Z.; Plun-Favreau, H.; Deas, E.; Klupsch, K.; Downward, J.; Latchman, D.S.; Tabrizi, S.; Wood, N.; et al. PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Mol. Cell. 2009, 33, 627–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trancikova, A.; Tsika, E.; Moore, D.J. Mitochondrial dysfunction in genetic animal models of Parkinson’s disease. Antioxid. Redox Signal. 2012, 16, 896–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Falkenburger, B.H.; Schulz, J.B.; Tieu, K.; Xu, Z.; Xia, X.G. Silencing of the Pink1 gene expression by conditional RNAi does not induce dopaminergic neuron death in mice. Int. J. Biol. Sci. 2007, 3, 242–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautier, C.A.; Kitada, T.; Shen, J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc. Natl. Acad. Sci. USA 2008, 105, 11364–11369. [Google Scholar] [CrossRef] [Green Version]
- Kitada, T.; Pisani, A.; Porter, D.R.; Yamaguchi, H.; Tscherter, A.; Martella, G.; Bonsi, P.; Zhang, C.; Pothos, E.; Shen, J. Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc. Natl. Acad. Sci. USA 2007, 104, 11441–11446. [Google Scholar] [CrossRef] [Green Version]
- Glasl, L.; Kloos, K.; Giesert, F.; Roethig, A.; Di Benedetto, B.; Kuhn, R. Pink1-deficiency in mice impairs gait, olfaction and serotonergic innervation of the olfactory bulb. Exp. Neurol. 2012, 235, 214–227. [Google Scholar] [CrossRef]
- Kelm-Nelson, C.A.; Brauer, A.F.L.; Barth, K.J.; Lake, J.M.; Sinnen, M.L.K.; Stehula, F.J. Characterization of early-onset motor deficits in the Pink1-/- mouse model of Parkinson disease. Brain Res. 2018, 1680, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gispert, S.; Ricciardi, F.; Kurz, A.; Azizov, M.; Hoepken, H.-H.; Becker, D.; Voos, W.; Leuner, K.; Müller, W.E.; Kudin, A.P.; et al. Parkinson phenotype in aged PINK1-deficient mice is accompanied by progressive mitochondrial dysfunction in absence of neurodegeneration. PLoS ONE 2009, 4, e5777. [Google Scholar] [CrossRef]
- Wood-Kaczmar, A.; Gandhi, S.; Yao, Z.; Abramov, A.Y.; Miljan, E.A.; Keen, G. PINK1 is necessary for long term survival and mitochondrial function in human dopaminergic neurons. PLoS ONE 2008, 3, e2455. [Google Scholar] [CrossRef]
- Sun, J.; Kouranova, E.; Cui, X.; Mach, R.H.; Xu, J. Regulation of dopamine presynaptic markers and receptors in the striatum of DJ-1 and Pink1 knockout rats. Neurosci. Lett. 2013, 557, 123–128. [Google Scholar] [CrossRef] [Green Version]
- Villeneuve, L.M.; Purnell, P.R.; Boska, M.D.; Fox, H.S. Early expression of Parkinson’s disease-related mitochondrial abnormalities in PINK1 knockout rats. Mol. Neurobiol. 2016, 53, 171–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, L.M.; Kelm-Nelson, C.; Hilby, B.L.; Blue, K.V.; Rajamanickam, E.S.P.; Pultorak, J.D.; Fleming, S.M.; Ciucci, M.R. Evidence for early and progressive ultrasonic vocalization and oromotor deficits in a PINK1 gene knockout rat model of Parkinson’s disease. J. Neurosci. Res. 2015, 93, 1713–1727. [Google Scholar] [CrossRef] [Green Version]
- Pultorak, J.D.; Kelm-Nelson, C.A.; Holt, L.R.; Blue, K.V.; Ciucci, M.R.; Johnson, A.M. Decreased approach behavior and nucleus accumbens immediate early gene expression in response to Parkinsonian ultrasonic vocalizations in rats. Soc. Neurosci. 2016, 11, 365–379. [Google Scholar] [CrossRef] [Green Version]
- Anichtchik, O.; Diekmann, H.; Fleming, A.; Roach, A.; Goldsmith, P.; Rubinsztein, D.C. Loss of PINK1 function affects development and results in neurodegeneration in zebrafish. J. Neurosci. 2008, 28, 8199–8207. [Google Scholar] [CrossRef]
- Priyadarshini, M.; Tuimala, J.; Chen, Y.C.; Panula, P. A zebrafish model of PINK1 deficiency reveals key pathway dysfunction including HIF signaling. Neurobiol. Dis. 2013, 54, 127–138. [Google Scholar] [CrossRef]
- Sallinen, V.; Kolehmainen, J.; Priyadarshini, M.; Toleikyte, G.; Chen, Y.C.; Panula, P. Dopaminergic cell damage and vulnerability to MPTP in Pink1 knockdown zebrafish. Neurobiol. Dis. 2010, 40, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Flinn, L.J.; Keatinge, M.; Bretaud, S.; Mortiboys, H.; Matsui, H.; De Felice, E.; Woodroof, H.I.; Brown, L.; McTighe, A.; Soellner, R.; et al. TigarB causes mitochondrial dysfunction and neuronal loss in PINK1 deficiency. Ann. Neurol. 2013, 74, 837–847. [Google Scholar] [CrossRef] [Green Version]
- Matsui, H.; Taniguchi, Y.; Inoue, H.; Kobayashi, Y.; Sakaki, Y.; Toyoda, A. Loss of PINK1 in medaka fish (Oryzias latipes) causes late-onset decrease in spontaneous movement. Neurosci. Res. 2010, 66, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lee, S.B.; Lee, S.; Kim, Y.; Song, S.; Kim, S. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 2006, 441, 1157–1161. [Google Scholar] [CrossRef] [PubMed]
- Clark, I.E.; Dodson, M.W.; Jiang, C.; Cao, J.H.; Huh, J.R.; Seol, J.H.; Yoo, S.J.; Hay, B.A.; Guo, M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 2006, 441, 1162–1166. [Google Scholar] [CrossRef] [PubMed]
- Morais, V.; Verstreken, P.; Roethig, A.; Smet, J.; Snellinx, A.; Vanbrabant, M.; Haddad, D.; Frezza, C.; Mandemakers, W.; Vogt-Weisenhorn, D.; et al. Parkinson’s disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. EMBO Mol. Med. 2009, 1, 99–111. [Google Scholar] [CrossRef]
- Liu, W.; Acin-Perez, R.; Geghman, K.D.; Manfredi, G.; Lu, B.; Li, C. Pink1 regulates the oxidative phosphorylation machinery via mitochondrial fission. Proc. Natl. Acad. Sci. USA 2011, 108, 12920–12924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Gehrke, S.; Imai, Y.; Huang, Z.; Ouyang, Y.; Wang, J.W. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc. Natl. Acad. Sci. USA 2006, 103, 10793–10798. [Google Scholar] [CrossRef] [Green Version]
- Samann, J.; Hegermann, J.; von Gromoff, E.; Eimer, S.; Baumeister, R.; Schmidt, E. Caenorhabditits elegans LRK-1 and PINK-1 act antagonistically in stress response and neurite outgrowth. J. Biol. Chem. 2009, 284, 16482–16491. [Google Scholar] [CrossRef] [Green Version]
- Luz, A.L.; Rooney, J.P.; Kubik, L.L.; Gonzalez, C.P.; Song, D.H.; Meyer, J.N. Mitochondrial morphology and fundamental parameters of the mitochondrial respiratory chain are altered in Caenorhabditis elegans strains deficient in mitochondrial dynamics and homeostasis processes. PLoS ONE 2015, 10, e0130940. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 2015, 521, 525–528. [Google Scholar] [CrossRef]
- Bess, A.S.; Crocker, T.L.; Ryde, I.T.; Meyer, J.N. Mitochondrial dynamics and autophagy aid in removal of persistent mitochondrial DNA damage in Caenorhabditis elegans. Nucleic Acids Res. 2012, 40, 7916–7931. [Google Scholar] [CrossRef] [Green Version]
- Bess, A.S.; Leung, M.C.; Ryde, I.T.; Rooney, J.P.; Hinton, D.E.; Meyer, J.N. Effects of mutations in mitochondrial dynamics-related genes on the mitochondrial response to ultraviolet C radiation in developing Caenorhabditis elegans. Worm 2013, 2, e23763. [Google Scholar] [CrossRef] [Green Version]
- Priyadarshini, M.; Orosco, L.A.; Panula, P.J. Oxidative stress and regulation of Pink1 in zebrafish (Danio rerio). PLoS ONE 2013, 8, e81851. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Ouyang, Y.; Yang, L.; Beal, M.F.; McQuibban, A.; Vogel, O.H.; Lu, B. Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc. Natl. Acad. Sci. USA 2008, 105, 7070–7075. [Google Scholar] [CrossRef] [Green Version]
- Deng, H.; Dodson, M.W.; Huang, H.; Guo, M. The Parkinson’s disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc. Natl. Acad. Sci. USA 2008, 105, 14503–14508. [Google Scholar] [CrossRef] [Green Version]
- Van Duijn, C.M.; Dekker, M.C.; Bonifati, V.; Galjaard, R.J.; Houwing-Duistermaat, J.J.; Snijders, P.J. Park7, a novel locus for autosomal recessive early-onset parkinsonism, on chromosome 1p36. Am. J. Hum. Genet. 2001, 69, 629–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abou-Sleiman, P.M.; Healy, D.G.; Quinn, N.; Lees, A.J.; Wood, N.W. The role of pathogenic DJ-1 mutations in Parkinson’s disease. Ann. Neurol. 2003, 54, 283–286. [Google Scholar] [CrossRef] [PubMed]
- Bonifati, V.; Rizzu, P.; Van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.J.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, M.S.; Pisani, A.; Haburcak, M.; Vortherms, T.A.; Kitada, T.; Costa, C.; Tong, Y.; Martella, G.; Tscherter, A.; Martins, A.; et al. Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ-1. Neuron 2005, 45, 489–496. [Google Scholar] [CrossRef] [Green Version]
- Kahle, P.J.; Waak, J.; Gasser, T. DJ-1 and prevention of oxidative stress in Parkinson’s disease and other age-related disorders. Free Radic. Biol. Med. 2009, 47, 1354–1361. [Google Scholar] [CrossRef]
- Van der Merwe, C.; Jalali Sefid Dashti, Z.; Christoffels, A.; Loos, B.; Bardien, S. Evidence for a common biological pathway linking three Parkinson’s disease-causing genes: Parkin, PINK1 and DJ-1. Eur. J. Neurosci. 2015, 41, 1113–1125. [Google Scholar] [CrossRef]
- Wilhelmus, M.M.; Nijland, P.G.; Drukarch, B.; de Vries, H.E.; van Horssen, J. Involvement and interplay of Parkin, PINK1, and DJ1 in neurodegenerative and neuroinflammatory disorders. Free Radic. Biol. Med. 2012, 53, 983–992. [Google Scholar] [CrossRef]
- Chen, L.; Cagniard, B.; Mathews, T.; Jones, S.; Koh, H.C.; Ding, Y.; Carvey, P.M.; Ling, Z.; Kang, U.; Zhuang, X. Age-dependent motor deficits and dopaminergic dysfunction in DJ-1 null mice. J. Biol. Chem. 2005, 280, 21418–21426. [Google Scholar] [CrossRef] [Green Version]
- Malgieri, G.; Eliezer, D. Structural effects of Parkinson’s disease linked DJ-1 mutations. Protein Sci. 2008, 17, 855–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cookson, M.R. DJ-1, PINK1, and their effects on mitochondrial pathways. Mov. Disord. 2010, 25, S44–S48. [Google Scholar] [CrossRef]
- Kim, R.H.; Smith, P.D.; Aleyasin, H.; Hayley, S.; Mount, M.P.; Pownall, S. Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative stress. Proc. Natl. Acad. Sci. USA 2005, 102, 5215–5220. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Barkow, J.C.; Freed, C.R. Running wheel exercise reduces alpha-synuclein aggregation and improves motor and cognitive function in a transgenic mouse model of Parkinson’s disease. PLoS ONE 2017, 12, e0190160. [Google Scholar] [CrossRef] [PubMed]
- Manning-Bog, A.B.; Caudle, W.M.; Perez, X.A.; Reaney, S.H.; Paletzki, R.; Isla, M.Z. Increased vulnerability of nigrostriatal terminals in DJ-1-deficient mice is mediated by the dopamine transporter. Neurobiol. Dis. 2007, 27, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Giangrasso, D.M.; Furlong, T.M.; Keefe, K.A. Characterization of striatum-mediated behavior and neurochemistry in the DJ-1 knock-out rat model of Parkinson’s disease. Neurobiol. Dis. 2020, 134, 104673. [Google Scholar] [CrossRef]
- Yang, K.M.; Blue, K.V.; Mulholland, H.M.; Kurup, M.P.; Kelm-Nelson, C.A.; Ciucci, M.R. Characterization of oromotor and limb motor dysfunction in the DJ1 -/- model of Parkinson disease. Behav. Brain Res. 2018, 339, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Kyser, T.L.; Dourson, A.J.; McGuire, J.L.; Hemmerle, A.M.; Williams, M.T.; Seroogy, K.B. Characterization of motor and non-motor behavioral alterations in the Dj-1 (PARK7) knockout rat. J. Mol. Neurosci. 2019, 69, 298–311. [Google Scholar] [CrossRef]
- Bretaud, S.; Allen, C.; Ingham, P.W.; Bandmann, O. p53-dependent neuronal cell death in a DJ-1-deficient zebrafish model of Parkinson’s disease. J. Neurochem. 2007, 100, 1626–1635. [Google Scholar] [CrossRef]
- Baulac, S.; Lu, H.; Strahle, J.; Yang, T.; Goldberg, M.S.; Shen, J.; Schlossmacher, M.G.; Lemere, C.A.; Lu, Q.; Xia, W. Increased DJ-1 expression under oxidative stress and in Alzheimer’s disease brains. Mol. Neurodegener. 2009, 4, 12. [Google Scholar] [CrossRef] [Green Version]
- Edson, A.J.; Hushagen, H.A.; Froyset, A.K.; Elda, I.; Khan, E.A.; Di Stefano, A. Dysregulation in the Brain protein profile of zebrafish lacking the Parkinson’s disease-related protein DJ-1. Mol. Neurobiol. 2019, 56, 8306–8322. [Google Scholar] [CrossRef] [PubMed]
- Hughes, G.L.; Lones, M.A.; Bedder, M.; Currie, P.D.; Smith, S.L.; Pownall, M.E. Machine learning discriminates a movement disorder in a zebrafish model of Parkinson’s disease. Dis. Model. Mech. 2020, 13. [Google Scholar] [CrossRef]
- Menzies, F.M.; Yenisetti, S.C.; Min, K.T. Roles of Drosophila DJ-1 in survival of dopaminergic neurons and oxidative stress. Curr. Biol. 2005, 15, 1578–1582. [Google Scholar] [CrossRef]
- Park, J.; Kim, S.Y.; Cha, G.H.; Lee, S.B.; Kim, S.; Chung, J. Drosophila DJ-1 mutants show oxidative stress-sensitive locomotive dysfunction. Gene 2005, 361, 133–139. [Google Scholar] [CrossRef]
- Poudel, S.; Lee, Y. Impaired taste associative memory and memory enhancement by feeding Omija in Parkinson’s disease fly model. Mol. Cells 2018, 41, 646–652. [Google Scholar] [PubMed]
- Meulener, M.; Whitworth, A.; Armstrong-Gold, C.E.; Rizzu, P.; Heutink, P.; Wes, P.; Pallanck, L.J.; Bonini, N. Drosophila DJ-1 mutants are selectively sensitive to environmental toxins associated with Parkinson’s disease. Curr. Biol. 2005, 15, 1572–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro, E.M.C.; Waak, J.; Weber, S.S.; Fiesel, F.C.; Oberhettinger, P.; Schütz, M.; Autenrieth, I.B.; Springer, W.; Kahle, P.J. Parkinson’s disease-associated DJ-1 modulates innate immunity signaling in Caenorhabditis elegans. J. Neural Transm. 2010, 117, 599–604. [Google Scholar] [CrossRef]
- Chen, P.; DeWitt, M.R.; Bornhorst, J.; Soares, F.; Mukhopadhyay, S.; Bowman, A.B.; Aschner, M. Age- and manganese-dependent modulation of dopaminergic phenotypes in a C. elegans DJ-1 genetic model of Parkinson’s disease. Metallomics 2015, 7, 289–298. [Google Scholar] [CrossRef] [Green Version]
- Baptista, M.A.; Dave, K.D.; Sheth, N.P.; De Silva, S.N.; Carlson, K.M.; Aziz, Y.N.; Fiske, B.K.; Sherer, T.B.; Frasier, M.A. A strategy for the generation, characterization and distribution of animal models by The Michael, J. Fox Foundation for Parkinson’s Research. Dis. Model. Mech. 2013, 6, 1316–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Number, S.; Harmuth, F.; Kohl, Z.; Adame, A.; Trejo, M.; Schonig, K. A progressive dopaminergic phenotype associated with neurotoxic conversion of alpha-synuclein in BAC-transgenic rats. Brain 2013, 136, 412–432. [Google Scholar] [CrossRef] [Green Version]
- Ellenbroek, B.; Youn, J. Rodent models in neuroscience research: Is it a rat race? Dis. Model. Mech. 2016, 9, 1079–1087. [Google Scholar] [CrossRef] [Green Version]
- Bai, Q.; Mullett, S.J.; Garver, J.A.; Hinkle, D.A.; Burton, E.A. Zebrafish DJ-1 is evolutionarily conserved and expressed in dopaminergic neurons. Brain Res. 2006, 1113, 33–44. [Google Scholar] [CrossRef]
- Najim al-Din, A.S.; Wriekat, A.; Mubaidin, A.; Dasouki, M.; Hiari, M. Pallido-pyramidal degeneration, supranuclear upgaze paresis and dementia: Kufor-Rakeb syndrome. Acta Neurol. Scand. 1994, 89, 347–352. [Google Scholar] [CrossRef]
- Park, J.S.; Blair, N.F.; Sue, C.M. The role of ATP13A2 in Parkinson’s disease: Clinical phenotypes and molecular mechanisms. Mov. Disord. 2015, 30, 770–779. [Google Scholar] [CrossRef] [PubMed]
- Demirsoy, S.; Martin, S.; Motamedi, S.; van Veen, S.; Holemans, T.; Van den Haute, C. ATP13A2/PARK9 regulates endo-/lysosomal cargo sorting and proteostasis through a novel PI(3, 5)P2-mediated scaffolding function. Hum. Mol. Genet. 2017, 26, 1656–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez, A.; Heimbach, A.; Grundemann, J.; Stiller, B.; Hampshire, D.; Cid, L.P. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 2006, 38, 1184–1191. [Google Scholar] [CrossRef]
- Van Veen, S.; Martin, S.; Van den Haute, C.; Benoy, V.; Lyons, J.; Vanhoutte, R. ATP13A2 deficiency disrupts lysosomal polyamine export. Nature 2020, 578, 419–424. [Google Scholar] [CrossRef]
- Dehay, B.; Ramirez, A.; Martinez-Vicente, M.; Perier, C.; Canron, M.-H.; Doudnikoff, E.; Vital, A.; Vila, M.; Klein, C.; Bezard, E. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc. Natl. Acad. Sci. USA 2012, 109, 9611–9616. [Google Scholar] [CrossRef] [Green Version]
- Gitler, A.D.; Chesi, A.; Geddie, M.L.; Strathearn, K.E.; Hamamichi, S.; Hill, K.J. Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat. Genet. 2009, 41, 308–315. [Google Scholar] [CrossRef] [Green Version]
- Kong, S.M.; Chan, B.K.; Park, J.-S.; Hill, K.J.; Aitken, J.B.; Cottle, L.; Farghaian, H.; Cole, A.R.; Lay, P.; Sue, C.M.; et al. Parkinson’s disease-linked human PARK9/ATP13A2 maintains zinc homeostasis and promotes alpha-Synuclein externalization via exosomes. Hum. Mol. Genet. 2014, 23, 2816–2833. [Google Scholar] [CrossRef] [Green Version]
- Gusdon, A.M.; Zhu, J.; Van Houten, B.; Chu, C.T. ATP13A2 regulates mitochondrial bioenergetics through macroautophagy. Neurobiol Dis. 2012, 45, 962–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usenovic, M.; Tresse, E.; Mazzulli, J.R.; Taylor, J.P.; Krainc, D. Deficiency of ATP13A2 leads to lysosomal dysfunction, alpha-synuclein accumulation, and neurotoxicity. J. Neurosci. 2012, 32, 4240–4246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultheis, P.J.; Fleming, S.M.; Clippinger, A.K.; Lewis, J.; Tsunemi, T.; Giasson, B. Atp13a2-deficient mice exhibit neuronal ceroid lipofuscinosis, limited alpha-synuclein accumulation and age-dependent sensorimotor deficits. Hum. Mol. Genet. 2013, 22, 2067–2082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kett, L.R.; Stiller, B.; Bernath, M.M.; Tasset, I.; Blesa, J.; Jackson-Lewis, V. Alpha-Synuclein-independent histopathological and motor deficits in mice lacking the endolysosomal Parkinsonism protein Atp13a2. J. Neurosci. 2015, 35, 5724–5742. [Google Scholar] [CrossRef] [Green Version]
- Lopes da Fonseca, T.; Correia, A.; Hasselaar, W.; van der Linde, H.C.; Willemsen, R.; Outeiro, T.F. The zebrafish homologue of Parkinson’s disease ATP13A2 is essential for embryonic survival. Brain Res. Bull. 2013, 90, 118–126. [Google Scholar] [CrossRef]
- Heins-Marroquin, U.; Jung, P.P.; Cordero-Maldonado, M.L.; Crawford, A.D.; Linster, C.L. Phenotypic assays in yeast and zebrafish reveal drugs that rescue ATP13A2 deficiency. Brain Commun. 2019, 1, fcz019. [Google Scholar] [CrossRef] [Green Version]
- Nyuzuki, H.; Ito, S.; Nagasaki, K.; Nitta, Y.; Matsui, N.; Saitoh, A. Degeneration of dopaminergic neurons and impaired intracellular trafficking in Atp13a2 deficient zebrafish. IBRO Rep. 2020, 9, 1–8. [Google Scholar] [CrossRef]
- Matsui, H.; Sato, F.; Sato, S.; Koike, M.; Taruno, Y.; Saiki, S. ATP13A2 deficiency induces a decrease in cathepsin D activity, fingerprint-like inclusion body formation, and selective degeneration of dopaminergic neurons. FEBS Lett. 2013, 587, 1316–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand, N.; Holcom, A.; Broussalian, M.; Schmidt, M.; Chinta, S.J.; Lithgow, G.J. Dysregulated iron metabolism in C. elegans catp-6/ATP13A2 mutant impairs mitochondrial function. Neurobiol Dis. 2020, 139, 104786. [Google Scholar] [CrossRef]
- Baesler, J.; Kopp, J.F.; Pohl, G.; Aschner, M.; Haase, H.; Schwerdtle, T.; Bornhorst, J. Zn homeostasis in genetic models of Parkinson’s disease in Caenorhabditis elegans. J. Trace Elem. Med. Biol. 2019, 55, 44–49. [Google Scholar] [CrossRef]
- Bakthavatsalam, S.; Das Sharma, S.; Sonawane, M.; Thirumalai, V.; Datta, A. A zebrafish model of manganism reveals reversible and treatable symptoms that are independent of neurotoxicity. Dis Model. Mech. 2014, 7, 1239–1251. [Google Scholar] [CrossRef] [Green Version]
- Polster, B.; Crosier, M.; Lindsay, S.; Hayflick, S. Expression of PLA2G6 in human fetal development: Implications for infantile neuroaxonal dystrophy. Brain Res. Bull. 2010, 83, 374–379. [Google Scholar] [CrossRef] [Green Version]
- Lin, G.; Lee, P.T.; Chen, K.; Mao, D.; Tan, K.L.; Zuo, Z. Phospholipase PLA2G6, a Parkinsonism-associated gene, affects Vps26 and Vps35, retromer function, and ceramide levels, similar to alpha-synuclein gain. Cell Metab. 2018, 28, 605–618.e6. [Google Scholar] [CrossRef] [Green Version]
- Schneider, S.A.; Bhatia, K.P. Rare causes of dystonia parkinsonism. Curr. Neurol. Neurosci. Rep. 2010, 10, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Paisan-Ruiz, C.; Bhatia, K.P.; Li, A.; Hernandez, D.; Davis, M.; Wood, N.; Hardy, J.; Houlden, H.; Singleton, A.B.; Schneider, S.A. Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann. Neurol. 2009, 65, 19–23. [Google Scholar] [CrossRef]
- Shi, C.-H.; Tang, B.-S.; Wang, L.; Lv, Z.-Y.; Wang, J.; Luo, L.-Z.; Shen, L.; Jiang, H.; Yan, X.-X.; Pan, Q.; et al. PLA2G6 gene mutation in autosomal recessive early-onset parkinsonism in a Chinese cohort. Neurology 2011, 77, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, H.; Tomiyama, H.; Tachibana, N.; Ogaki, K.; Li, Y.; Funayama, M.; Hashimoto, T.; Takashima, S.; Hattori, N. Phenotypic spectrum of patients with PLA2G6 mutation and PARK14-linked parkinsonism. Neurology 2010, 75, 1356–1361. [Google Scholar] [CrossRef]
- Guo, Y.P.; Tang, B.S.; Guo, J.F. PLA2G6-associated neurodegeneration (PLAN): Review of clinical phenotypes and genotypes. Front. Neurol. 2018, 9, 1100. [Google Scholar] [CrossRef] [Green Version]
- Shinzawa, K.; Sumi, H.; Ikawa, M.; Matsuoka, Y.; Okabe, M.; Sakoda, S.; Tsujimoto, Y. Neuroaxonal dystrophy caused by group VIA phospholipase A2 deficiency in mice: A model of human neurodegenerative disease. J. Neurosci. 2008, 28, 2212–2220. [Google Scholar] [CrossRef] [Green Version]
- Beck, G.; Shinzawa, K.; Hayakawa, H.; Baba, K.; Sumi-Akamaru, H.; Tsujimoto, Y. Progressive axonal degeneration of nigrostriatal dopaminergic neurons in calcium-independent phospholipase A2beta knockout mice. PLoS ONE 2016, 11, e0153789. [Google Scholar] [CrossRef] [Green Version]
- Sumi-Akamaru, H.; Beck, G.; Shinzawa, K.; Kato, S.; Riku, Y.; Yoshida, M.; Fujimura, H.; Tsujimoto, Y.; Sakoda, S.; Mochizuki, H. High expression of alpha-synuclein in damaged mitochondria with PLA2G6 dysfunction. Acta Neuropathol. Commun. 2016, 4, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basselin, M.; Rosa, A.O.; Ramadan, E.; Cheon, Y.; Chang, L.; Chen, M. Imaging decreased brain docosahexaenoic acid metabolism and signaling in iPLAbeta (VIA)-deficient mice. J. Lipid Res. 2010, 51, 3166–3173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Yen, A.; Rymarczyk, G.; Asai, H.; Trengrove, C.; Aziz, N. Impairment of PARK14-dependent Ca(2+) signalling is a novel determinant of Parkinson’s disease. Nat. Commun. 2016, 7, 10332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, C.-C.; Lu, C.-S.; Weng, Y.-H.; Chen, Y.-L.; Huang, Y.-Z.; Chen, R.-S.; Cheng, Y.-C.; Huang, Y.-C.; Liu, Y.-C.; Lai, S.-C.; et al. PARK14 (D331Y) PLA2G6 causes early-onset degeneration of substantia nigra dopaminergic neurons by inducing mitochondrial dysfunction, ER stress, mitophagy impairment and transcriptional dysregulation in a knockin mouse model. Mol. Neurobiol. 2019, 56, 3835–3853. [Google Scholar] [CrossRef]
- Mori, A.; Hatano, T.; Inoshita, T.; Shiba-Fukushima, K.; Koinuma, T.; Meng, H.; Kubo, S.-I.; Spratt, S.; Cui, C.; Yamashita, C.; et al. Parkinson’s disease-associated iPLA2-VIA/PLA2G6 regulates neuronal functions and alpha-synuclein stability through membrane remodeling. Proc. Natl. Acad. Sci. USA 2019, 116, 20689–20699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinghorn, K.J.; Castillo-Quan, J.I.; Bartolome, F.; Angelova, P.R.; Li, L.; Pope, S. Loss of PLA2G6 leads to elevated mitochondrial lipid peroxidation and mitochondrial dysfunction. Brain 2015, 138, 1801–1816. [Google Scholar] [CrossRef] [Green Version]
- Shojaee, S.; Sina, F.; Banihosseini, S.S.; Kazemi, M.H.; Kalhor, R.; Shahidi, G.A. Genome-wide linkage analysis of a Parkinsonian-pyramidal syndrome pedigree by 500 K SNP arrays. Am. J. Hum. Genet. 2008, 82, 1375–1384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Fonzo, A.; Dekker, M.; Montagna, P.; Baruzzi, A.; Yonova, E.H.; Guedes, L.C.; Szczerbinska, A.; Zhao, T.; Dubbel-Hulsman, L.O.; Wouters, C.; et al. FBXO7 mutations cause autosomal recessive, early-onset parkinsonian-pyramidal syndrome. Neurology 2009, 72, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, E.; Coquel, A.-S.; Honoré, A.; Gurvit, H.; Hanagasi, H.; Emre, M.; Leutenegger, A.-L.; Drouet, V.; Sahbatou, M.; Guven, G.; et al. A new F-box protein 7 gene mutation causing typical Parkinson’s disease. Mov. Disord. 2015, 30, 1130–1133. [Google Scholar] [CrossRef]
- Wei, L.; Ding, L.; Li, H.; Lin, Y.; Dai, Y.; Xu, X.; Dong, Q.; Lin, Y.; Long, L. Juvenile-onset parkinsonism with pyramidal signs due to compound heterozygous mutations in the F-Box only protein 7 gene. Parkinsonism Relat. Disord. 2018, 47, 76–79. [Google Scholar] [CrossRef]
- Zhao, T.; De Graaff, E.; Breedveld, G.J.; Loda, A.; Severijnen, L.A.; Wouters, C.H. Loss of nuclear activity of the FBXO7 protein in patients with parkinsonian-pyramidal syndrome (PARK15). PLoS ONE 2011, 6, e16983. [Google Scholar] [CrossRef] [Green Version]
- Delgado-Camprubi, M.; Esteras, N.; Soutar, M.P.; Plun-Favreau, H.; Abramov, A.Y. Deficiency of Parkinson’s disease-related gene Fbxo7 is associated with impaired mitochondrial metabolism by PARP activation. Cell Death Differ. 2017, 24, 2210. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.D.; Xie, S.P.; Sathiyamoorthy, S.; Saw, W.T.; Sing, T.Y.; Ng, S.H.; Chua, H.P.H.; Tang, A.M.Y.; Shaffra, F.; Li, Z.; et al. F-box protein 7 mutations promote protein aggregation in mitochondria and inhibit mitophagy. Hum. Mol. Genet. 2015, 24, 6314–6330. [Google Scholar] [CrossRef] [Green Version]
- Vingill, S.; Brockelt, D.; Lancelin, C.; Tatenhorst, L.; Dontcheva, G.; Preisinger, C.; Schwedhelm-Domeyer, N.; Joseph, S.; Mitkovski, M.; Goebbels, S.; et al. Loss of FBXO7 (PARK15) results in reduced proteasome activity and models a parkinsonism-like phenotype in mice. EMBO J. 2016, 35, 2008–2025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, T.; Zondervan-van der Linde, H.; Severijnen, L.A.; Oostra, B.A.; Willemsen, R.; Bonifati, V. Dopaminergic neuronal loss and dopamine-dependent locomotor defects in Fbxo7-deficient zebrafish. PLoS ONE 2012, 7, e48911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goebbels, S.; Bormuth, I.; Bode, U.; Hermanson, O.; Schwab, M.H.; Nave, K.A. Genetic targeting of principal neurons in neocortex and hippocampus of NEX-Cre mice. Genesis 2006, 44, 611–621. [Google Scholar] [CrossRef]
- Savitt, J.M.; Jang, S.S.; Mu, W.; Dawson, V.L.; Dawson, T.M. Bcl-x is required for proper development of the mouse substantia nigra. J. Neurosci. 2005, 25, 6721–6728. [Google Scholar] [CrossRef] [Green Version]
- Olgiati, S.; Quadri, M.; Fang, M.; Rood, J.P.; Saute, J.A.; Chien, H.F. DNAJC6 mutations associated with early-onset Parkinson’s disease. Ann. Neurol. 2016, 79, 244–256. [Google Scholar] [CrossRef]
- Edvardson, S.; Cinnamon, Y.; Ta-Shma, A.; Shaag, A.; Yim, Y.I.; Zenvirt, S. A deleterious mutation in DNAJC6 encoding the neuronal-specific clathrin-uncoating co-chaperone auxilin, is associated with juvenile parkinsonism. PLoS ONE 2012, 7, e36458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koroglu, C.; Baysal, L.; Cetinkaya, M.; Karasoy, H.; Tolun, A. DNAJC6 is responsible for juvenile parkinsonism with phenotypic variability. Parkinsonism Relat. Disord. 2013, 19, 320–324. [Google Scholar] [CrossRef]
- Shi, C.; Li, F.; Yang, J.; Zhang, S.; Mao, C.; Wang, H. DNAJC6 mutations are not common causes of early onset Parkinson’s disease in Chinese Han population. Neurosci. Lett. 2016, 634, 60–62. [Google Scholar] [CrossRef]
- Kanaoka, Y.; Kimura, S.H.; Okazaki, I.; Ikeda, M.; Nojima, H. GAK: A cyclin G associated kinase contains a tensin/auxilin-like domain. FEBS Lett. 1997, 402, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Ahle, S.; Ungewickell, E. Auxilin, a newly identified clathrin-associated protein in coated vesicles from bovine brain. J. Cell Biol. 1990, 111, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Yim, Y.I.; Sun, T.; Wu, L.G.; Raimondi, A.; De Camilli, P.; Eisenberg, E. Endocytosis and clathrin-uncoating defects at synapses of auxilin knockout mice. Proc. Natl. Acad. Sci. USA 2010, 107, 4412–4417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, L.; He, Y.; Ou, J.; Zhao, Y.; Li, R.; Cheng, J. Auxilin underlies progressive locomotor deficits and dopaminergic neuron loss in a Drosophila model of Parkinson’s disease. Cell Rep. 2017, 18, 1132–1143. [Google Scholar] [CrossRef] [Green Version]
- Krebs, C.E.; Karkheiran, S.; Powell, J.C.; Cao, M.; Makarov, V.; Darvish, H. The Sac1 domain of SYNJ1 identified mutated in a family with early-onset progressive Parkinsonism with generalized seizures. Hum. Mutat. 2013, 34, 1200–1207. [Google Scholar] [CrossRef] [Green Version]
- Quadri, M.; Fang, M.; Picillo, M.; Olgiati, S.; Breedveld, G.J.; Graafland, J.; Wu, B.; Xu, F.; Erro, R.; Amboni, M.; et al. Mutation in the SYNJ1 gene associated with autosomal recessive, early-onset Parkinsonism. Hum. Mutat. 2013, 34, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Kirola, L.; Behari, M.; Shishir, C.; Thelma, B.K. Identification of a novel homozygous mutation Arg459Pro in SYNJ1 gene of an Indian family with autosomal recessive juvenile Parkinsonism. Parkinsonism Relat. Disord. 2016, 31, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Rauschendorf, M.A.; Jost, M.; Stock, F.; Zimmer, A.; Rosler, B.; Rijntjes, M. Novel compound heterozygous synaptojanin-1 mutation causes l-dopa-responsive dystonia-parkinsonism syndrome. Mov. Disord. 2017, 32, 478–480. [Google Scholar] [CrossRef] [PubMed]
- Taghavi, S.; Chaouni, R.; Tafakhori, A.; Azcona, L.J.; Firouzabadi, S.G.; Omrani, M.D.; Jamshidi, J.; Emamalizadeh, B.; Shahidi, G.A.; Ahmadi, M.; et al. A clinical and molecular genetic study of 50 families with autosomal recessive Parkinsonism revealed known and novel gene mutations. Mol. Neurobiol. 2018, 55, 3477–3489. [Google Scholar] [CrossRef] [PubMed]
- McPherson, P.S.; Garcia, E.P.; Slepnev, V.I.; David, C.; Zhang, X.; Grabs, D.; Sossini, W.S.; Bauerfeind, R.; Nemoto, Y.; De Camilli, P. A presynaptic inositol-5-phosphatase. Nature 1996, 379, 353–357. [Google Scholar] [CrossRef]
- Cremona, O.; Di Paolo, G.; Wenk, M.R.; Luthi, A.; Kim, W.T.; Takei, K.; Daniell, L.; Nemoto, Y.; Shears, S.; Flavell, R.A.; et al. Essential role of phosphoinositide metabolism in synaptic vesicle recycling. Cell 1999, 99, 179–188. [Google Scholar] [CrossRef] [Green Version]
- Pan, P.Y.; Sheehan, P.; Wang, Q.; Zhu, X.; Zhang, Y.; Choi, I. Synj1 haploinsufficiency causes dopamine neuron vulnerability and alpha-synuclein accumulation in mice. Hum. Mol. Genet. 2020, 29, 2300–2312. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Thorsness, M.K.; Policastro, R.; McGoldrick, L.L.; Hollingsworth, N.M.; Thorsness, P.E. Yeast Vps13 promotes mitochondrial function and is localized at membrane contact sites. Mol. Biol Cell. 2016, 27, 2435–2449. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, E.M.; Schmid, E.; Munzer, P.; Hermann, A.; Eyrich, A.K.; Russo, A. Chorein sensitivity of cytoskeletal organization and degranulation of platelets. FASEB J. 2013, 27, 2799–2806. [Google Scholar] [CrossRef]
- Kumar, N.; Leonzino, M.; Hancock-Cerutti, W.; Horenkamp, F.A.; Li, P.; Lees, J.A.; Wheeler, H.; Reinisch, K.M.; De Camilli, P. VPS13A and VPS13C are lipid transport proteins differentially localized at ER contact sites. J. Cell Biol. 2018, 217, 3625–3639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peikert, K.; Danek, A.; Hermann, A. Current state of knowledge in Chorea-Acanthocytosis as core Neuroacanthocytosis syndrome. Eur. J. Med. Genet. 2018, 61, 699–705. [Google Scholar] [CrossRef]
- Tomiyasu, A.; Nakamura, M.; Ichiba, M.; Ueno, S.; Saiki, S.; Morimoto, M.; Kobal, J.; Kageyama, Y.; Inui, T.; Wakabayashi, K.; et al. Novel pathogenic mutations and copy number variations in the VPS13A gene in patients with chorea-acanthocytosis. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2011, 156B, 620–631. [Google Scholar] [CrossRef] [PubMed]
- Dulski, J.; Soltan, W.; Schinwelski, M.; Rudzinska, M.; Wojcik-Pedziwiatr, M.; Wictor, L. Clinical variability of neuroacanthocytosis syndromes-a series of six patients with long follow-up. Clin. Neurol. Neurosurg. 2016, 147, 78–83. [Google Scholar] [CrossRef]
- Lesage, S.; Drouet, V.; Majounie, E.; Deramecourt, V.; Jacoupy, M.; Nicolas, A. Loss of VPS13C function in autosomal-recessive Parkinsonism causes mitochondrial dysfunction and increases PINK1/Parkin-dependent mitophagy. Am. J. Hum. Genet. 2016, 98, 500–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nalls, M.A.; Pankratz, N.; Lill, C.M.; Do, C.B.; Hernandez, D.G.; Saad, M.; DeStefano, A.L.; Kara, E.; Bras, J.; Sharma, M.; et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 2014, 46, 989–993. [Google Scholar] [CrossRef]
- Schormair, B.; Kemlink, D.; Mollenhauer, B.; Fiala, O.; Machetanz, G.; Roth, J. Diagnostic exome sequencing in early-onset Parkinson’s disease confirms VPS13C as a rare cause of autosomal-recessive Parkinson’s disease. Clin. Genet. 2018, 93, 603–612. [Google Scholar] [CrossRef] [Green Version]
- Darvish, H.; Bravo, P.; Tafakhori, A.; Azcona, L.J.; Ranji-Burachaloo, S.; Johari, A.H. Identification of a large homozygous VPS13C deletion in a patient with early-onset Parkinsonism. Mov. Disord. 2018, 33, 1968–1970. [Google Scholar] [CrossRef]
- Gu, X.; Li, C.; Chen, Y.; Ou, R.; Cao, B.; Wei, Q.; Hou, Y.; Zhang, L.; Song, W.; Zhao, B.; et al. Mutation screening and burden analysis of VPS13C in Chinese patients with early-onset Parkinson’s disease. Neurobiol. Aging 2020, 94, 311.e1–311.e4. [Google Scholar] [CrossRef] [PubMed]
- Bolus, H.; Crocker, K.; Boekhoff-Falk, G.; Chtarbanova, S. Modeling neurodegenerative disorders in Drosophila melanogaster. Int. J. Mol. Sci. 2020, 21, 3055. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Model | Phenotype | Mitochondrial Morphology | Mitochondrial Activity | Sensitivity to Oxidative Stress | Accumulation of Parkin SUBSTRATES | Reference(s) |
|---|---|---|---|---|---|---|
| Exon 3-deleted Park2 KO mouse (B6;129S2-Park2tm1Roo) | Normal brain morphology, weight and size ⇩ body weight and temperature Absence of neurodegeneration No behavioral changes ⇩ DAT and VMAT2 striatal levels at 15 months of age Deficit in learning and memory | N.D. | N.D. | N.D. | N.D. | [22] |
| Exon 3-deleted Park2 KO mouse (B6;129S4-Park2tm1Shn) | Normal brain morphology Absence of neurodegeneration ⇧ in extracellular DA concentration in the striatum ⇩ in synaptic excitability ⇩ behavioral performance in tests sensitive to nigrostriatal dysfunction | N.D. | ⇩ levels of proteins involved in mitochondrial function | ⇩ levels of proteins involved in oxidative stress | N.D. | [23,24] |
| Exon 3-deleted Park2 KO mouse (NeoR cassette) | Absence of neurodegeneration in the SNcNo motor disabilities | Electron dense inclusion bodies in mitochondria Dilated and disorganized cristae | ⇩ respiratory complex I activity in the SNc | N.D. | N.D. | [25] |
| Exon 2-deleted Park2 KO mouse (B6;129S4-Park2tm1Rpa) | Absence of DA neuron degeneration No abnormalities in locomotor activities No changes in catecholamine levels | N.D. | N.D. | N.D. | N.D. | [26] |
| Exon 2-deleted Park2 KO mouse (MC1-NeoR + in-frame insertion of tauGFP fusion protein) | No abnormalities in locomotor activities Absence of DA neuron degeneration No alteration in DAT level ⇩ release of DA | N.D. | N.D. | N.D. | N.D. | [27] |
| Exon 7-deleted Park2 KO mouse (B6;129S7/S4-Park2tm1Tmd) | Absence of DA neuron degeneration Early loss of catecholaminergic neurons in LC ⇩ of norepinephrine-dependent acoustic startle response ⇩ of norepinephrine concentration in olfactory bulb and spinal cord | N.D. | N.D. | N.D. | N.D. | [28] |
| Conditional exon 7-deleted Park2 KO mouse using lentiviral delivery of GFP-tagged Cre-recombinase in midbrain of adult mice | Progressive loss of DA neurons | ⇩ in mitochondrial size, number and protein markers in ventral midbrain Defect in mitochondrial biogenesis | N.D. | N.D. | ⇧ levels of PARIS | [29,30] |
| Spontaneus mouse mutant quakingviable (qkv) | Normal cellular conformation in the grey matter Alteration in DA metabolism ⇩ locomotor and exploratory activity Tremor in the caudal part of the trunk and proximal portions of the hind extremities Absence of DA neuron degeneration in the SNc Dysmyelination phenotype | N.D. | N.D. | N.D. | [31,32] | |
| BAC transgenic mouse expressing the C-terminal truncated human parkinQ311X mutation in DA neurons | Multiple late onset and progressive hypokinetic motor deficits Age-dependent DA neuron degeneration in the SNc and loss of striatal DA neuron terminals ⇩ in striatal DA level | N.D. | N.D. | ⇧ levels of nitrotyrosine | Age-dependent accumulation of proteinase K-resistant endogenous α-syn in the SNc | [33] |
| Exon 4-deleted Park2 KO rat | Small reduction in DA neurons at 8 months of age Unaltered core body temperature ⇧ body weightNo neurochemical changes ⇩ activities of MAOs at 2 months of age ⇧ of β-PEA levels at 2 months of ageNormal behavior and locomotor activity in basal condition ⇩ locomotor activity upon low dose of methamphetamine | Mitochondrial pathological alteration | No mitochondrial dysfunctions | N.D. | N.D. | [34,35,36] |
| Human Park2 T240R overexpression mutant rat | Progressive DA neuron death starting 8 weeks after rAAV2/8 injection | N.D. | N.D. | N.D. | N.D. | [37] |
| park2 KO zebrafish | ⇩ in ascending DA neuron number in the posterior tuberculum | Normal mitochondrial morphology | Respiratory complex I deficiency | ⇧ sensitivity to MPP+ ROS production | N.D. | [38] |
| Parkin KO Drosophila | Absence of neurodegeneration ⇩ lifespan Male sterility Severe disruption of muscle integrity Locomotion defects Myofibril degeneration | Swollen mitochondria with degenerated cristae | N.D. | N.D. | N.D. | [39,40] |
| Parkin KO Drosophila using P-element mutagenesis | Absence of DA neuron degeneration Male and female infertility ⇩ lifespan Severe disruption of muscle integrity Locomotion defects Myofibril degeneration | N.D. | N.D. | ⇧ sensitivity to chemical and environmental stress | N.D. | [41] |
| Human ParkinR275W overexpression mutant Drosophila | Age-dependent degeneration of DA neuronal clusters Locomotor deficits that accelerate with age | Mitochondrial abnormalities in flight muscles | ⇧ sensitivity to rotenone | [42] | ||
| N-terminal deleted parkin and parkinK71P mutant Drosophila | ⇩ longevity Drooped wing phenotype Locomotor dysfunction Muscle degeneration accompanied by apoptosis Severe loss of DA neurons Shrunken morphology of DA neurons Rescue of the phenotype by overexpression of WT parkin | N.D. | N.D. | N.D. | N.D. | [43] |
| ParkinQ311X and parkin T240R mutant Drosophila | Age-dependent DA neuron degeneration ⇩ in climbing ability Severe motor deficits 2–3 weeks after eclosion | N.D. | N.D. | N.D. | N.D. | [44] |
| PARKIN KO C. elegans | Normal development Shorter lifespan | N.D. | Mitochondrial complex I vulnerability | ⇧ sensitivity to mitochondrial complex I inhibitors | N.D. | [45] |
| PARK2-pdr1(gk448) III (CGC) mutant C. elegans | No Mn-induced degeneration of the CEP dopaminergic neurons | N.D. | N.D. | ⇧ hypersensitivity to Mn-induced lethality Time-dependent ⇧ in Mn-induced RONS | N.D. | [46] |
| Model | Phenotype | Mitochondrial Morphology | Mitochondrial Activity | Sensitivity to Oxidative Stress | Reference(s) |
|---|---|---|---|---|---|
| Pink1 RNAi knockdown mouse | Absence of DA neuron degeneration in the SNc No alteration in DA level in the striatum No abnormalities in spontaneous locomotor activities | N.D. | N.D. | N.D. | [67] |
| Exon 4-7-deleted Pink1 KO mouse | Absence of DA neuron degeneration No alteration in striatal DA levels No alteration in DA synthesis or DA receptors levels No abnormalities in spontaneous locomotor activities ⇩ in evoked DA release in striatal slices ⇩ in the quantal size and release frequency of catecholamine in dissociated chromaffin cells ⇩ in corticostriatal LTP and LTD | No gross ultrastructural alterations No changes in the total number of mitochondria ⇧ number of larger mitochondria in the striatum at 3–4 and 24 months | Age-dependent impairment of mitochondrial function ⇩ in respiratory complex I and II activity in the striatum (young and old mice) and cerebral cortex (old mice) ⇩ in aconitase activity in the striatum (young and old mice) and cerebral cortex (old mice) | Mitochondria in the cortex are more sensitivity to oxidative stress | [68,69] |
| Exon 2-3-deleted Pink1 KO mouse | Absence of DA neuron degeneration at 6 and 19 months of age No alteration in striatal DA content at 6 and 19 months of age No abnormalities in the spontaneous locomotor activity and normal motor coordination at 3 and 24 months Gait alterations and olfactory dysfunctions at 26 months ⇩ of the density of serotoninergic fibers in the glomerular layer of the olfactory bulb at 26 monthsAged males Pink1 KO mice showed a significant deficit in the fine olfactory discrimination and in smell sensitivity | Less fragmented mitochondria | N.D. | N.D. | [70] |
| Pink1 KO mouse (Pink1tm1Shn) | Absence of DA neuron degeneration in the SNc No alteration in TH optical density in the striatum Vocalization’s impairments Impairment in limb motor skills with fewer hindlimb and forelimb steps ⇩ rearing and landing on the cylinder test Impairment during the pole test | N.D. | N.D. | N.D. | [71] |
| G309D-Pink1 transgenic mouse | Age-dependent ⇩ of DA level ⇩ spontaneous locomotor activity Progressive ⇩ of body weight from middle age Absence of nigrostriatal degeneration No Lewy Bodies formation | ⇩ of Mtp18 Normal mitochondrial morphology and mass | ⇩ ATP levels ⇩ mitochondrial membrane potential ⇩ respiratory complex activity | ⇧ sensitivity to proteasomal stress | [72] |
| Exon 2-5-deleted Pink1 KO mouse (in vitro studies on primary neuronal cultures) | Age-dependent ⇩ in long-term viability of cortical neuron cultures | N.D. | Mitochondrial calcium overload in primary cortical and midbrain neurons Loss of mitochondrial membrane potential ⇩ respiratory complex activity | ⇧ cytotoxicity indices in cortical neuron cultures ⇧ ROS production in primary cortical and midbrain neurons | [65,73] |
| Pink1 KO rat | ⇧ of striatal densities of DA D2 and D3 receptors at 6 months of age No changes in striatal density of DA D1 receptors, VMAT2 and DAT at 6 months of ageMotor impairments in movement, strength and coordination starting from 4 months of age Loss of SNc DA neurons starting from 6 months No change in striatal TH or α-syn immunoreactivity within the SNc and striatum ⇧ of DA and 5-HT striatal content at 8 months of age ⇧ glycolysis in the striatum Early and progressive vocalization impairment and oromotor deficits Compromised communication Sensorimotor deficits with a ⇩ in spontaneous activity starting from 8 months No change in TH immunoreactivity in the striatum and in the SNc at 8 months of age ⇩ in TH immunoreactivity in LC at 8 months of age α-syn aggregates in PAG, SNc and LC at 8 months | ⇧ DRP1 and ⇩ MFN2 in the striatum at 4 months of age ⇧ mitochondrial fission and fragmentation | ⇩ in the level of respiratory complex I, III, IV, V in the striatum ⇧ proton leak at 4 and 9 months of age | ⇧ ROS generation Altered stress pathway in the striatum at 4 months of age | [34,74,75,76,77] |
| Zebrafish MO-mediated pink1 knockdown | Structural alterations in the axonal scaffold ⇩ number of central DA neurons Lower heart rate ⇧ of VEGF and erythropoiesis ⇩ th1 and th2 mRNA, but normal levels of dat mRNA Locomotor dysfunctions Weak or absent response to tactile stimuli ⇩ swimming behavior ⇧ susceptibility to MPTP-induced motor disturbances | N.D. | ⇩ mitochondrial membrane potential ⇧ GSK-3β activity Alteration of mitochondria biogenesis | ⇧ ROS levels ⇧ oxidative stress ⇧ caspase-3 activity ⇩ hif1α mRNA level ⇩ catalase enzyme activity ⇩ catalase and SOD3 transcript ⇧ susceptibility to MPTP ⇧ susceptibility to H2O2 | [8,78,79,80] |
| Y431* pink1 transgenic zebrafish | Progressive loss of DA neurons from 5 dpf to 18 months of age No obvious behavioral abnormalities Microglial activation | Enlarged mitochondria | ⇩ mitochondrial complex I and III activity ⇧ TigarB expression | N.D. | [55,81] |
| Q178X pink1 transgenic Medaka | Late-onset motor deficits ⇩ in the frequency of spontaneous swimming movements Shortened lifespan Absence of neurodegeneration ⇩ DOPAC | Normal mitochondrial morphology | N.D. | N.D. | [82] |
| pink1 KO Drosophila | Male sterility Degeneration of flight muscles Mild loss of DA neurons Impaired flight abilityS evere ⇩ climbing rate Abnormally positioned wings Crushed thorax Disorganized muscle fibers Muscle cell apoptosis Impaired mobilization of synaptic vesicle reserve pool during rapid stimulation Synaptic ATP depletion | Enlarged and swollen mitochondria with loss of the outer membrane Fragmented cristae | ⇩ ATP levels and synthesis ⇩ mitochondrial complex I and IV activity Deficit in mitochondrial membrane potential | ⇧ oxidative stress | [83,84,85,86] |
| pink1 RNAi knockdown Drosophila | ⇩ lifespan Abnormal wing posture Disruption of muscle integrity Degeneration of indirect flight muscles Impaired flight ability (limited to the early days of life) ⇩ in climbing ability Degeneration of TH-positive neurons ⇩ of brain DA content | Grossly swollen mitochondria lacking electron-dense material Disintegrated cristae | ⇩ ATP levels | N.D. | [87] |
| C. elegans pink-1(tm1779) mutant | Defects in axonal outgrowth of CAN ⇩ of lifespan | ⇩ in mitochondrial cristae length ⇧ fused mitochondrial network Fragmented mitochondria | No difference in basal OCR ⇧ OCR after FCCP exposure ⇩ OCR after DCCD exposure ⇧ proton leak ⇩ ATP levels ⇩ Mitochondrial membrane potential Loss of mitophagy ⇩ in mitochondrial turnover | ⇧ paraquat sensitivity ⇩ oxidative stress response ⇧ DCCD and FCCP sensitivity ⇧ mitochondrial ROS production | [88,89,90,91,92] |
| Model | Phenotype | Sensitivity to Oxidative Stress | Mitochondrial Activity | Reference(s) |
|---|---|---|---|---|
| Exons 3-5-deleted Dj-1 KO mouse | Absence of DA neuron degeneration No motor deficits observed by pole test, open field, adhesive removal test No changes in the density of striatal TH fibers and DAT level No alteration in DA striatal levels Motor deficit observed on running wheels and rotarod | ⇧ susceptibility to MPTP-induced neuron loss | N.D. | [106,107] |
| Exons 1-5-deletd Dj-1 KO mouse | Absence of DA neuron degeneration No motor deficits observed by rotarod test at any age Motor impairments at 11 months of age with open field test Nigrostriatal dysfunction with tape removal task at 5 and 11 months of age ⇧ evoked release of DA in dorsal striatum Age-dependent ⇧ in striatal DA content No differences in TH, DAT and VMAT2 protein levels No α-syn- or ubiquitin-positive inclusions in the SNc | N.D. | N.D. | [103] |
| Exon 2-deleted Dj-1 KO mouse | Absence of DA neuron degeneration No alteration in DA levels in basal ganglia Normal TH activity No α-syn- or ubiquitin-positive inclusions in the SNc ⇩ evoked release of DA with amperometry analysis Normal LTP induction, but absence of LTD Motor impairments at 3 months of age with open field, rotarod and startle tests | N.D. | N.D. | [99] |
| Dj-1 mouse with deletion of intron between exons 6 and 7 | No change in the number of TH-positive and Nissl-stained nigral cells No change in DA, DOPAC, HVA No behavioral impairment Alteration in striatal DA transmission ⇧ in striatal DAT in synaptosomal fraction | N.D. | N.D. | [108] |
| Dj-1 KO rats | Loss of DA neurons in the SNc and LC at 8 months No change in TH immunoreactivity in the striatum ⇧ in striatal DA and 5-HT content at 8 months of age No change in DAT density in striatum ⇧ in VMAT2 and D1, D2 and D3 DA receptor density in striatum between 4 and 8 months of age ⇧ in body weight Motor impairments in movement, strength and coordination between 6 and 8 months Deficit in cylinder test from 4 to 13 months of age No alterations in sensorimotor functions with adhesive removal test No anxiety or depression at 4, 8 or 17 months of age Abnormality in the neuroendocrine system | N.D. | N.D. | [34,74,109,110,111] |
| Zebrafish MO-mediated dj-1 KO | No loss of DA neurons in basal condition Loss of DA neurons after exposure to H2O2 or to proteasome inhibitor MG132⇧ levels of p53 and Bax | ⇧ susceptibility to H2O2 | N.D. | [112,113] |
| CRISPR-Cas9 dj-1 KO Zebrafish | Smaller size and ⇩ in body mass starting from 3 months of age ⇩ in TH levels and DA content at 16 months of age Locomotor deficits (bradykinesia): reduction in distance travelled, velocity, time spent moving and duration of a swimming episode | N.D. | ⇩ mitochondrial complex I activity in skeletal muscle at 16 months of age | [114,115] |
| dj-1β KO Drosophila | No loss of DA neurons ⇩ taste sensitivity Defective in ability to form associative memories ⇩ climbing ability and further loss of climbing activity after repeated paraquat exposure | ⇧ susceptibility to H2O2 Resistance to oxidative stress induced by paraquat | N.D. | [116,117,118] |
| dj-1α and dj-1β double KO Drosophila | No loss of DA neurons Normal lifespan | ⇧ sensitivity to H2O2, paraquat and rotenone | N.D. | [119] |
| djr-1.1 KO C. elegans | N.D. | ⇧ susceptibility to rotenone | N.D. | [45] |
| djr-1.1 and djr-1.2 double KO C. elegans | ⇧ inflammatory signaling after exposure to Pseudomonas aeruginosa | N.D. | N.D. | [120] |
| djr KO C. elegans | ⇩ survival and lifespan after acute Mn exposure in djr-1.2 or djr double deletion mutants ⇧ dauer movement in djr-1.2 deletion mutant | ⇧ sensitivity to Mn in djr-1.2 or djr double deletion mutants | N.D. | [121] |
| Model | Phenotype | Sensitivity to Oxidative Stress | Mitochondrial Activity | Reference(s) |
|---|---|---|---|---|
| Atp13a2 KO mouse | Late onset sensorimotor and cognitive deficit (20–29 months of age) Late onset motor impairments (20–29 months of age) No loss of DA neurons in the SNc No change in striatal DA levels Gliosis at 1 month of age Lipofuscinosis at 3 months of age Accumulation of LAMP1, LAMP2 and BMP at 6 months of age Aggregation of ubiquitinated proteins and p62 at 12 months of age Aberrant processing of the lysosomal protease CATD at 12 months of age Accumulation of insoluble α-syn in the hippocampus at 18–20 months of age | N.D. | N.D. | [131,135,136,137] |
| Zebrafish MO-mediated atp13a2 knockdown | Complete abrogation of atp13a2 led to embryonic lethality Partial abrogation of atp13a2 allowed offspring survival Curved phenotype at 48 hpf Movement latency and abnormal response to environmental stimulus at 7 dpf | N.D. | N.D. | [138] |
| Zebrafish atp13a2sa18624 and atp13a2sa14250 mutant | No obvious morphological or behavioral abnormality At 5 dpf, pericardial oedemas, movement loss, spine curvature and underdevelopment of the swimming bladder in homozygous atp13a2 mutants exposed to Mn2+ Apoptotic areas throughout the CNS after exposure to Mn2+ in atp13a2sa18624−/− larvae | ⇧ susceptibility to Mn2+ | N.D. | [139] |
| CRISPR-Cas9 KO atp13a2 Zebrafish | Loss of DA neurons in posterior tuberculum and norepinephrine neurons in LC at both 4 and 12 months of age CATD deficiency Lysosomal and intracellular vesicle trafficking dysfunction | N.D. | N.D. | [140] |
| atp13a2 “IVS13, T-C, +2” mutant medaka | Shorter lifespan ⇧ spontaneous swimming movement at 4 months, but no differences in swimming at 12 months Age-dependent and progressive loss of DA neuron in the middle diencephalon at 8 and 12 months of age ⇩ density of TH-positive fibers in the telencephalon at 8 and 12 months ⇩ of noradrenergic neurons in the medulla oblongata at 8 and 12 months of age ⇩ in DA content at 12 months of age ⇩ CATD protein level and activity Fingerprint-like subcellular structures in the brain | N.D. | N.D. | [141] |
| catp-6(ok3473) C. elegans mutant | ⇩ locomotion Delay in the rate of development Higher mortality rate in midlife age Alteration in iron homeostasis Down-regulation of the core genes required for metabolizing iron Altered Zn homeostasis after Zn exposure ⇩ of cleaved LGG1/LC3-II protein levels ⇧ P-CATD levels ⇩ mRNA levels of genes required for autophagy and lysosomal function | N.D. | ⇩ of mitochondrial membrane potential ⇩ of maximal respiration rate ⇧ sensitivity to rotenone | [142,143] |
| Model | Phenotype | Mitochondrial Activity | Sensitivity to Oxidative Stress | Reference(s) |
|---|---|---|---|---|
| Pla2g6 KO mouse | Degeneration of nigrostriatal DA neurons and loss of striatal TH and nerve terminal DAT from 56 weeks of age Axonal degeneration and atrophic axonsLate onset motor dysfunctions (2 years of age) Presence of spheroids and vacuoles throughout the CNS and the PNS ⇧ expression of α-syn and phosphorylated α-syn in mitochondria ⇩ male fertility ⇩ in DHA metabolism at 4 months of age | N.D. | N.D. | [152,153,154,155] |
| Exon2-deleted Pla2g6 KO mouse (Pla2g6 ex2KO) | Progressive loss of SNc DA neurons starting from 16 months of age Progressive age-dependent L-DOPA-sensitive motor dysfunctions starting from 16 months of age Impairment of PLA2G6-dependent Ca2+ signaling and depletion of intracellular Ca2+ stores in MEFs Accumulation of LC3 and increased autophagosome numbers in the SNc DA neurons | N.D. | N.D. | [156] |
| D331Y KI Pla2g6 mouse | Loss of DA neurons in the SNc at 6 and 9 months Degeneration of nigrostriatal dopaminergic terminals at 9 months Presence of Lewy Bodies in the SNc at 9 months ⇧ α-syn and phosphorylated α-syn expression in the SNc at 9 months Early-onset and progressive PD phenotype from 6 to 12 months: slowness of movement, hypoactivity, impaired motor coordination and performance | Disrupted structure of mitochondria cristae ⇩ of mitochondrial size ⇩ mitochondrial complex I and III activity ⇩ ATP level ⇧ cytosolic level of cytochrome c Activation of mitochondrial apoptotic pathway Mitophagy impairment | ⇧ endoplasmic reticulum stress ⇧ ROS production in the SNc ⇧ lipid peroxidation | [157] |
| pla2g6 KO Drosophila (iPLA2-VIA KO) | Progressive degeneration of DA neurons Impaired synaptic transmission ⇧ ceramide production Shorter lifespan α-syn aggregates | N.D. | N.D. | [146,158] |
| Model | Phenotype | Mitochondrial Activity | Reference(s) |
|---|---|---|---|
| Exon4-deleted Fbx07 KO mouse | No difference in body or brain weight at P5 Lower body and brain weight at P18 Early-onset motor deficits at P18 Premature death in the 4th postnatal week Moderate ⇧ in cell death in the cortex at P18 Absence of DA neuron degeneration in the SNc at P18 No change in the levels of DA and its metabolites in the striatum at P18 Absence of α-syn protein deposits at P18 Astrogliosis in the cortex at P18 | N.D. | [167] |
| Fbx07 conditional KO mouse (Nex-Cre;fl/fl). Deletion of Fbx07 from pyramidal neurons of the cortex and hippocampus | Longer lifespan (at least up to 4 months) Spasticity and progressive motor coordination deficits at 2 and 4 months of age Normal body weight at 2 months of age, with successive stagnation Astrogliosis in the cortex | N.D. | [167] |
| Fbx07 conditional KO mouse (TH-Cre;fl/fl). Deletion of Fbx07 gene from TH-expressing neurons | Longer lifespan (at least up to 12 months) Absence of motor deficit at 2 months of age Progressive motor deficit at 6 and 12 months of age: slow movements, reduced mobility, alteration in several fine gait parameters Absence of DA neuron degeneration at 2 and 12 months of age ⇩ of DA content (50%) in the striatum at 2 and 12 months ⇧ of astrogliosis at 12 months in the SNc | N.D. | [167] |
| Zebrafish MO-mediated zFbxo7 knockdown | Knockdown of zFbxo7 protein expression Mild and severe morphological phenotype correlated with the silencing of zFbxo7 protein at 72 hpf: curly tails, heart edema, heart malformations Locomotor impairments in the ATG-MO-Mild and SP-MO-Mild morphants ⇩ of number of DA neurons in SP-MO-severe morphants ⇩ of number of DAT-positive neurons in ATG-MO and SP-MO morphants | N.D. | [168] |
| hFBXO7 WT overexpression Drosophila mutants | Locomotor deficits in flight and climbing ability starting from 30–40 days Loss of DA neurons in PPM1/2 and PPM3 DA neuron cluster at 40 days FBXO7 protein aggregation | Mitochondrial deficit Swelling of muscle mitochondria Broken mitochondria cristae Accumulation of high-density materials in the swollen mitochondria | [166] |
| Model | Phenotype | Mitochondrial Morphology | Sensitivity to Oxidative Stress | Reference(s) |
|---|---|---|---|---|
| Dnajc6 KO mouse | High rate of early postnatal mortality, with an apparently normal lifespan of the surviving pups ⇩ body weight Delayed female sexual maturity ⇧ GAK levels in the brain in embryos and in 3–5-week-old mice | N.D. | N.D. | [177] |
| dnajc6 RNAi knockdown Drosophila | ⇩ lifespan Age-dependent ⇩ of DA neuron number Locomotor deficit in climbing ability | No alterations | ⇧ sensitivity to paraquat-induced oxidative stress | [178] |
| Model | Phenotype | Reference(s) |
|---|---|---|
| Synj1 KO mouse | Severe reduction of milk in their stomachs few hours after birth Higher post-natal mortality rate (85% within 24 h and 15% within 15 days) Severe weakness, ataxia and generalized convulsions on stimulation by nociceptive stimuli at 10 days of age ⇩ body weight at 10 days of age ⇧ number of clathrin-coated vesicles in the cytomatrix-rich area Impairment in synaptic vesicle recycling ⇩ of synaptic transmission after prolonged high-frequency stimulation and delayed recovery after interruption of the stimulus in CA1 area | [185] |
| Synj1 heterozygous mouse | Normal body weight Normal lifespan Age-dependent motor impairments ⇩ of striatal DA, DOPAC and HVA content at 7 and 12 months Progressive ⇩ of DA terminals from 3 to 18 months Accumulation of pS129 α-syn in the cortex, striatum and midbrain at 18 months | [186] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bastioli, G.; Regoni, M.; Cazzaniga, F.; De Luca, C.M.G.; Bistaffa, E.; Zanetti, L.; Moda, F.; Valtorta, F.; Sassone, J. Animal Models of Autosomal Recessive Parkinsonism. Biomedicines 2021, 9, 812. https://doi.org/10.3390/biomedicines9070812
Bastioli G, Regoni M, Cazzaniga F, De Luca CMG, Bistaffa E, Zanetti L, Moda F, Valtorta F, Sassone J. Animal Models of Autosomal Recessive Parkinsonism. Biomedicines. 2021; 9(7):812. https://doi.org/10.3390/biomedicines9070812
Chicago/Turabian StyleBastioli, Guendalina, Maria Regoni, Federico Cazzaniga, Chiara Maria Giulia De Luca, Edoardo Bistaffa, Letizia Zanetti, Fabio Moda, Flavia Valtorta, and Jenny Sassone. 2021. "Animal Models of Autosomal Recessive Parkinsonism" Biomedicines 9, no. 7: 812. https://doi.org/10.3390/biomedicines9070812