1. Introduction
Splice-switching therapy with splice-switching oligonucleotides (SSOs) has recently gained significant momentum for the treatment of various mis-splice disorders [
1]. Treatments with SSOs usually lead to the restoration of correct splicing, production of a novel splice-variant or directing from one splice-variant to another. Recent successful clinical trials for neuromuscular disorders, such as for Duchenne muscular dystrophy (DMD) and spinal muscular atrophy (SMA), have demonstrated the excellent potential that SSO therapeutics can offer for the treatment of human disease. Despite these major breakthroughs, issues with the efficacy of SSO therapies persists and this is primarily attributed to the limited bioavailability of SSOs in the target tissues/cells. In fact, it has been estimated that as low as 0.1% of the total administered SSO dose is ultimately available at their target site for engagement, while the majority of material is lost due to limited tissue/cellular uptake, clearance by liver and excretion by kidneys, but also caused by unspecific binding to proteins in both extra- and intracellular compartments [
2]. Therefore, there is clearly enormous room for improving the delivery and efficacy of SSO-based therapeutic modalities.
One way to improve the delivery and bioavailability of SSOs in tissues/cells is by utilizing various drug delivery systems (DDSs). A group of DDSs that has lately gained increasing attention are short delivery peptides called cell-penetrating peptides (CPPs) [
3]. CPPs are cationic and/or amphipathic peptides up to 30 amino acids in length [
4] that have been successfully applied for the delivery of a wide variety of nucleic acid-based molecules, ranging from short oligonucleotides (ONs) to larger plasmid molecules [
5]. CPPs can be used to formulate SSOs by two main strategies, either via covalent conjugation or by formation of discrete nanoparticles. Compared to other delivery vectors, CPPs are known for their high cellular permeability and ability to enter a broad range of cell types and are considered to have a favorable toxicity and immunological safety profile compared to other synthetic vectors, such as cationic lipids or polymers [
6]. CPPs are known to enter cells by using various energy-dependent endocytic mechanisms, while there are also reports on the direct translocation mechanism for cellular entry [
3].
The hydrophobic nature of the CPPs, as well as the inclusion of basic amino acids, have been reported to play a vital role in the cellular uptake and the subsequent release from endosomes [
7]. Over the years, many attempts have been made to enhance the delivery properties of CPPs or CPP-nucleic acid complexes. These include various strategies which aim to improve the structural features of the CPP sequences. For example, fine-tuning of their secondary structure, especially the α-helical motif, has been shown to have a significant impact on protein-protein interactions and cellular internalization of some of the CPPs [
8]. To this end, several approaches have been developed to stabilize the α-helical secondary structure in peptides via side-chain crosslinking or peptide stapling in order to improve their cellular uptake and stability to proteases [
9]. One such approach is ruthenium-catalyzed ring-closing metathesis (RCM), where α,α-disubstituted amino acids modified with hydrocarbon (alkenyl) side-chains of varying lengths are introduced to the peptide backbone and the α-helical structure locked by covalently joining the alkenyl side-chains after one or two helical turns [
10].
Another commonly used approach for improving their delivery efficacy is the N-terminal modification of CPPs with fatty acids to increase their hydrophobicity. For example, increased hydrophobicity by stearylation has been shown to increase the stability of CPP/nucleic acid complexes [
11], enhance the association of complexes with the cell membrane [
11,
12] and increase the cellular uptake and endosomal escape [
13]. On the other hand, N-terminal modification of CPPs with fatty acids can be a double-edged sword, as it also increases their membrane activity and hemolytic activity [
12], which can lead to possible adverse effects. Hence, we propose a strategy where shorter hydrophobic modifications are distributed evenly over the peptide sequence to yield the same net hydrophobicity as longer fatty acids would.
To this end, we have recently developed several amphipathic peptide analogs based on a sequence called hPep1. In this study, we repurpose the hydrophobic alkenyl-alanines used in RCM to orthogonally introduce hydrophobicity into hPep1 peptide in order to improve its ability to efficiently formulate and deliver SSOs. Here we evaluate the viability of this approach by varying the number and length of these hydrocarbon modifications for improving the in vitro and in vivo delivery of SSO therapeutics.
2. Methods and Materials
2.1. Cell-Penetrating Peptides
All peptides were ordered from Pepscan Presto (Lelystad, The Netherlands) with >90% purity. The peptides are C-terminally amidated and have a free amine in the N-terminus. The purities of the peptides were obtained by UPLC (Ultra Performance Liquid Chromatography) with a linear gradient system (gradient: 5–55% B in 2 min, flow: 1 mL/min, eluent A: 100% H2O + 0.05% TFA; eluent B: 100% ACN + 0.05% TFA) using an C18 RP-HPLC column and detection at 215 nm. All peptides were provided as white powders. The hydrophobicity of the peptides was evaluated by UPLC with the following ACN gradient: from 5 to 90% in 13 min, 90–100% in 1.5 min followed by 2.5 min at 100% ACN in a C18 column.
2.2. Splice-Switching Oligonucleotides
In cell culture studies we used SSO705 with or without 5′-AlexaFluor568 fluorescent label (Seq: 5′-ccucuuaccucaguuaca-3′, small letters are indicating phosphorothioate 2′-O-methyl modified RNA). Interleukin 6 Signal Transducer (IL6ST) SSO: 5′-accuuccacacgaguuguac-3′ was used as a negative control. For evaluating the in vivo biodistribution of peptide/oligonucleotide complexes, fluorescently labelled SSO targeting IL6ST was used (Seq: 5′-Cy5-ggTcugGaugGuccTa, small letters are indicating phosphorothioate 2′-O-methyl modified RNA and capital letters are indicating locked nucleic acid modified RNA). Oligonucleotides were synthesized by Sigma Aldrich (Tokyo, Japan).
2.3. Peptide/Oligonucleotide Complex Formulation
Peptide/SSO complexes were prepared in HEPES-buffered glucose (HBG) (20 mM HEPES, pH = 7.4, 5% glucose) at different peptide-to-SSO molar ratios (MRs) 1:1 to 10:1. For that, appropriate amounts of peptide (100 µM) and SSO (10 µM) working solutions where first diluted to an equal volume with HBG and then pipetted together and left for complex formation at RT for 30 min.
2.4. Complex Stability Assay
Peptide/SSO complexes were formed at MR5 using three different peptides: hPep1, hPep2, and hPep3. The complexes were then transferred to black 96-well plates containing SYBR™ Gold dye (Invitrogen™, Waltham, MA, USA) to measure the amount of accessible SSO in the complexes with SynergyMX fluorometer (BioTek, Winooski, VT, USA). For complex stability evaluation, competitive anion binder hexametaphosphate (HMP) was added at different concentrations from 0.04 to 40 µg/mL and the amount of released oligonucleotide was measured after the HMP addition. Measurements were done in duplicates and results show the mean of three independent experiments (mean ± SEM, n = 3).
2.5. Cell Cultures
HeLa pLuc705 reporter cells, first developed by Prof R. Kole and colleagues [
14], were cultivated in DMEM (Dulbecco’s Modified Eagle’s Medium; Thermo Fisher Scientific, Waltham, MA, USA) with 10% FBS (fetal bovine serum) with 1% (Penicillin: 100 U/mL, Streptomycin: 100 μg/mL) and maintained in a water-jacketed incubator at 37 °C and 5% CO
2 atmosphere.
2.6. Splice-Switching Assay
For the splice-switching activity measurements either 7000 or 35,000 cells were seeded 24 h prior to treatment into 24 or 96-well plates, respectively. Then complexes were added to the cells in 1/10 of the final volume of the cell media at 24 h post seeding. For varying the cell treatment, concentration complexes were diluted prior the treatment to appropriate concentration to retain the 1/10 of the final volume of the cell media during treatment. After 24 h of treatment, media was removed and cells were lysed in 0.1% Triton X-100 (Sigma-Aldrich) for 30 min at room temperature. The luciferase activity was measured from the lysates with the Luciferase Assay Kit (Promega, Madison, WI, USA) under GLOMAX 96 microplate luminometer (Promega) and normalized to the protein content (Bio-Rad Protein Assay Kit II, BioRad, Hercules, CA, USA). The data were further normalized to untreated cells and the splice switching activity was presented as a fold-increase over untreated cells.
For the chloroquine-induced endosomal release, the cells were treated at 200 nM hPep3/SSO complexes for 4 h, followed by 2 h challenging with 50 μM chloroquine. All stages included an intermediate washing step. Luciferase expression was measured after 18 h.
2.7. Quantitative Uptake
20,000 cells per well were seeded on 96-well plates 24 h prior to treatment. Cells were treated with peptide/AF568-SSO complexes (using peptides hPep1, hPep2, hPep3) with final concentrations of 25–100 nM at MR 5 in both serum-free and serum-containing conditions for 4 h. Complexes were formed in MQ water. After treatment, cells were washed two times with 100 µg/mL heparin (in DPBS). Cells were then lysed with 50 µL of 0.1% Triton X-100 per well and the fluorescence was measured at ex 568 nm, em 603 nm with SynergyMX fluorometer (BioTek) and normalized to the protein content (Bio-Rad Protein Assay Kit II, BioRad). The values represent the mean of at least three independent experiments done in duplicates (mean ± SEM).
2.8. Confocal Microscopy
20,000 HeLa pLuc705 cells/well were seeded in Nunc® Lab-Tek II® glass-bottom chamber slides (Thermo Scientific™, Waltham, MA, USA). After 24 h of adherence, hPep3/AF568-SSO complexes were added to cells at a 200nM final concentration of SSO in serum-containing media. After 4 h, the nuclei were stained with Hoechst 33342 (Thermo Scientific™) for 10 min, then media was removed and cells washed 3 times with PBS. Live-cell imaging was performed in a stage-top incubator at 37 °C with 5% CO2. Imaging was carried out using a confocal microscope (A1R confocal, Nikon, Tokyo, Japan) and analyzed by the NIS-Elements software (Nikon, Tokyo, Japan).
2.9. Nanoparticle Size Measurements
The size of the peptide/SSO complex was measured by Nanoparticle Tracking Analysis (NTA). The complexes were formulated as described above and measured on the NS500 instrument (Malvern Instruments, Malvern, Worcestershire, UK). The movement of nanoparticles for each sample was recorded five times (30 s each video). In order to process the videos, the camera gain and minimum detection threshold was set to 14 and 7, respectively.
2.10. Cytotoxicity Evaluation
Metabolic activity of the cells was observed by WST-1 assay (Roche, Clifton, NJ, USA) as instructed by the manufacturer’s protocol. HeLa pLuc705 cells were seeded in 96-well plates and treated with complexes for 24 h at 12.5–200 nM final SSO concentration. Thereafter, the media was changed, and WST-1 reagent was added for 4 h. The toxicity was evaluated by measuring the absorbance of the formazan product at 450 nm and the background at 650 nm on Spectra Max (Molecular Devices, Silicon Valley, CA, USA). The results were normalized to untreated cells (UT).
2.11. RNA Expression Analysis of Splice-Switching
The corrected percentage of luciferase mRNA was determined by total RNA isolation from the cells using the standard phenol-chloroform extraction protocol with Trizol (Thermo Fisher Scientific, Waltham, MA, USA). A high-capacity cDNA reverse transcription kit was used to perform each RT-PCR reaction with 500 ng of isolated RNA (Applied Biosystems, Waltham, MA, USA). The total volume was 20 µL and the primers had a following sequence: Fwd-5′-TTGATATGTGGATTTCGAGTCGTC; Rev-5′-TGTCAATCAGAGTGCTTTTGGCG. The RT-PCR protocol was as follows: 55 °C for 35 min, followed by 15 min at 95 °C for reverse transcription. Secondary nested PCR was performed using two microliters of the RT-PCR using Hot Taq plus DNA polymerase (Qiagen, Germantown, MD, USA). The PCR program was performed for 30 cycles (94 °C for 30 s, then 55 °C for 30 s, then 72 °C for 30 s, and lastly 72 °C for 10 min). A 2% agarose gel for the PCR products was used in 1 × TAE buffer and detected by SYBR Gold staining (Invitrogen). The Versadoc imaging system provided with a CCD camera was used to analyze gels (BioRad, Hercules, CA, USA). Quantity One software was used to analyze band intensities (BioRad, Hercules, CA, USA). The percentage of splice-correction was expressed as [Band intensity of corrected RNA × 100/(The sum of corrected and uncorrected RNA band intensities)].
2.12. Endocytosis Inhibition Assay
50,000 cells per well were seeded on 24-well plates in serum-containing DMEM 24 h prior to treating them for 30 min with the following inhibitors: 4 µM cytochalasin D for macropinocytosis inhibition; 15 µM chlorpromazine for clathrin-mediated endocytosis inhibition; 12.5 µM nystatin for caveolae-mediated endocytosis inhibition; 6 mM 2-Deoxy-D-Glucose and 10 mM NaN3 for ATP depletion (-ATP). Thereafter, cells were treated with hPep3/AF568-SSO complexes at 200 nM final concentration for 4 h in the presence of the inhibitors. Cells were washed twice with cold DPBS containing 100 µg/mL of heparin and detached with 0.05% trypsin-EDTA. The cell pellet was further washed with cold DPBS, and thereafter lysed in 0.1% Triton X-100. The endocytosis inhibition was evaluated by fluorescence spectroscopy and evaluated over hPep3/AF568-SSO treatments without inhibitors.
2.13. Animal Study
20-g female NMRI mice were subjected to intravenous injection with the formulations through the tails to compare the naked Cy5-SSO and hPep3/Cy5-SSO complexes in vivo at a total of 50 µg of Cy5-SSO at MR10 (3 animals per group). After 24 h, the animals were sacrificed, and organs harvested and imaged with fluorescence analysis using IVIS Spectrum (Perkin Elmer, Waltham, MA, USA). Data were processed by IVIS software (Living Image Software). Adobe Photoshop CS4 and Adobe Illustrator were used to crop out and align the organ images. The in vivo experiments were approved by the Swedish local laboratory animal research ethics committee (approval no. S4-16, approved in 5 January 2020) and all experiments were performed in accordance with relevant guidelines and regulations.
2.14. Statistical Analysis
Data are presented as mean with a standard error of the (mean ± SEM) of at least three independent experiments. Significant differences were evaluated by analysis of variance (ANOVA) with Dunnetts’s multiple comparison test, Bonferroni′s multiple comparison test or unpaired t test (GraphPad Prism 8 or 9; GraphPad Software, Inc., San Diego, CA, USA). In all cases, differences with p < 0.05 were deemed to be significant (* p < 0.05, ** p < 0.01, *** p < 0.001, and **** p < 0.0001).
4. Discussion
The numerous ON-based methods for gene function modulation all face the same challenge of effective delivery to target cells. This inherent limitation is shared by antisense ONs and SSOs, implying that improving the delivery of this class of therapeutic molecules is very important for the wider clinical translation of ON therapeutics. Broader interest in the development of ON-based therapies has also intensified efforts in finding more effective and safe delivery vectors for both in vitro and in vivo applications. The use of CPPs to enhance cellular uptake and bioactivity of different types of ONs represents one approach that offers great promise for improving the bioavailability of ON-based compounds. Different chemically modified ONs, such as locked nucleic acids (LNAs), peptide nucleic acids (PNAs), and 2′-O-Methyl RNA are being used to modify aberrant splicing patterns. SSOs have been successfully applied to modulate splicing patterns in the context of various splicing disorders, such as for spinal muscular atrophy and Duchenne muscular dystrophy [
16,
17,
18]. These findings emphasize the therapeutic potential of SSOs in human diseases; however, further development is necessary to enhance efficiency and systemic delivery of SSOs. To enhance delivery, SSOs are usually complexed with positively charged DDSs to form nanoparticles [
19]. CPPs are one such versatile class of nucleic acid DDSs, which are able to formulate therapeutic oligonucleotides of different sizes, including SSOs, into nanoparticles. However, many challenges remain, such as low colloidal stability and poor endosomal release of the nanoparticles.
A common way to increase the cellular uptake of CPP/SSO nanoparticles is by increasing the hydrophobic interactions with the membrane via lipidation of the CPPs [
13,
20,
21]. This is usually achieved by adding hydrocarbon chains with varying length to the N-terminus of the CPPs. While this approach comes with many advantages it can also bring unwanted side-effects such as increased hemolytic activity [
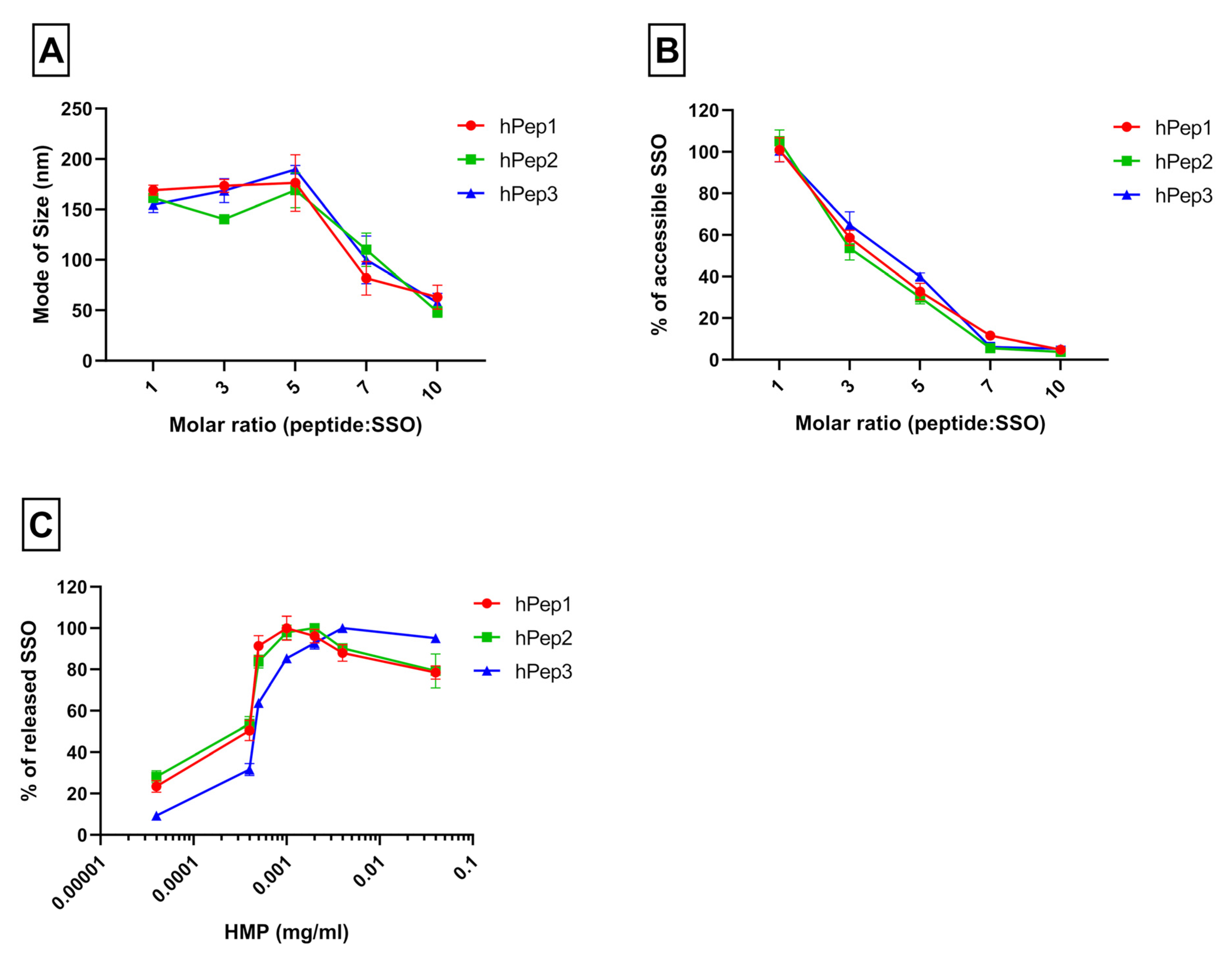
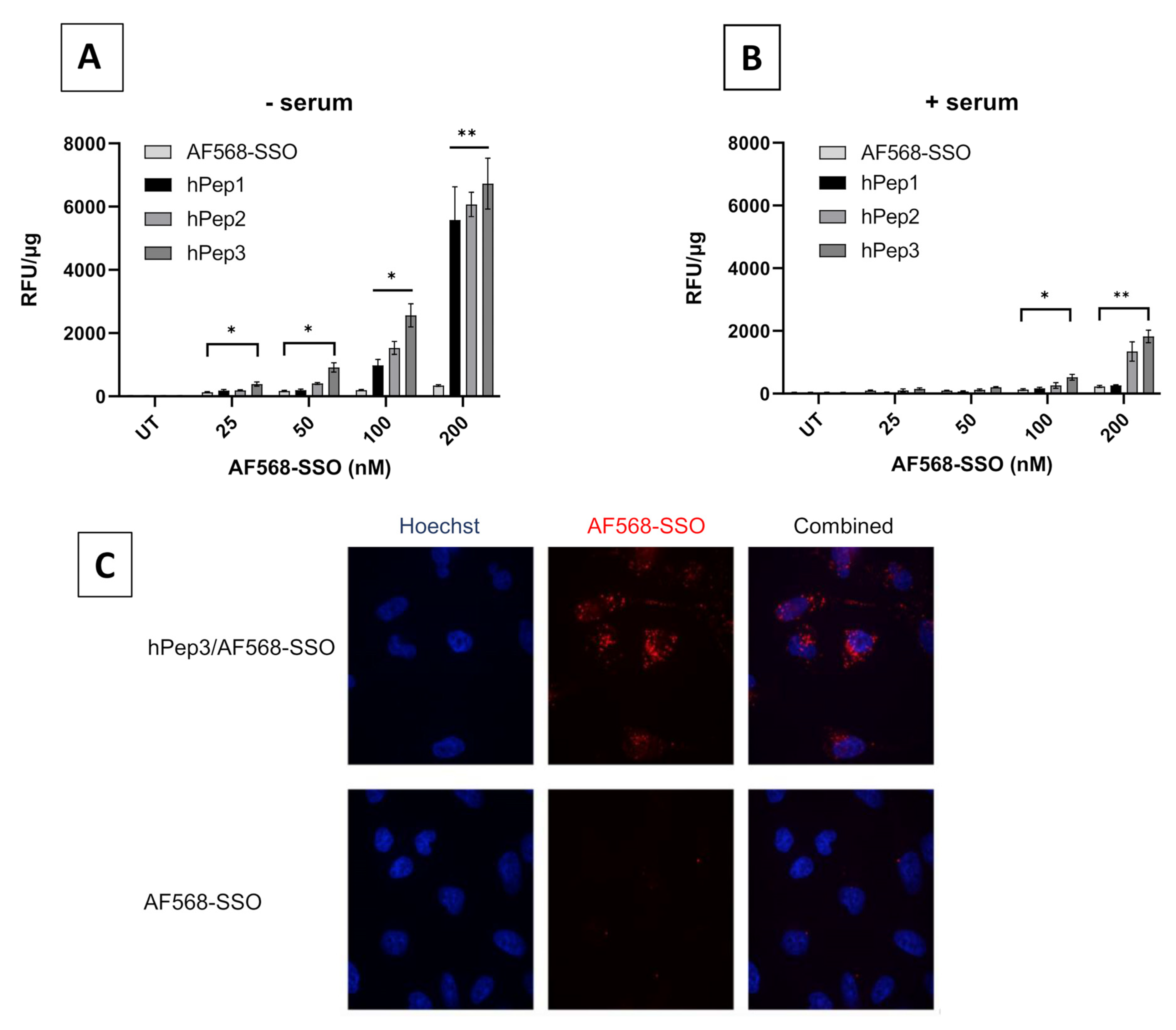
12]. In the current study, we aimed to find an alternative chemical approach for CPP lipidation and designed new chemically modified CPPs called hPep′s using orthogonal hydrocarbon modifications with alkenyl-alanines to introduce hydrophobicity into the peptide backbone. The advantage of this approach is that the alkenyl-alanines can be incorporated into the peptide backbone at different locations during the automated synthesis stage. Therefore, it makes this approach very versatile and fine-tunable as it gives the opportunity to choose the number and length of the hydrocarbon modifications based on the application. In fact, we find that complexes formulated with hPep3 peptide with three octenyl modifications (24 added carbons) have higher uptake than hPep1 and hPep2 with 13 and 16 carbons (
Figure 5), while all of them form almost identically sized nanoparticles, in the 50–200 nm range. This is similar to our previous findings with PepFect/SSO nanoparticles, where it was found that at least 12 carbon atoms were required for the complexes to start associating with the cell membrane, and the higher the number of carbon atoms the more effectively the complexes associated with the cells [
12].
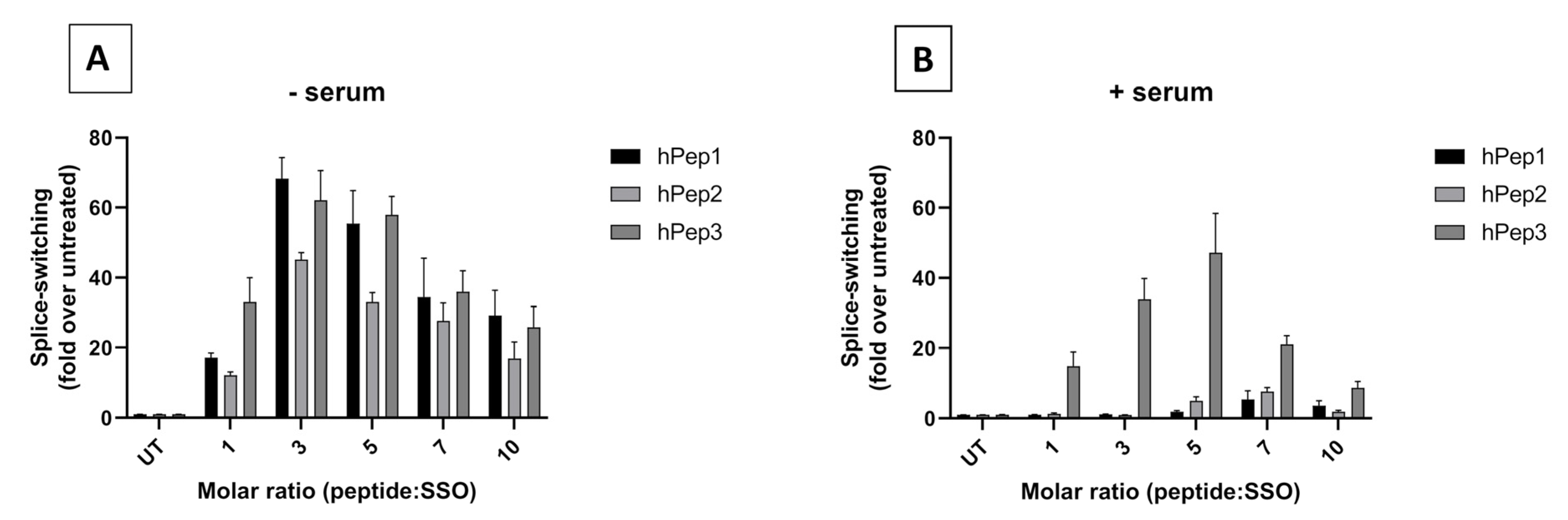
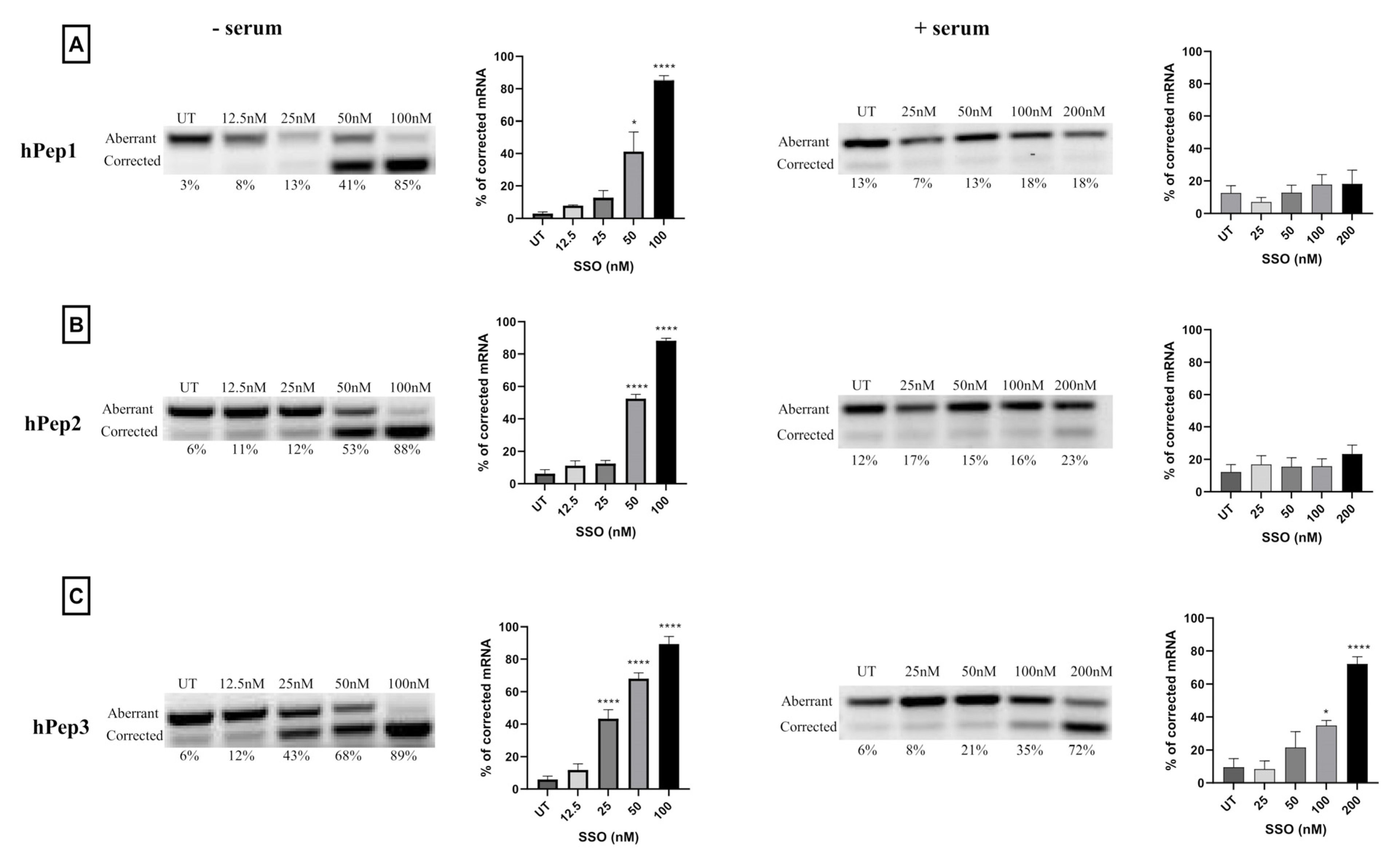
In order to determine the optimal peptide/SSO molar ratio, we performed screening of different MRs in the HeLa 705 cell line. We found that splice-switching activity was the highest between MRs 3 and 7 in both serum-free and serum-containing conditions (
Figure 2). Hence, we chose MR5 for subsequent detailed studies. Another aspect supporting choosing the MR5 is that it has been shown for different cationic DDSs that complexation of ONs with excessive amounts of delivery vectors leads to unwanted toxicity caused by the unbound free fraction of DDS due to its membrane active properties [
12,
22,
23].
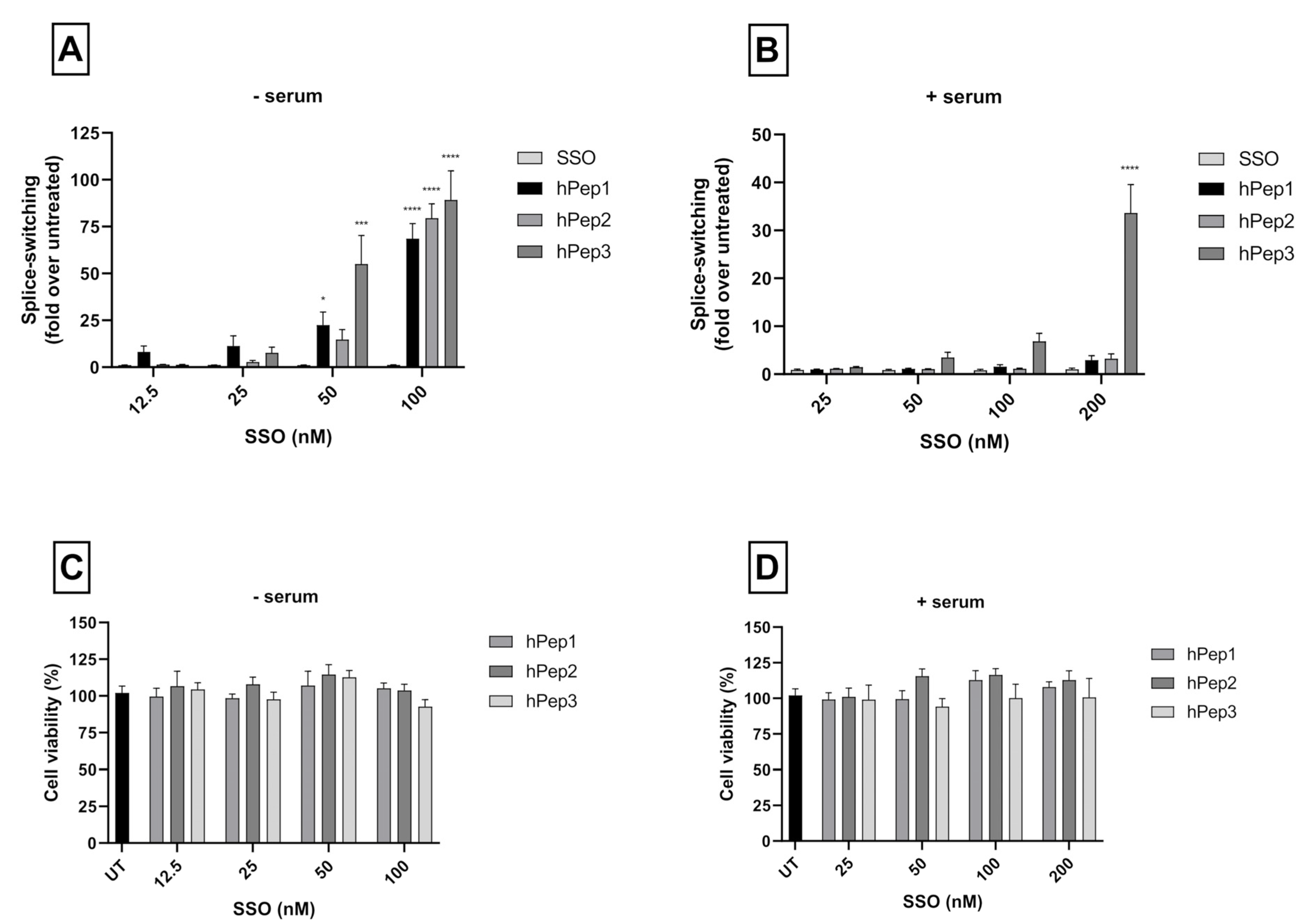
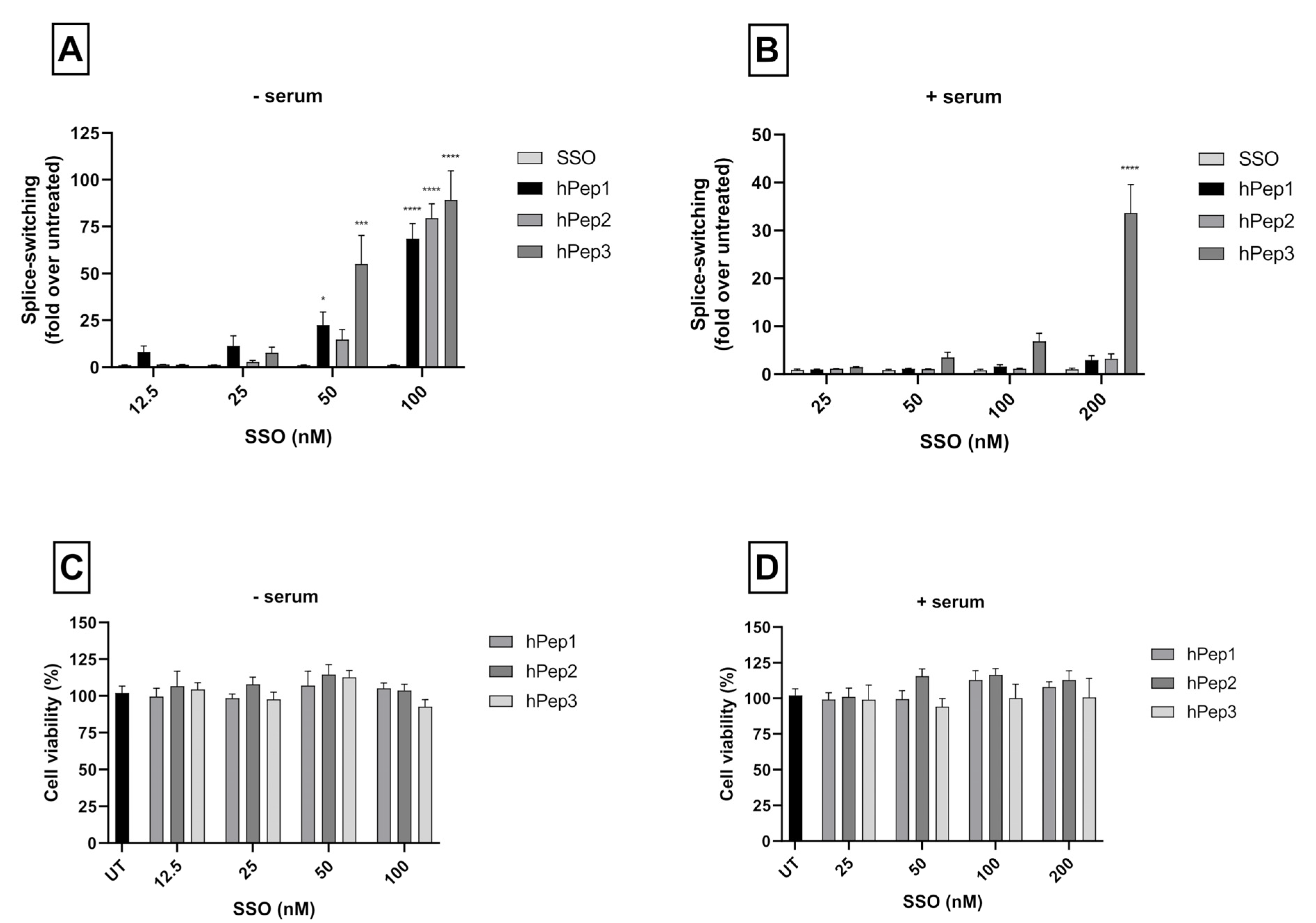
More detailed cell culture studies with hPep/SSO complexes showed that out of all the tested peptides, only the most hydrophobic hPep3 was able to induce significant splice-switching activity both at the luciferase as well as the mRNA (EC50 = 104 nM) level at serum-containing conditions. To our surprise, these results are very well in line with our previous findings with N-terminal fatty acid analogs of PepFect14, where we showed, by varying the hydrocarbon chain length, that by introducing ≥12 carbons, splice-correction activity of the SSO complexes increased linearly with each added carbon [
12]. We hypothesize that the superior activity of the hPep3 could be due to the higher stability, as hPep3 nanoparticles were much more stable towards competitive polyanion treatment than the rest of the peptides (
Figure 1C), correlating well with other similar findings on the importance of hydrophobic interactions for the stability of CPP/nucleic acid complexes [
11,
24]. Furthermore, none of the tested peptides showed any adverse effect on the cell viability, even at the 200 nM SSO treatment concentration. Together these data imply that introduction of orthogonal hydrocarbon modifications into the peptide to increase its hydrophobicity can be used to generate a highly efficient and well-tolerated drug delivery system for SSOs in cell cultures.
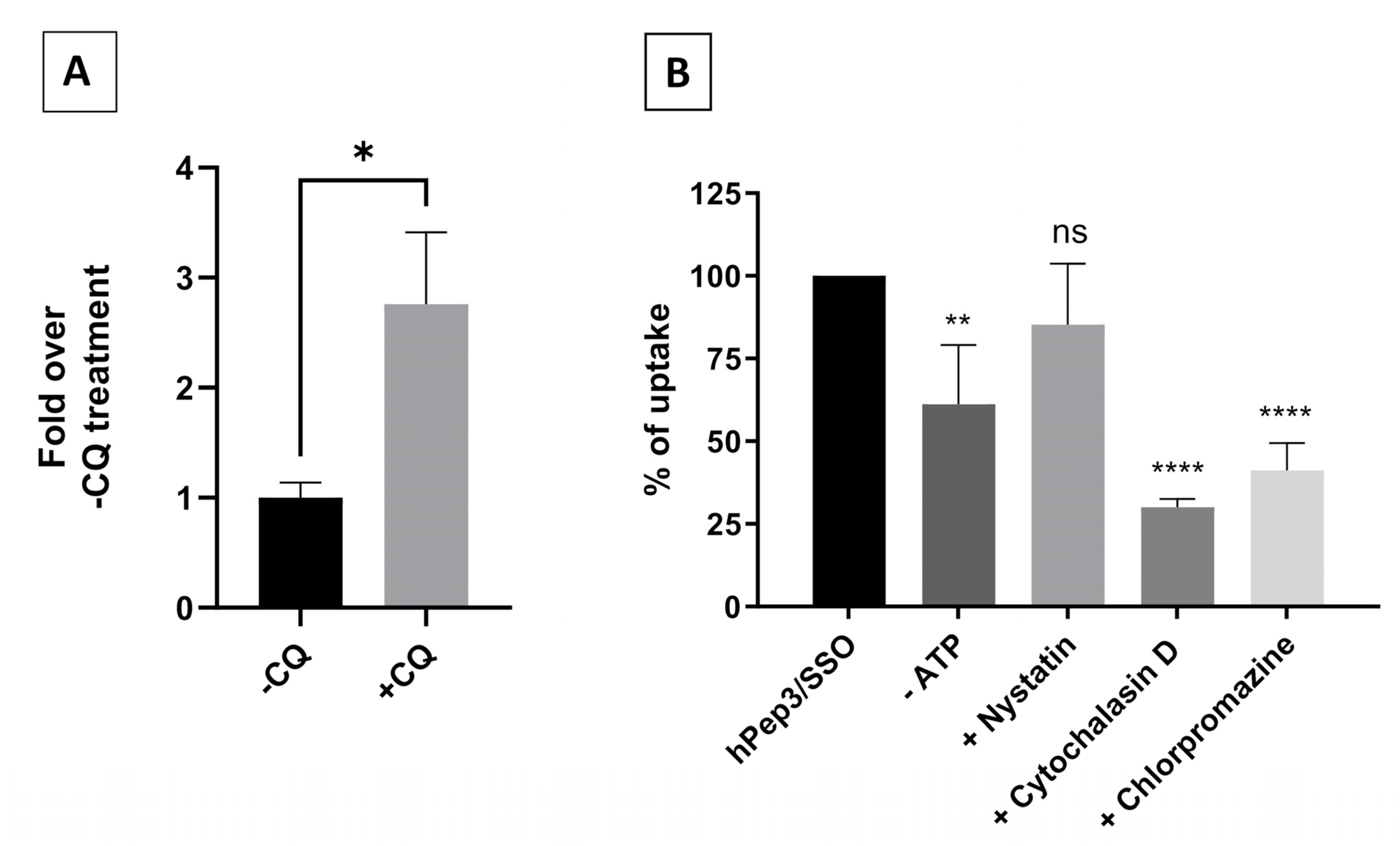
It is widely recognized that nanoparticle-based DDSs mainly enter cells via endocytosis and end up in endocytic vesicles. When endosomes mature into lysosomes the pH in these vesicles drops, which activates lysosomal enzymes that will degrade the contents inside the vesicles. Therefore, DDSs have to escape the endosomes with their payload before they get degraded. Endosomal entrapment is considered one of the major bottlenecks for all DDSs. Similarly to other DDSs, we found that the hPep3/SSO nanoparticles to a high extent get entrapped in the endosomes, as treatment with endosomolytic agent chloroquine could be used to release the endosome-bound material (
Figure 6A). When we delved more into the cellular uptake mechanisms with specific pharmacological inhibitors it was clear that hPep3/SSO NPs enter cells via energy-dependent pathways, mainly, by macropinocytosis and clathrin-mediated endocytosis. The use of several endocytic pathways has also been reported for other peptide-based ON delivery systems. For example, PepFect14/SSO NPs have been shown to enter cells via macropinocytosis and caveolin-dependent endocytosis while involvement of scavenger receptors during these processes has been documented in several studies [
25,
26,
27].
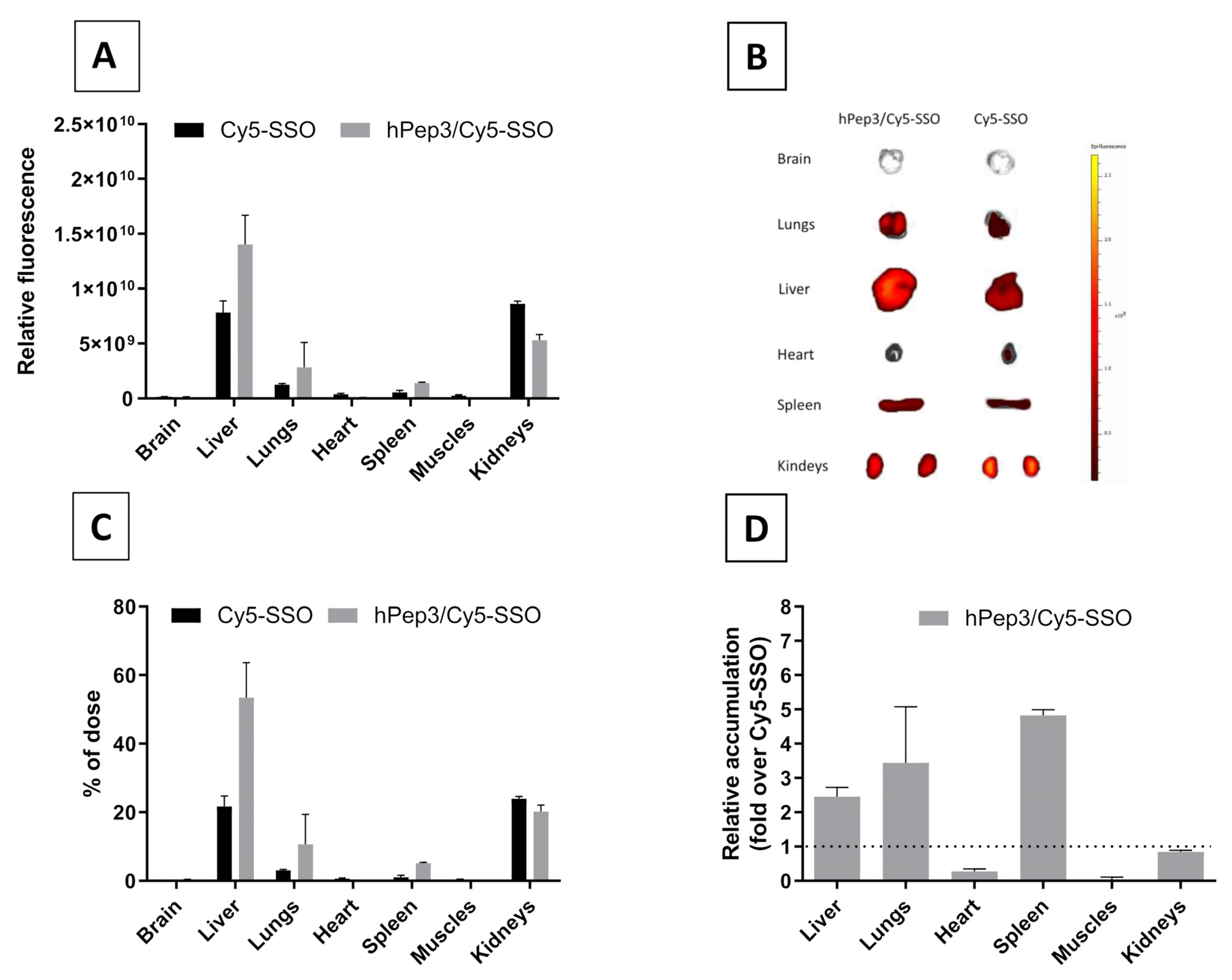
After confirming the safety and efficacy of hPep3/SSO NPs in vitro we went on to study its delivery capabilities in vivo. Biodistribution studies in mice with the fluorescently labelled hPep3/SSO nanoparticles indicated that hPep3 could increase the accumulation of SSOs in liver, lungs and spleen by up to several folds as compared to the treatments with free SSO (
Figure 7D). Furthermore, the i.v.-administered complexes were very well tolerated by the animals.


 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}