
Linking Immunity with Genomics in Sarcomas: Is Genomic Complexity an Immunogenic Trigger?

 , ,
, ,  ,
,  and
and 
Abstract
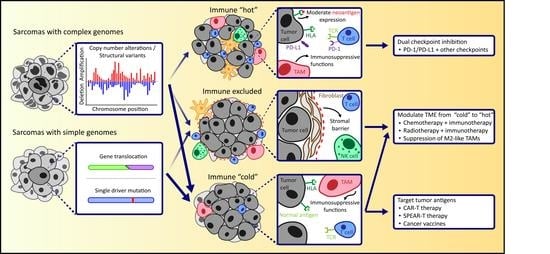
:
1. Introduction
2. The Tumor Microenvironment in the Context of Genomic Complexity
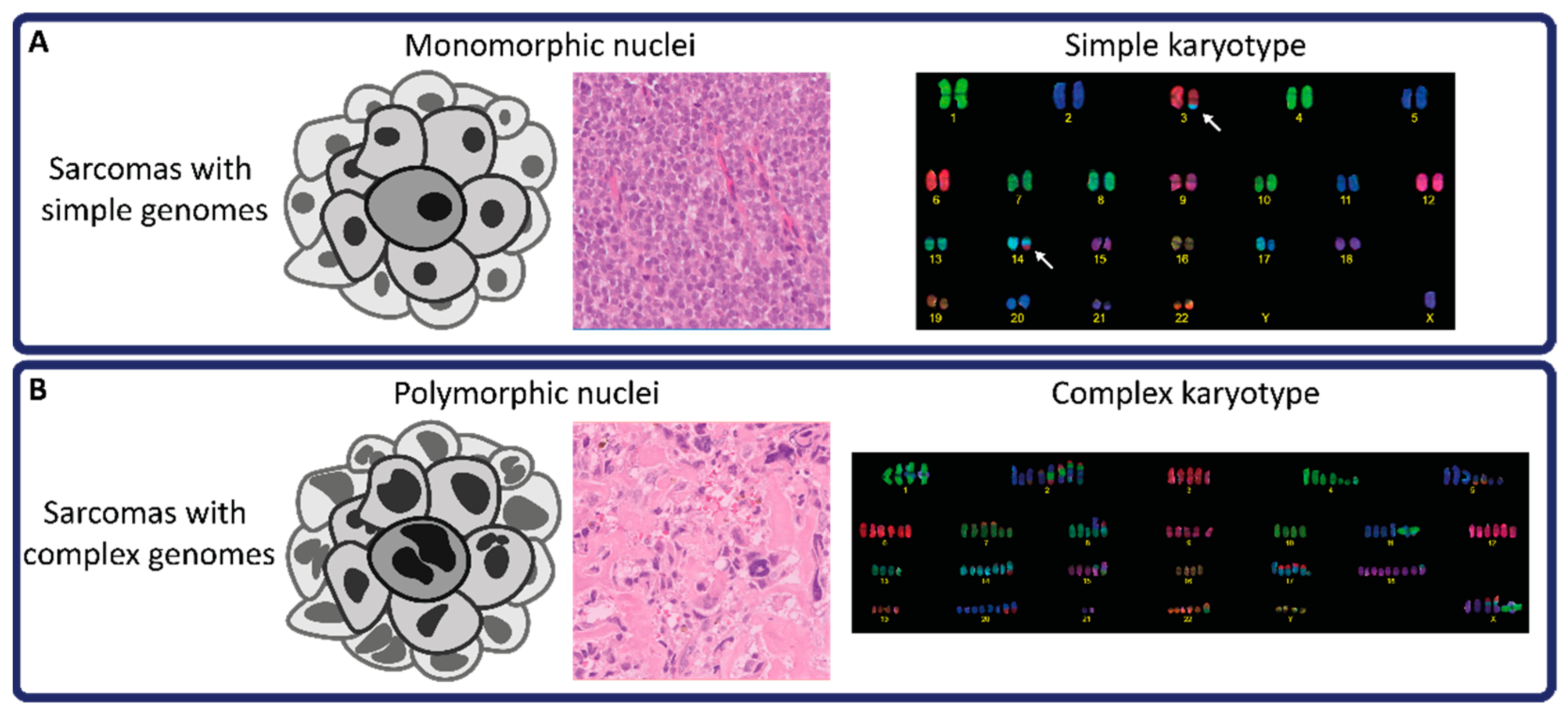
2.1. The Tumor Microenvironment of Sarcomas with Simple Genomes
2.2. The Tumor Microenvironment in Sarcomas with Complex Genomes
3. Clinical Responses to Immunotherapy in Relation to the Immunogenomics of Sarcomas
3.1. Response to T Cell Checkpoint Blockade
3.2. Response to Other Immunotherapeutic Agents
4. Cues from the Tumor Microenvironment for the Development of Novel (Immune) Therapeutic Approaches
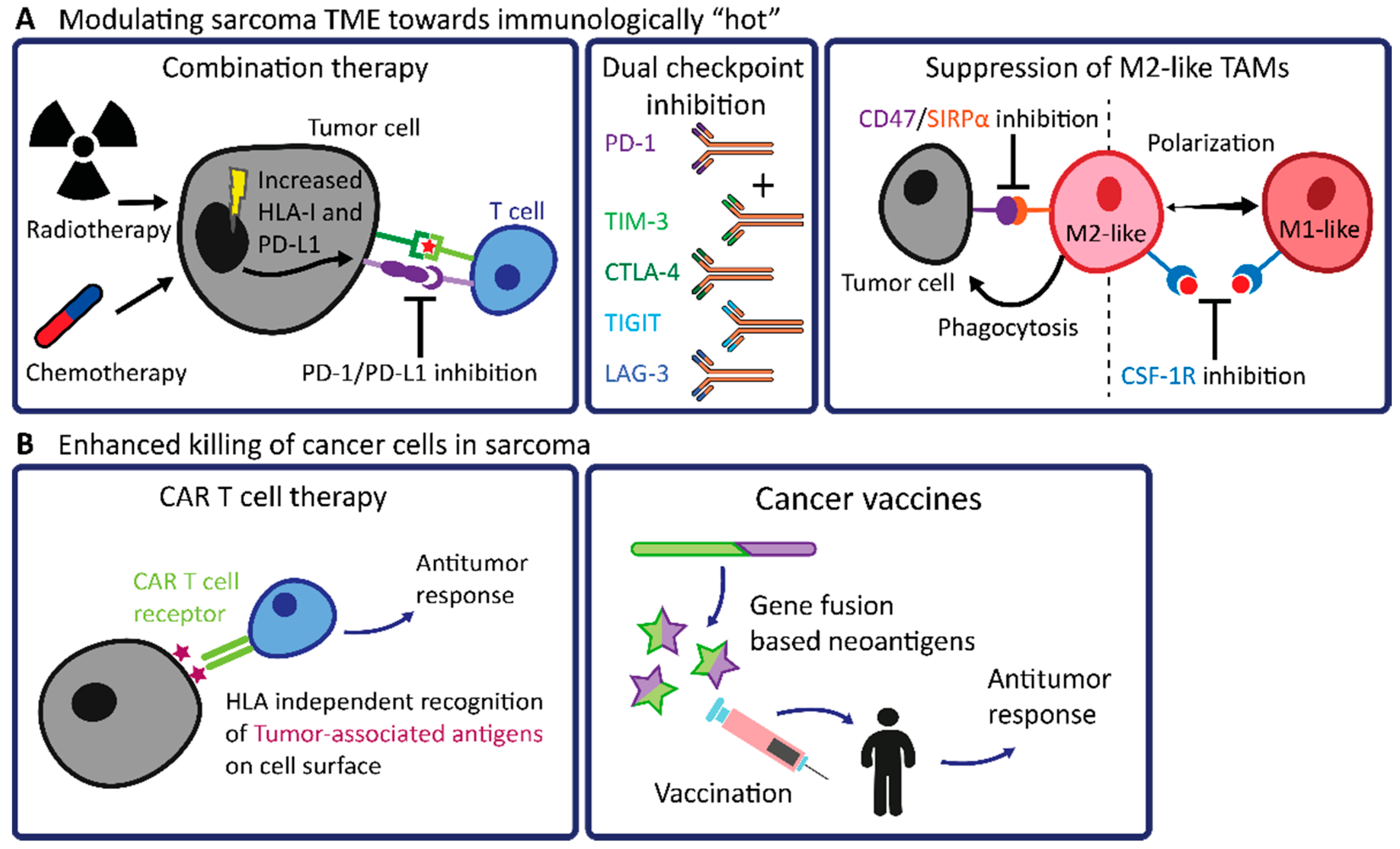
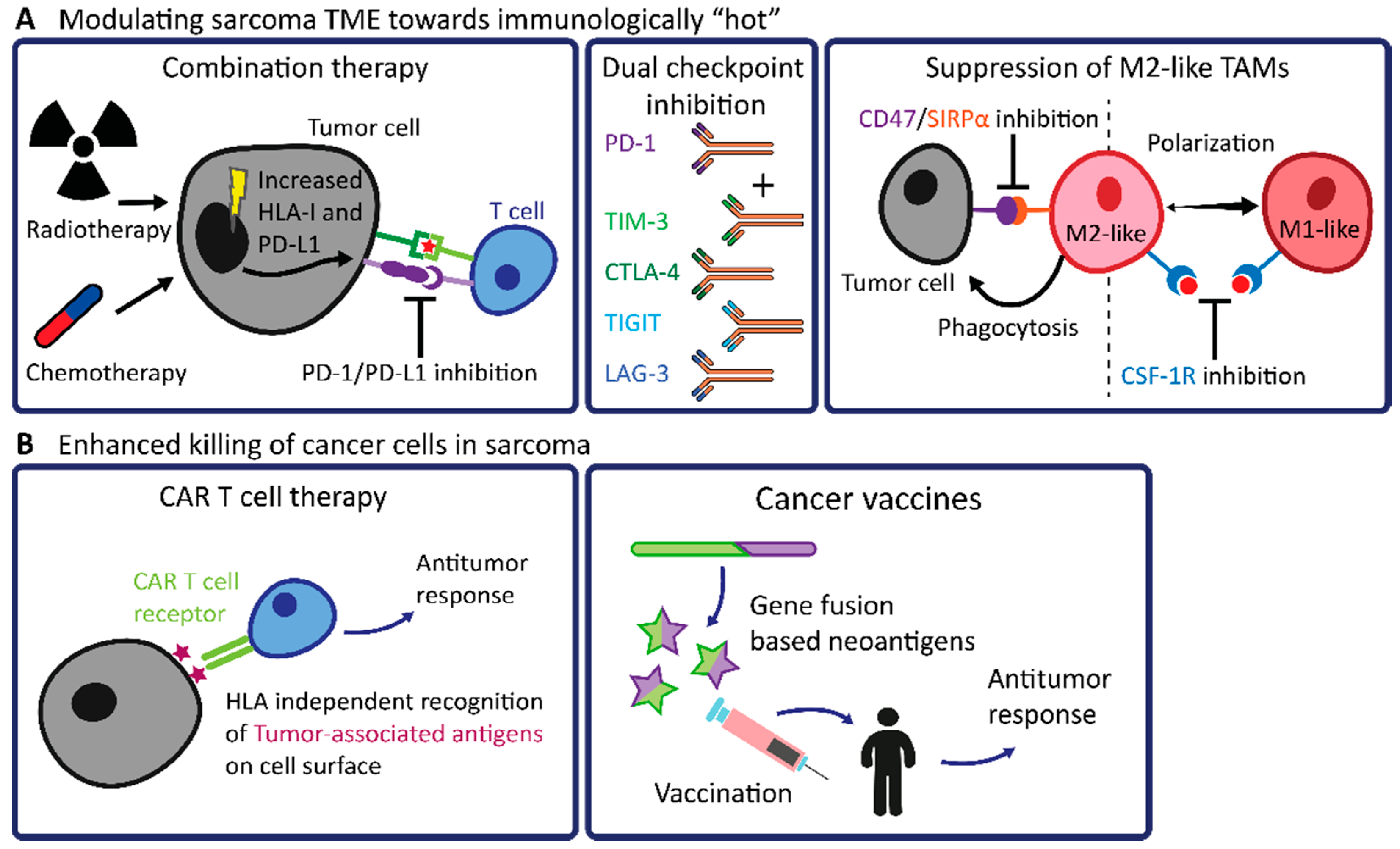
4.1. Modulating the TME of Sarcomas towards Immunologically “Hot”
4.2. Future Prospects in Engineered T Cell Therapy and Cancer Vaccines in Sarcomas
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Antonescu, C.R.; WHO Classification of Tumours Editorial Board. WHO Classification of Tumours of Soft Tissue and Bone, 5th ed.; IARC Press: Lyon, France, 2020. [Google Scholar]
- Grünewald, T.G.; Cidre-Aranaz, F.; Surdez, D.; Tomazou, E.M.; de Álava, E.; Kovar, H.; Dirksen, U. Ewing sarcoma. Nat. Rev. Dis. Primers 2018, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Pedeutour, F.; Forus, A.; Coindre, J.M.; Berner, J.M.; Nicolo, G.; Michiels, J.F.; Turc-Carel, C. Structure of the supernumerary ring and giant rod chromosomes in adipose tissue tumors. Genes Chromosomes Cancer 1999, 24, 30–41. [Google Scholar] [CrossRef]
- Lam, S.W.; van IJzendoorn, D.G.P.; Cleton-Jansen, A.M.; Szuhai, K.; Bovée, J.V.M.G. Molecular pathology of bone tumors. J. Mol. Diagn. 2019, 21, 171–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortés-Ciriano, I.; Lee, J.J.-K.; Xi, R.; Jain, D.; Jung, Y.L.; Yang, L.; Gordenin, D.; Klimczak, L.J.; Zhang, C.-Z.; Pellman, D.S.; et al. Comprehensive analysis of chromothripsis in 2658 human cancers using whole-genome sequencing. Nat. Genet. 2020, 52, 331–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chibon, F.; Lagarde, P.; Salas, S.; Pérot, G.; Brouste, V.; Tirode, F.; Lucchesi, C.; De Reynies, A.; Kauffmann, A.; Bui, B.; et al. Validated prediction of clinical outcome in sarcomas and multiple types of cancer on the basis of a gene expression signature related to genome complexity. Nat. Med. 2010, 16, 781–787. [Google Scholar] [CrossRef]
- Nicolle, R.; Ayadi, M.; Gomez-Brouchet, A.; Armenoult, L.; Banneau, G.; Elarouci, N.; Tallegas, M.; Decouvelaere, A.-V.; Aubert, S.; Rédini, F.; et al. Integrated molecular characterization of chondrosarcoma reveals critical determinants of disease progression. Nat. Commun. 2019, 10, 4622. [Google Scholar] [CrossRef]
- Jhunjhunwala, S.; Hammer, C.; Delamarre, L. Antigen presentation in cancer: Insights into tumour immunogenicity and immune evasion. Nat. Rev. Cancer 2021, 21, 298–312. [Google Scholar] [CrossRef]
- Bakhoum, S.F.; Cantley, L.C. The multifaceted role of chromosomal instability in cancer and its microenvironment. Cell 2018, 174, 1347–1360. [Google Scholar] [CrossRef] [Green Version]
- Samstein, R.M.; Lee, C.-H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; PCAWG Mutational Signatures Working Group; Kim, J.; Haradhvala, N.J.; Huang, N.M.; Ng, A.W.T.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; et al. The repertoire of mutational signatures in human cancer. Nat. Cell Biol. 2020, 578, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Santaguida, S.; Richardson, A.; Iyer, D.R.; M’Saad, O.; Zasadil, L.; Knouse, K.A.; Wong, Y.L.; Rhind, N.; Desai, A.; Amon, A. Chromosome mis-segregation generates cell-cycle-arrested cells with complex karyotypes that are eliminated by the immune system. Dev. Cell 2017, 41, 638.e5–651.e5. [Google Scholar] [CrossRef] [Green Version]
- Kwon, J.; Bakhoum, S.F. The cytosolic DNA-sensing cGAS-STING pathway in cancer. Cancer Discov. 2020, 10, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Davoli, T.; Uno, H.; Wooten, E.C.; Elledge, S.J. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 2017, 355, eaaf8399. [Google Scholar] [CrossRef] [Green Version]
- Siozopoulou, V.; Domen, A.; Zwaenepoel, K.; Van Beeck, A.; Smits, E.; Pauwels, P.; Marcq, E. Immune checkpoint inhibitory therapy in sarcomas: Is there light at the end of the tunnel? Cancers 2021, 13, 360. [Google Scholar] [CrossRef]
- van den Bulk, J.; de Miranda, N.F.; Ten Dijke, P. Therapeutic targeting of TGF-β in cancer: Hacking a master switch of immune suppression. Clin. Sci. 2021, 135, 35–52. [Google Scholar] [CrossRef]
- Fridman, W.H.; Zitvogel, L.; Sautes-Fridman, C.; Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whiteside, T.L.; Demaria, S.; Rodriguez-Ruiz, M.; Zarour, H.M.; Melero, I. Emerging opportunities and challenges in cancer immunotherapy. Clin. Cancer Res. 2016, 22, 1845–1855. [Google Scholar] [CrossRef] [Green Version]
- Montesion, M.; Murugesan, K.; Jin, D.X.; Sharaf, R.; Sanchez, N.; Guria, A.; Minker, M.; Li, G.; Fisher, V.; Sokol, E.S.; et al. Somatic HLA class I loss is a widespread mechanism of immune evasion which refines the use of tumor mutational burden as a biomarker of checkpoint inhibitor response. Cancer Discov. 2020, 11, 282–292. [Google Scholar] [CrossRef]
- Ijsselsteijn, M.E.; Petitprez, F.; Lacroix, L.; Ruano, D.; Van Der Breggen, R.; Julie, C.; Morreau, H.; Sautes-Fridman, C.; Fridman, W.H.; Miranda, N.F. Revisiting immune escape in colorectal cancer in the era of immunotherapy. Br. J. Cancer 2019, 120, 815–818. [Google Scholar] [CrossRef] [Green Version]
- Koumarianou, A.; Duran-Moreno, J. The sarcoma immune landscape: Emerging challenges, prognostic significance and prospective impact for immunotherapy approaches. Cancers 2021, 13, 363. [Google Scholar] [CrossRef]
- Petitprez, F.; Reyniès, A.; Keung, E.Z.; Chen, T.W.-W.; Sun, C.-M.; Calderaro, J.; Jeng, Y.-M.; Hsiao, L.-P.; Lacroix, L.; Bougoüin, A.; et al. B cells are associated with survival and immunotherapy response in sarcoma. Nat. Cell Biol. 2020, 577, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Ogose, A.; Kawashima, H.; Hotta, T.; Ariizumi, T.; Yamagishi, T.; Oike, N.; Sasaki, T.; Hatano, H.; Umezu, H.; Endo, N. Frequent expression of human leukocyte antigen class I and the status of intratumoral immune cells in alveolar soft part sarcoma. Oncol. Lett. 2017, 13, 2169–2176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilky, B.A.; Trucco, M.M.; Subhawong, T.K.; Florou, V.; Park, W.; Kwon, D.; Trent, J.C. Axitinib plus pembrolizumab in patients with advanced sarcomas including alveolar soft-part sarcoma: A single-centre, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 837–848. [Google Scholar] [CrossRef]
- Shi, Y.; Cai, Q.; Jiang, Y.; Huang, G.; Bi, M.; Wang, B.; Zhou, Y.; Wang, G.; Ying, H.; Tao, Z.; et al. Activity and safety of geptanolimab (GB226) for patients with unresectable, recurrent, or metastatic alveolar soft part sarcoma: A phase II, single-arm study. Clin. Cancer Res. 2020, 26, 6445–6452. [Google Scholar] [CrossRef]
- Simard, F.A.; Richert, I.; Vandermoeten, A.; Decouvelaere, A.-V.; Michot, J.-M.; Caux, C.; Blay, J.-Y.; Dutour, A. Description of the immune microenvironment of chondrosarcoma and contribution to progression. Oncoimmunology 2016, 6, e1265716. [Google Scholar] [CrossRef] [Green Version]
- Iseulys, R.; Anne, G.B.; Corinne, B.; Gonzague, D.P.; Marie, K.; Jean-Yves, B.; Aurélie, D. The immune landscape of chondrosarcoma reveals an immunosuppressive environment in the dedifferentiated subtypes and exposes CSFR1+ macrophages as a promising therapeutic target. J. Bone Oncol. 2020, 20, 100271. [Google Scholar]
- Kostine, M.; Cleven, A.H.; de Miranda, N.; Italiano, A.; Cleton-Jansen, A.-M.; Bovee, J. Analysis of PD-L1, T-cell infiltrate and HLA expression in chondrosarcoma indicates potential for response to immunotherapy specifically in the dedifferentiated subtype. Mod. Pathol. 2016, 29, 1028–1037. [Google Scholar] [CrossRef] [Green Version]
- Tseng, W.W.; Malu, S.; Zhang, M.; Chen, J.; Sim, G.C.; Wei, W.; Hwu, P. Analysis of the intratumoral adaptive immune response in well differentiated and dedifferentiated retroperitoneal liposarcoma. Sarcoma 2015, 2015, 547460. [Google Scholar] [CrossRef]
- Pollack, S.M.; He, Q.; Yearley, J.H.; Emerson, R.; Vignali, M.; Zhang, Y.; Redman, M.W.; Baker, K.K.; Cooper, S.; Donahue, B.; et al. T-cell infiltration and clonality correlate with programmed cell death protein 1 and programmed death-ligand 1 expression in patients with soft tissue sarcomas. Cancer 2017, 123, 3291–3304. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.R.; Moon, Y.J.; Kwon, K.S.; Bae, J.S.; Wagle, S.; Kim, K.M.; Park, H.S.; Lee, H.; Moon, W.S.; Chung, M.J.; et al. Tumor infiltrating PD1-positive lymphocytes and the expression of PD-L1 predict Poor prognosis of soft tissue sarcomas. PLoS ONE 2013, 8, e82870. [Google Scholar] [CrossRef]
- Vargas, A.C.; MacLean, F.M.; Sioson, L.; Tran, D.; Bonar, F.; Mahar, A.; Cheah, A.L.; Russell, P.; Grimison, P.; Richardson, L.; et al. Prevalence of PD-L1 expression in matched recurrent and/or metastatic sarcoma samples and in a range of selected sarcomas subtypes. PLoS ONE 2020, 15, e0222551. [Google Scholar] [CrossRef] [Green Version]
- van Erp, A.E.; Versleijen-Jonkers, Y.M.; Hillebrandt-Roeffen, M.H.; van Houdt, L.; Gorris, M.A.; van Dam, L.S.; Meyer-Wentrup, F.A. Expression and clinical association of programmed cell death-1, programmed death-ligand-1 and CD8+ lymphocytes in primary sarcomas is subtype dependent. Oncotarget 2017, 8, 71371. [Google Scholar] [CrossRef] [Green Version]
- Hingorani, P.; Maas, M.L.; Gustafson, M.P.; Dickman, P.; Adams, R.H.; Watanabe, M.; Dietz, A.B. Increased CTLA-4+ T cells and an increased ratio of monocytes with loss of class II (CD14+ HLA-DR lo/neg) found in aggressive pediatric sarcoma patients. J. Immunother. Cancer 2015, 3, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Handl, M.; Hermanova, M.; Hotarkova, S.; Jarkovsky, J.; Mudry, P.; Shatokhina, T.; Vesela, M.; Sterba, J.; Zambo, I. Clinicopathological correlation of tumor-associated macrophages in Ewing sarcoma. Biomed. Pap. 2018, 162, 54–60. [Google Scholar] [CrossRef] [Green Version]
- Oike, N.; Kawashima, H.; Ogose, A.; Hotta, T.; Hatano, H.; Ariizumi, T.; Sasaki, T.; Yamagishi, T.; Umezu, H.; Endo, N. Prognostic impact of the tumor immune microenvironment in synovial sarcoma. Cancer Sci. 2018, 109, 3043–3054. [Google Scholar] [CrossRef] [Green Version]
- Dancsok, A.R.; Gao, D.; Lee, A.; Steigen, S.E.; Blay, J.-Y.; Thomas, D.M.; Maki, R.G.; Nielsen, T.O.; Demicco, E.G. Tumor-associated macrophages and macrophage-related immune checkpoint expression in sarcomas. Oncoimmunology 2020, 9, 1747340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dridi, M.; Krebs-Drouot, L.; Meyronet, D.; Dumollard, J.; Vassal, F.; Jouanneau, E.; Jacquesson, T.; Barrey, C.; Grange, S.; Boutonnat, J.; et al. The immune microenvironment of chordomas: An immunohistochemical analysis. Cancers 2021, 13, 3335. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.-X.; Lv, G.-H.; Wang, X.-B.; Huang, W.; Li, J.; Jiang, Y.; She, X.-L. Clinical impact of the immune microenvironment in spinal chordoma: Immunoscore as an independent favorable prognostic factor. Neurosurgery 2018, 84, E318–E333. [Google Scholar] [CrossRef]
- Zou, M.-X.; Pan, Y.; Huang, W.; Zhang, T.; Escobar, D.; Wang, X.; Jiang, Y.; She, X.; Lv, G.; Li, J. A four-factor immune risk score signature predicts the clinical outcome of patients with spinal chordoma. Clin. Transl. Med. 2020, 10, 224–237. [Google Scholar] [CrossRef]
- Smolle, M.; Herbsthofer, L.; Goda, M.; Granegger, B.; Brcic, I.; Bergovec, M.; Scheipl, S.; Prietl, B.; El-Heliebi, A.; Pichler, M.; et al. Influence of tumor-infiltrating immune cells on local control rate, distant metastasis, and survival in patients with soft tissue sarcoma. Oncoimmunology 2021, 10, 1896658. [Google Scholar] [CrossRef]
- Dancsok, A.R.; Setsu, N.; Gao, D.; Blay, J.-Y.; Thomas, D.; Maki, R.G.; Nielsen, T.O.; Demicco, E.G. Expression of lymphocyte immunoregulatory biomarkers in bone and soft-tissue sarcomas. Mod. Pathol. 2019, 32, 1772–1785. [Google Scholar] [CrossRef]
- Wunder, J.S.; Lee, M.J.; Nam, J.; Lau, B.Y.; Dickson, B.C.; Pinnaduwage, D.; Bull, S.B.; Ferguson, P.C.; Seto, A.; Gokgoz, N.; et al. Osteosarcoma and soft-tissue sarcomas with an immune infiltrate express PD-L1: Relation to clinical outcome and Th1 pathway activation. Oncoimmunology 2020, 9, 1737385. [Google Scholar] [CrossRef] [Green Version]
- Koirala, P.; Roth, M.E.; Gill, J.; Piperdi, S.; Chinai, J.M.; Geller, D.S.; Hoang, B.H.; Park, A.; Fremed, M.A.; Zang, X.; et al. Immune infiltration and PD-L1 expression in the tumor microenvironment are prognostic in osteosarcoma. Sci. Rep. 2016, 6, 30093. [Google Scholar] [CrossRef]
- Palmerini, E.; Agostinelli, C.; Picci, P.; Pileri, S.; Marafioti, T.; Lollini, P.L.; Ferrari, S. Tumoral immune-infiltrate (IF), PD-L1 expression and role of CD8/TIA-1 lymphocytes in localized osteosarcoma patients treated within protocol ISG-OS1. Oncotarget 2017, 8, 111836. [Google Scholar] [CrossRef] [Green Version]
- Zheng, B.; Ren, T.; Huang, Y.; Guo, W. Apatinib inhibits migration and invasion as well as PD-L1 expression in osteosarcoma by targeting STAT3. Biochem. Biophys. Res. Commun. 2018, 495, 1695–1701. [Google Scholar] [CrossRef]
- Kostine, M.; Bruijn, I.H.B.-D.; Cleven, A.H.G.; Vervat, C.; Corver, W.E.; Schilham, M.W.; Van Beelen, E.; Van Boven, H.; Haas, R.L.; Italiano, A.; et al. Increased infiltration of M2-macrophages, T-cells and PD-L1 expression in high grade leiomyosarcomas supports immunotherapeutic strategies. Oncoimmunology 2017, 7, e1386828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keung, E.Z.; Tsai, J.-W.; Ali, A.M.; Cormier, J.N.; Bishop, A.; Guadagnolo, B.A.; Torres, K.E.; Somaiah, N.; Hunt, K.K.; Wargo, J.A.; et al. Analysis of the immune infiltrate in undifferentiated pleomorphic sarcoma of the extremity and trunk in response to radiotherapy: Rationale for combination neoadjuvant immune checkpoint inhibition and radiotherapy. Oncoimmunology 2017, 7, e1385689. [Google Scholar] [CrossRef]
- Toulmonde, M.; Lucchesi, C.; Verbeke, S.; Crombe, A.; Adam, J.; Geneste, D.; Chaire, V.; Laroche-Clary, A.; Perret, R.; Bertucci, F.; et al. High throughput profiling of undifferentiated pleomorphic sarcomas identifies two main subgroups with distinct immune profile, clinical outcome and sensitivity to targeted therapies. EBioMedicine 2020, 62, 103131. [Google Scholar] [CrossRef]
- Manzoni, M.; Bolognesi, M.M.; Antoranz, A.; Mancari, R.; Carinelli, S.; Faretta, M.; Bosisio, F.M.; Cattoretti, G. The adaptive and innate immune cell landscape of uterine leiomyosarcomas. Sci. Rep. 2020, 10, 702. [Google Scholar] [CrossRef] [Green Version]
- Shanes, E.D.; Friedman, L.A.; Mills, A.M. PD-L1 Expression and tumor-infiltrating lymphocytes in uterine smooth muscle tumors. Am. J. Surg. Pathol. 2019, 43, 780–792. [Google Scholar] [CrossRef] [PubMed]
- Yabe, H.; Tsukahara, T.; Kawaguchi, S.; Wada, T.; Torigoe, T.; Sato, N.; Yabe, H. Prognostic significance of HLA class I expression in Ewing’s sarcoma family of tumors. J. Surg. Oncol. 2011, 103, 380–385. [Google Scholar] [CrossRef]
- Spurny, C.; Kailayangiri, S.; Altvater, B.; Jamitzky, S.; Hartmann, W.; Wardelmann, E.; Ranft, A.; Dirksen, U.; Amler, S.; Hardes, J.; et al. T cell infiltration into Ewing sarcomas is associated with local expression of immune-inhibitory HLA-G. Oncotarget 2017, 9, 6536–6549. [Google Scholar] [CrossRef] [Green Version]
- Berghuis, D.; Santos, S.J.; Baelde, H.J.; Taminiau, A.H.; Egeler, R.M.; Schilham, M.W.; Hogendoorn, P.C.; Lankester, A.C. Pro-inflammatory chemokine-chemokine receptor interactions within the Ewing sarcoma microenvironment determine CD8+ T-lymphocyte infiltration and affect tumour progression. J. Pathol. 2010, 223, 347–357. [Google Scholar] [CrossRef]
- Boxberg, M.; Steiger, K.; Lenze, U.; Rechl, H.; Von Eisenhart-Rothe, R.; Wörtler, K.; Weichert, W.; Langer, R.; Specht, K. PD-L1 and PD-1 and characterization of tumor-infiltrating lymphocytes in high grade sarcomas of soft tissue—Prognostic implications and rationale for immunotherapy. Oncoimmunology 2017, 7, e1389366. [Google Scholar] [CrossRef] [Green Version]
- He, M.; Abro, B.; Kaushal, M.; Chen, L.; Chen, T.; Gondim, M.; Yan, W.; Neidich, J.; Dehner, L.P.; Pfeifer, J.D. Tumor mutation burden and checkpoint immunotherapy markers in primary and metastatic synovial sarcoma. Hum. Pathol. 2020, 100, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Nowicki, T.; Akiyama, R.; Huang, R.R.; Shintaku, I.P.; Wang, X.; Tumeh, P.C.; Singh, A.; Chmielowski, B.; Denny, C.; Federman, N.; et al. Infiltration of CD8 T Cells and expression of PD-1 and PD-L1 in synovial sarcoma. Cancer Immunol. Res. 2016, 5, 118–126. [Google Scholar] [CrossRef] [Green Version]
- Machado, I.; López-Guerrero, J.A.; Scotlandi, K.; Picci, P.; Llombart-Bosch, A. Immunohistochemical analysis and prognostic significance of PD-L1, PD-1, and CD8+ tumor-infiltrating lymphocytes in Ewing’s sarcoma family of tumors (ESFT). Virchows Arch. 2018, 472, 815–824. [Google Scholar] [CrossRef]
- Sautès-Fridman, C.; Petitprez, F.; Calderaro, J.; Fridman, W.H. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat. Rev. Cancer 2019, 19, 307–325. [Google Scholar] [CrossRef]
- Chen, L.; Oke, T.; Siegel, N.; Cojocaru, G.; Tam, A.J.; Blosser, R.L.; Swailes, J.; Ligon, J.; Lebid, A.; Morris, C.; et al. The immunosuppressive niche of soft-tissue sarcomas is sustained by tumor-associated macrophages and characterized by intratumoral tertiary lymphoid structures. Clin. Cancer Res. 2020, 26, 4018–4030. [Google Scholar] [CrossRef] [Green Version]
- Lazar, A.; Das, P.; Tuvin, D.; Korchin, B.; Zhu, Q.; Jin, Z.; Warneke, C.L.; Zhang, P.S.; Hernandez, V.; Lopez-Terrada, D.; et al. Angiogenesis-promoting gene patterns in alveolar soft part sarcoma. Clin. Cancer Res. 2007, 13, 7314–7321. [Google Scholar] [CrossRef] [Green Version]
- Balan, M.; Teran, E.M.Y.; Waaga-Gasser, A.M.; Gasser, M.; Choueiri, T.K.; Freeman, G.; Pal, S. Novel roles of C-met in the survival of renal cancer cells through the regulation of HO-1 and PD-L1 expression. J. Biol. Chem. 2015, 290, 8110–8120. [Google Scholar] [CrossRef] [Green Version]
- McGrail, D.J.; Federico, L.; Li, Y.; Dai, H.; Lu, Y.; Mills, G.B.; Yi, S.; Lin, S.-Y.; Sahni, N. Multi-omics analysis reveals neoantigen-independent immune cell infiltration in copy-number driven cancers. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chudasama, P.; Mughal, S.S.; Sanders, M.A.; Huebschmann, D.; Chung, I.; Deeg, K.I.; Wong, S.-H.; Rabe, S.; Hlevnjak, M.; Zapatka, M.; et al. Integrative genomic and transcriptomic analysis of leiomyosarcoma. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewin, J.; Garg, S.; Lau, B.Y.; Dickson, B.C.; Traub, F.; Gokgoz, N.; Razak, A.R. Identifying actionable variants using next generation sequencing in patients with a historical diagnosis of undifferentiated pleomorphic sarcoma. Int. J. Cancer 2018, 142, 57–65. [Google Scholar] [CrossRef] [Green Version]
- Ng, K.W.; Marshall, E.A.; Bell, J.C.; Lam, W.L. cGAS–STING and cancer: Dichotomous roles in tumor immunity and development. Trends Immunol. 2018, 39, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; Konno, H.; Barber, G.N. Recurrent loss of STING signaling in melanoma correlates with susceptibility to viral oncolysis. Cancer Res. 2016, 76, 6747–6759. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Niu, X.; Wang, Z.; Song, C.-L.; Huang, Z.; Chen, K.-N.; Duan, J.; Bai, H.; Xu, J.; Wang, Y.; et al. Multiregion sequencing reveals the genetic heterogeneity and evolutionary history of osteosarcoma and matched pulmonary metastases. Cancer Res. 2018, 79, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Sundara, Y.T.; Kostine, M.; Cleven, A.H.G.; Bovée, J.V.M.G.; Schilham, M.W.; Cleton-Jansen, A.-M. Increased PD-L1 and T-cell infiltration in the presence of HLA class I expression in metastatic high-grade osteosarcoma: A rationale for T-cell-based immunotherapy. Cancer Immunol. Immunother. 2016, 66, 119–128. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-C.; Beird, H.C.; Livingston, J.A.; Advani, S.; Mitra, A.; Cao, S.; Reuben, A.; Ingram, D.; Wang, W.-L.; Ju, Z.; et al. Immuno-genomic landscape of osteosarcoma. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Song, Y.; Deng, C.; Xu, Y.; Xu, H.; Zhu, X.; Song, G.; Tang, Q.; Lu, J.; Wang, J. Comprehensive analysis of immune infiltration and gene expression for predicting survival in patients with sarcomas. Aging 2020, 13, 2168–2183. [Google Scholar] [CrossRef]
- Campbell, B.B.; Light, N.; Fabrizio, D.; Zatzman, M.; Fuligni, F.; De Borja, R.; Davidson, S.; Edwards, M.; Elvin, J.A.; Hodel, K.P.; et al. Comprehensive analysis of hypermutation in human cancer. Cell 2017, 171, 1042.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Painter, C.A.; Jain, E.; Tomson, B.N.; Dunphy, M.; Stoddard, R.E.; Thomas, B.S.; Damon, A.L.; Shah, S.; Kim, D.; Zañudo, J.G.T.; et al. The angiosarcoma project: Enabling genomic and clinical discoveries in a rare cancer through patient-partnered research. Nat. Med. 2020, 26, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Cheung, L.S.; Chen, L.; Oke, T.F.; Schaffer, T.B.; Boudadi, K.; Ngo, J.T.; Gross, J.M.; Kemberling, H.; Diaz, L.; Lipson, E.; et al. Anti-PD-1 elicits regression of undifferentiated pleomorphic sarcomas with UV-mutation signatures. J. Immunother. Cancer 2021, 9, e002345. [Google Scholar] [CrossRef]
- Chan, J.Y.; Lim, J.Q.; Yeong, J.; Ravi, V.; Guan, P.; Boot, A.; Tay, T.K.Y.; Selvarajan, S.; Nasir, N.D.M.; Loh, J.H.; et al. Multiomic analysis and immunoprofiling reveal distinct subtypes of human angiosarcoma. J. Clin. Investig. 2020, 130, 5833–5846. [Google Scholar] [CrossRef]
- Choi, J.; Manzano, A.; Dong, W.; Bellone, S.; Bonazzoli, E.; Zammataro, L.; Yao, X.; Deshpande, A.; Zaidi, S.; Guglielmi, A.; et al. Integrated mutational landscape analysis of uterine leiomyosarcomas. Proc. Natl. Acad. Sci. USA 2021, 118, e2025182118. [Google Scholar] [CrossRef] [PubMed]
- Yatsenko, S.A.; Mittal, P.; Wood-Trageser, M.A.; Jones, M.W.; Surti, U.; Edwards, R.P.; Sood, A.K.; Rajkovic, A. Highly heterogeneous genomic landscape of uterine leiomyomas by whole exome sequencing and genome-wide arrays. Fertil. Steril. 2016, 107, 457–466.e9. [Google Scholar] [CrossRef] [Green Version]
- Karpathiou, G.; Dumollard, J.M.; Dridi, M.; Col, P.D.; Barral, F.-G.; Boutonnat, J.; Peoc’H, M. Chordomas: A review with emphasis on their pathophysiology, pathology, molecular biology, and genetics. Pathol. Res. Pract. 2020, 216, 153089. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.S.; Nota, S.P.; Sabbatino, F.; Nielsen, G.P.; Deshpande, V.; Wang, X.; Ferrone, S.; Schwab, J.H. Defective HLA class I expression and patterns of lymphocyte infiltration in chordoma tumors. Clin. Orthop. Relat. Res. 2020, 479, 1373–1382. [Google Scholar] [CrossRef]
- Bai, J.; Shi, J.; Li, C.; Wang, S.; Zhang, T.; Hua, X.; Yang, X.R. Whole genome sequencing of skull-base chordoma reveals genomic alterations associated with recurrence and chordoma-specific survival. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Tarpey, P.S.; Behjati, S.; Young, M.D.; Martincorena, I.; Alexandrov, L.B.; Farndon, S.J.; Guzzo, C.; Hardy, C.; Latimer, C.; Butler, A.P.; et al. The driver landscape of sporadic chordoma. Nat. Commun. 2017, 8, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakiryan, N.H.; Hajiran, A.; Kim, Y.; Aydin, A.M.; Zemp, L.; Katende, E.; Nguyen, J.; Fan, W.; Cheng, C.-H.; Lopez-Blanco, N.; et al. Correlating immune cell infiltration patterns with recurrent somatic mutations in advanced clear cell renal cell carcinoma. Eur. Urol. Focus 2021. [Google Scholar] [CrossRef] [PubMed]
- Miao, D.; Margolis, C.A.; Gao, W.; Voss, M.H.; Kaelin, W.G.; Martini, D.; Norton, C.; Bossé, D.; Wankowicz, S.; Cullen, D.; et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 2018, 359, 801–806. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Shen, R.; Xu, H.; Shi, X.; Xu, L.; Zhang, L.; Jin, X. Comprehensive analyses of PBRM1 in multiple cancer types and its association with clinical response to immunotherapy and immune infiltrates. Ann. Transl. Med. 2021, 9, 465. [Google Scholar] [CrossRef]
- Bulk, J.V.D.; Verdegaal, E.M.; de Miranda, N.F. Cancer immunotherapy: Broadening the scope of targetable tumours. Open Biol. 2018, 8, 180037. [Google Scholar] [CrossRef] [Green Version]
- Chew, H.Y.; Chan, V.; Simpson, F.; Dolcetti, R. Will next-generation immunotherapy overcome the intrinsic diversity and low immunogenicity of sarcomas to improve clinical benefit? Cancers 2020, 12, 3392. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 1–14. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Shoushtari, A.N.; Agaram, N.P.; Kuk, D.; Qin, L.X.; Carvajal, R.D.; Tap, W.D. Prevalence of tumor-infiltrating lymphocytes and PD-L1 expression in the soft tissue sarcoma microenvironment. Hum. Pathol. 2015, 46, 357–365. [Google Scholar] [CrossRef] [Green Version]
- Lam, S.W.; Kostine, M.; de Miranda, N.F.; Schöffski, P.; Lee, C.; Morreau, H.; Bovée, J.V. Mismatch repair deficiency is rare in bone and soft tissue tumors. Histopathology 2021. [Google Scholar] [CrossRef]
- Lewin, J.; Davidson, S.; Anderson, N.D.; Lau, B.Y.; Kelly, J.; Tabori, U.; Salah, S.; Butler, M.O.; Aung, K.L.; Shlien, A.; et al. Response to immune checkpoint inhibition in two patients with alveolar soft-part sarcoma. Cancer Immunol. Res. 2018, 6, 1001–1007. [Google Scholar] [CrossRef] [Green Version]
- Modi, M.B.; Patel, P.N.; Modi, V.M.; Mehta, S.P.; Nilkanthe, R.G.; Patel, P.H.; Trivedi, P.P.; Jetly, D.H. First reported case of alveolar soft part sarcoma in constitutional mismatch repair deficiency syndrome tumor spectrum diagnosed in one of the siblings with constitutional mismatch repair deficiency. South Asian J. Cancer 2017, 6, 041–043. [Google Scholar] [CrossRef] [PubMed]
- Tawbi, H.; Burgess, M.; Bolejack, V.; Van Tine, A.B.; Schuetze, S.M.; Hu, J.; D’Angelo, S.; Attia, S.; Riedel, R.F.; Priebat, A.D.; et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): A multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1493–1501. [Google Scholar] [CrossRef]
- Wu, X.; Lin, X.; Chen, Y.; Kong, W.; Xu, J.; Yu, Z. Response of metastatic chordoma to the immune checkpoint inhibitor pembrolizumab: A case report. Front Oncol. 2020, 10, 2848. [Google Scholar] [CrossRef] [PubMed]
- Doyle, L.A.; Nowak, J.A.; Nathenson, M.J.; Thornton, K.; Wagner, A.J.; Johnson, J.M.; Albrayak, A.; George, S.; Sholl, L.M. Characteristics of mismatch repair deficiency in sarcomas. Mod. Pathol. 2019, 32, 977–987. [Google Scholar] [CrossRef] [PubMed]
- Boichard, A.; Wagner, M.J.; Kurzrock, R. Angiosarcoma heterogeneity and potential therapeutic vulnerability to immune checkpoint blockade: Insights from genomic sequencing. Genome Med. 2020, 12, 1–6. [Google Scholar] [CrossRef]
- Luo, Y.; Min, L.; Zhou, Y.; Tang, F.; Lu, M.; Xie, H.; Tu, C. Remarkable response to anti-PD1 immunotherapy in refractory metastatic high-grade myxofibrosarcoma patient: A case report. Medicine 2021, 100, e25262. [Google Scholar] [CrossRef] [PubMed]
- Lazar, A.J.; McLellan, M.D.; Bailey, M.H.; Miller, C.A.; Appelbaum, E.L.; Cordes, M.G.; Lichtenberg, T.M. Comprehensive and integrated genomic characterization of adult soft tissue sarcomas. Cell 2017, 171, 950–965. [Google Scholar]
- D’Angelo, S.P.; Mahoney, M.R.; Van Tine, B.; Atkins, J.; Milhem, M.; Jahagirdar, B.N.; Antonescu, C.R.; Horvath, E.; Tap, W.D.; Schwartz, G.K.; et al. Nivolumab with or without ipilimumab treatment for metastatic sarcoma (Alliance A091401): Two open-label, non-comparative, randomised, phase 2 trials. Lancet Oncol. 2018, 19, 416–426. [Google Scholar] [CrossRef]
- Tsimberidou, A.-M.; Van Morris, K.; Vo, H.H.; Eck, S.; Lin, Y.-F.; Rivas, J.M.; Andersson, B.S. T-cell receptor-based therapy: An innovative therapeutic approach for solid tumors. J. Hematol. Oncol. 2021, 14, 1–22. [Google Scholar] [CrossRef]
- Dallos, M.; Tap, W.D.; D’Angelo, S.P. Current status of engineered T-cell therapy for synovial sarcoma. Immunotherapy 2016, 8, 1073–1080. [Google Scholar] [CrossRef] [Green Version]
- Endo, M.; De Graaff, M.; Ingram, D.R.; Lim, S.; Lev, D.C.; Bruijn, I.H.B.-D.; Somaiah, N.; Bovée, J.V.; Lazar, A.J.; O Nielsen, T. NY-ESO-1 (CTAG1B) expression in mesenchymal tumors. Mod. Pathol. 2014, 28, 587–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran, I.; Lowther, D.E.; Dryer-Minnerly, R.; Wang, R.; Fayngerts, S.; Nunez, D.; Betts, G.; Bath, N.; Tipping, A.J.; Melchiori, L.; et al. Systemic and local immunity following adoptive transfer of NY-ESO-1 SPEAR T cells in synovial sarcoma. J. Immunother. Cancer 2019, 7, 1–14. [Google Scholar] [CrossRef]
- Luk, S.J.; Van Der Steen, D.M.; Hagedoorn, R.S.; Jordanova, E.S.; Schilham, M.W.; Bovée, J.V.; Cleven, A.H.; Falkenburg, J.F.; Szuhai, K.; Heemskerk, M.H. PRAME and HLA Class I expression patterns make synovial sarcoma a suitable target for PRAME specific T-cell receptor gene therapy. Oncoimmunology 2018, 7, e1507600. [Google Scholar] [CrossRef] [PubMed]
- Melief, C.J.; van Hall, T.; Arens, R.; Ossendorp, F.; Van Der Burg, S.H. Therapeutic cancer vaccines. J. Clin. Investig. 2015, 125, 3401–3412. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Türeci, Ö. Personalized vaccines for cancer immunotherapy. Science 2018, 59, 1355–1360. [Google Scholar] [CrossRef] [Green Version]
- Albershardt, T.C.; Campbell, D.J.; Parsons, A.J.; Slough, M.M.; Ter Meulen, J.; Berglund, P. LV305, a dendritic cell-targeting integration-deficient ZVexTM-based lentiviral vector encoding NY-ESO-1, induces potent anti-tumor immune response. Mol. Ther. Oncolytics 2016, 3, 16010. [Google Scholar] [CrossRef] [Green Version]
- Pollack, S.M. The potential of the CMB305 vaccine regimen to target NY-ESO-1 and improve outcomes for synovial sarcoma and myxoid/round cell liposarcoma patients. Expert Rev. Vaccines 2017, 17, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Chawla, S.P.; Van Tine, B.A.; Pollack, S.M.; Ganjoo, K.N.; Elias, A.D.; Riedel, R.F.; Attia, S.; Choy, E.; Okuno, S.H.; Agulnik, M.; et al. Phase II randomized study of CMB305 and atezolizumab compared with atezolizumab alone in soft-tissue sarcomas expressing NY-ESO-1. J. Clin. Oncol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Miwa, S.; Nishida, H.; Tanzawa, Y.; Takeuchi, A.; Hayashi, K.; Yamamoto, N.; Tsuchiya, H. Phase 1/2 study of immunotherapy with dendritic cells pulsed with autologous tumor lysate in patients with refractory bone and soft tissue sarcoma. Cancer 2017, 123, 1576–1584. [Google Scholar] [CrossRef] [Green Version]
- DeMaria, P.J.; Bilusic, M.; Park, D.M.; Heery, C.R.; Donahue, R.N.; Madan, R.A.; Gulley, J.L. Randomized, double-blind, placebo-controlled phase II study of yeast-brachyury vaccine (GI-6301) in combination with standard-of-care radiotherapy in locally advanced, unresectable chordoma. Oncologist 2021, 26, e847–e858. [Google Scholar] [CrossRef]
- Deng, C.; Xu, Y.; Fu, J.; Zhu, X.; Chen, H.; Xu, H.; Wang, G.; Song, Y.; Song, G.; Lu, J.; et al. Reprograming the tumor immunologic microenvironment using neoadjuvant chemotherapy in osteosarcoma. Cancer Sci. 2020, 111, 1899–1909. [Google Scholar] [CrossRef] [PubMed]
- Toulmonde, M.; Penel, N.; Adam, J.; Chevreau, C.; Blay, J.Y.; Le Cesne, A.; Italiano, A. Use of PD-1 targeting, macrophage infiltration, and IDO pathway activation in sarcomas: A phase 2 clinical trial. JAMA Oncol. 2018, 4, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, G.C.; Malachowski, W.J.; Mondal, A.; Scherle, P.; Muller, A.J. Indoleamine 2,3-dioxygenase and its therapeutic inhibition in cancer. Int. Rev. Cell Mol. Biol. 2018, 336, 175–203. [Google Scholar] [CrossRef] [PubMed]
- Yum, S.; Li, M.; Chen, Z.J. Old dogs, new trick: Classic cancer therapies activate cGAS. Cell Res. 2020, 30, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Lhuillier, C.; Rudqvist, N.-P.; Elemento, O.; Formenti, S.C.; DeMaria, S. Radiation therapy and anti-tumor immunity: Exposing immunogenic mutations to the immune system. Genome Med. 2019, 11, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Kohli, K.; Black, R.G.; Yao, L.; Spadinger, S.M.; He, Q.; Pollack, S.M. Systemic interferon-γ increases MHC class I expression and T-cell infiltration in cold tumors: Results of a phase 0 clinical trial. Cancer Immunol. Res. 2019, 7, 1237–1243. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.M.; Shenasa, E.; Nielsen, T.O. Sarcomas: Immune biomarker expression and checkpoint inhibitor trials. Cancer Treat. Rev. 2020, 91, 102115. [Google Scholar] [CrossRef]
- Klaver, Y.; Rijnders, M.; Oostvogels, A.; Wijers, R.; Smid, M.; Grünhagen, D.; Verhoef, K.; Sleijfer, S.; Lamers, C.; Debets, R. Differential quantities of immune checkpoint-expressing CD8 T cells in soft tissue sarcoma subtypes. J. Immunother. Cancer 2020, 8, e000271. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, D.; Yang, Q.; Lv, X.; Huang, W.; Zhou, Z.; Wang, Y.; Zhang, Z.; Yuan, T.; Ding, X.; et al. Single-cell RNA landscape of intratumoral heterogeneity and immunosuppressive microenvironment in advanced osteosarcoma. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Bono, P.; Bhatia, S.; Melero, I.; Nyakas, M.S.; Svane, I.M.; Couselo, E.M. Efficacy of BMS-986016, a monoclonal antibody that targets lymphocyte activation gene-3 (LAG-3), in combination with nivolumab in pts with melanoma who progressed during prior anti–PD-1/PD-L1 therapy (mel prior IO) in all-comer and biomarker-enriched populations. Ann. Oncol. 2017, 28, v611–v612. [Google Scholar]
- Logtenberg, M.E.; Scheeren, F.A.; Schumacher, T.N. The CD47-SIRPα immune checkpoint. Immunity 2020, 52, 742–752. [Google Scholar] [CrossRef]
- Stanley, E.R.; Chitu, V. CSF-1 receptor signaling in myeloid cells. Cold Spring Harb. Perspect. Biol. 2014, 6, a021857. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Brouchet, A.; Gilhodes, J.; Acker, N.V.; Brion, R.; Bouvier, C.; Assemat, P.; Rédini, F. Characterization of macrophages and osteoclasts in the osteosarcoma tumor microenvironment at diagnosis: New perspective for osteosarcoma treatment? Cancers 2021, 13, 423. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, I.; Beck, A.H.; Lee, C.H.; Zhu, S.; Montgomery, K.D.; Marinelli, R.J.; van de Rijn, M. Coordinate expression of colony-stimulating factor-1 and colony-stimulating factor-1-related proteins is associated with poor prognosis in gynecological and nongynecological leiomyosarcoma. Am. J. Pathol. 2009, 174, 2347–2356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.J.; Xu, Y.; Zhu, X.; Fu, J.; Deng, C.; Chen, H.; Wang, J. Immune landscape of the tumor microenvironment identifies prognostic gene signature CD4/CD68/CSF1R in osteosarcoma. Front. Oncol. 2020, 10, 1198. [Google Scholar] [CrossRef]
- Buddingh, E.; Kuijjer, M.; Duim, R.A.; Bürger, H.; Agelopoulos, K.; Myklebost, O.; Serra, M.; Mertens, F.; Hogendoorn, P.; Lankester, A.C.; et al. Tumor-infiltrating macrophages are associated with metastasis suppression in high-grade osteosarcoma: A rationale for treatment with macrophage activating agents. Clin. Cancer Res. 2011, 17, 2110–2119. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, T.; Yakoub, M.A.; Chandler, A.; Christ, A.B.; Yang, G.; Ouerfelli, O.; Healey, J.H. CSF-1/CSF-1R signaling inhibitor pexidartinib (PLX3397) reprograms tumor-associated macrophages and stimulates T-cell infiltration in the sarcoma microenvironment. Mol. Cancer Ther. 2021, 20, 1388–1399. [Google Scholar] [CrossRef] [PubMed]
- Querfeld, C.; Thompson, J.A.; Taylor, M.; Pillai, R.; Johnson, L.D.; Catalano, T.; Petrova, P.S.; Uger, B.A.; Irwin, M.; Thompson, T.; et al. Intralesional injection of the CD47-blocking immune checkpoint inhibitor TTI-621 (SIRPaFc) induces antitumor activity in patients with relapsed/refractory mycosis fungoides and Sézary syndrome: Interim results of a multicenter phase 1 trial. Eur. J. Cancer 2018, 101, S34. [Google Scholar] [CrossRef]
- Thanindratarn, P.; Dean, D.C.; Nelson, S.D.; Hornicek, F.J.; Duan, Z. Chimeric antigen receptor T (CAR-T) cell immunotherapy for sarcomas: From mechanisms to potential clinical applications. Cancer Treat. Rev. 2019, 82, 101934. [Google Scholar] [CrossRef] [Green Version]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [Green Version]
- Chulanetra, M.; Morchang, A.; Sayour, E.; Eldjerou, L.; Milner, R.; Lagmay, J.; Cascio, M.; Stover, B.; Slayton, W.; Chaicumpa, W.; et al. GD2 chimeric antigen receptor modified T cells in synergy with sub-toxic level of doxorubicin targeting osteosarcomas. Am. J. Cancer Res. 2020, 10, 674–687. [Google Scholar]
- Majzner, R.G.; Theruvath, J.L.; Nellan, A.; Heitzeneder, S.; Cui, Y.; Mount, C.W.; Rietberg, S.P.; Linde, M.H.; Xu, P.; Rota, C.; et al. Car T cells targeting B7-H3, a pan-cancer antigen, demonstrate potent preclinical activity against pediatric solid tumors and brain tumors. Clin. Cancer Res. 2019, 25, 2560–2574. [Google Scholar] [CrossRef]
- Hegde, M.; Derenzo, C.C.; Zhang, H.; Mata, M.; Gerken, C.; Shree, A.; Yi, Z.; Brawley, V.; Dakhova, O.; Wu, M.-F.; et al. Expansion of HER2-CAR T cells after lymphodepletion and clinical responses in patients with advanced sarcoma. J. Clin. Oncol. 2017, 35, 10508. [Google Scholar] [CrossRef]
- Marofi, F.; Motavalli, R.; Safonov, V.A.; Thangavelu, L.; Yumashev, A.V.; Alexander, M.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; Jarahian, M.; et al. CAR T cells in solid tumors: Challenges and opportunities. Stem Cell Res. Ther. 2021, 12, 1–16. [Google Scholar] [CrossRef]
- Long, A.H.; Highfill, S.L.; Cui, Y.; Smith, J.P.; Walker, A.J.; Ramakrishna, S.; El-Etriby, R.; Galli, S.; Tsokos, M.G.; Orentas, R.J.; et al. Reduction of MDSCs with all-trans retinoic acid improves CAR therapy efficacy for sarcomas. Cancer Immunol. Res. 2016, 4, 869–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Siriwon, N.; Zhang, X.; Yang, S.; Jin, T.; He, F.; Kim, Y.J.; Mac, J.; Lu, Z.; Wang, S.; et al. Enhanced cancer immunotherapy by chimeric antigen receptor-modified T cells engineered to secrete checkpoint inhibitors. Clin. Cancer Res. 2017, 23, 6982–6992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Lee, K.-W.; Srivastava, R.M.; Kuo, F.; Krishna, C.; Chowell, D.; Makarov, V.; Hoen, D.; Dalin, M.G.; Wexler, L.; et al. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat. Med. 2019, 25, 767–775. [Google Scholar] [CrossRef]
- Worley, B.S.; Broeke, L.T.V.D.; Goletz, T.J.; Pendleton, C.D.; Daschbach, E.M.; Thomas, E.K.; Marincola, F.M.; Helman, L.J.; Berzofsky, J. Antigenicity of fusion proteins from sarcoma-associated chromosomal translocations. Cancer Res. 2001, 61, 6868–6875. [Google Scholar]
- Kawaguchi, S.; Wada, T.; Ida, K.; Sato, Y.; Nagoya, S.; Tsukahara, T.; Kimura, S.; Sahara, H.; Ikeda, H.; Shimozawa, K.; et al. Phase I vaccination trial of SYT-SSX junction peptide in patients with disseminated synovial sarcoma. J. Transl. Med. 2005, 3, 1. [Google Scholar] [CrossRef] [Green Version]
- Kawaguchi, S. SYT-SSX breakpoint peptide vaccines in patients with synovial sarcoma: A study from the Japanese Musculoskeletal Oncology Group. Cancer Sci. 2012, 103, 1625–1630. [Google Scholar] [CrossRef] [PubMed]
- Suehara, Y. Identification of a novel MAN1A1-ROS1 fusion gene through mRNA-based screening for tyrosine kinase gene aberrations in a patient with leiomyosarcoma. Clin. Orthop. Relat. Res. 2021, 479, 838–852. [Google Scholar] [CrossRef] [PubMed]
- Hirose, K.; Usami, Y.; Kohara, M.; Sato, S.; Iwamoto, Y.; Murakami, S.; Uchihashi, T.; Oya, K.; Fukuda, Y.; Hori, Y.; et al. Clear cell carcinoma of palatal minor salivary gland harboring a novel EWSR1-ATF1 fusion gene: Report of a case and review of the literature. Head. Neck. Pathol. 2020, 15, 676–681. [Google Scholar] [CrossRef]
- Lin, D.I.; Hemmerich, A.; Edgerly, C.; Duncan, D.; Severson, E.A.; Huang, R.S.; Ramkissoon, S.H.; Connor, Y.D.; Shea, M.; Hecht, J.L.; et al. Genomic profiling of BCOR-rearranged uterine sarcomas reveals novel gene fusion partners, frequent CDK4 amplification and CDKN2A loss. Gynecol. Oncol. 2020, 157, 357–366. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Sarcoma Type | DNA Alterations | Immune Infiltration 1 | PD-L1 Expression 2 | Genomic/TME Heterogeneity 1 | General TME Composition |
|---|---|---|---|---|---|
| Simple genome | |||||
| Alveolar soft-part sarcoma | TFE3-ASPCR1 | − | 29.7–100% | −/− | CD8 T cells; TAMs [25,26,27] |
| Chondrosarcoma (low-grade) | IDH, COL2A1 | − | 0% | −/± | CD4 and CD8 T cells; TAMs [28,29,30] |
| Well-differentiated liposarcoma | MDM2, CDK4, CNA | −/± | 0–50% | −/− | CD4 Th and CD8 T cells; B cells, DCs; TAMs [31,32,33,34] |
| Ewing sarcoma | EWSR1-ETS | − | 0% | −/− | M2-like TAMs; CD4 and CD8 T cells [34,35,36,37] |
| Synovial sarcoma | SS18-SSX | − | 0% | −/± | TAMs; CD4 FOXP3 Tregs and CD8 T cells [34,35,38] |
| Complex genome | |||||
| Chondrosarcoma (dedifferentiated) | IDH, COL2A1, CNA | ± | 41–52% | +/± | CD4 and CD8 T cells; TAMs [29,30] |
| Chordoma | CDKN2A, PBRM1, SMARCB1, CNA | + | 0–68.5% | ±/± | CD4 FOXP3 Tregs and CD8 T cells; M1-like and M2-like TAMs [39,40,41,42] |
| Dedifferentiated liposarcoma | MDM2, CDK4, CNA | + | 10–67% | +/+ | CD4 Th and CD8 T cells; B cells, DCs; TAMs [24,31,32,33,34] |
| Myxofibrosarcoma | CIN | + | 16–20% | +/+ | CD4 Th, CD4 Treg and CD8 T cells; B cells; DCs; M1-like and M2-like TAMs [34,39,43,44,45] |
| Osteosarcoma | CIN | ± | 0–25% | +/+ | CD4 and CD8 T cells; M1-like and M2-like TAMs, NK cells; DCs [36,46,47,48] |
| Soft tissue leiomyosarcoma | CNA | ± | 34–59% | +/+ | M2-like TAMs, CD4 T cells [32,39,43,49] |
| Undifferentiated soft tissue sarcoma | CIN, SNV | + | 0–33% | +/+ | CD4 Th, CD4 Treg and CD8 T cells; B cells; DCs; M2-like TAMs [24,34,39,43,45,50,51] |
| Uterine leiomyosarcoma | CNA | ± | 0–70% | +/+ | M2-like TAMs; CD4 T cells; NK cells [39,43,49,52,53] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Oost, S.; Meijer, D.M.; Kuijjer, M.L.; Bovée, J.V.M.G.; de Miranda, N.F.C.C. Linking Immunity with Genomics in Sarcomas: Is Genomic Complexity an Immunogenic Trigger? Biomedicines 2021, 9, 1048. https://doi.org/10.3390/biomedicines9081048
van Oost S, Meijer DM, Kuijjer ML, Bovée JVMG, de Miranda NFCC. Linking Immunity with Genomics in Sarcomas: Is Genomic Complexity an Immunogenic Trigger? Biomedicines. 2021; 9(8):1048. https://doi.org/10.3390/biomedicines9081048
Chicago/Turabian Stylevan Oost, Siddh, Debora M. Meijer, Marieke L. Kuijjer, Judith V. M. G. Bovée, and Noel F. C. C. de Miranda. 2021. "Linking Immunity with Genomics in Sarcomas: Is Genomic Complexity an Immunogenic Trigger?" Biomedicines 9, no. 8: 1048. https://doi.org/10.3390/biomedicines9081048
APA Stylevan Oost, S., Meijer, D. M., Kuijjer, M. L., Bovée, J. V. M. G., & de Miranda, N. F. C. C. (2021). Linking Immunity with Genomics in Sarcomas: Is Genomic Complexity an Immunogenic Trigger? Biomedicines, 9(8), 1048. https://doi.org/10.3390/biomedicines9081048