
Oligonucleotide Therapies in the Treatment of Arthritis: A Narrative Review


Abstract
:1. Introduction
1.1. Rheumatoid Arthritis
1.2. Osteoarthritis
2. Methods
3. Oligonucleotides Targeting Synovial Inflammation
4. Oligonucleotides Targeting Subchondral Bone Pathology
5. Oligonucleotides Targeting Cartilage Degeneration
6. Optimisation of Oligonucleotide Method of Delivery into Joint Tissues
6.1. Viral Delivery Systems in Inflammatory Joint Disease
6.2. Biomaterial Delivery Systems in Inflammatory Joint Disease
6.3. Bioconjugation and Cell-Penetrating Peptide Delivery
6.4. Nanoparticle Delivery Systems in Inflammatory Joint Disease
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Guo, Q.; Wang, Y.; Xu, D.; Nossent, J.; Pavlos, N.J.; Xu, J. Rheumatoid arthritis: Pathological mechanisms and modern pharmacologic therapies. Bone Res. 2018, 6, 15. [Google Scholar] [CrossRef]
- Crowson, C.S.; Matteson, E.L.; Myasoedova, E.; Michet, C.J.; Ernste, F.C.; Warrington, K.J.; Davis, J.M.; Hunder, G.G.; Therneau, T.M.; Gabriel, S.E. The lifetime risk of adult-onset rheumatoid arthritis and other inflammatory autoimmune rheumatic diseases. Arthritis Rheum. 2011, 63, 633–639. [Google Scholar] [CrossRef] [Green Version]
- Crane, M.M.; Juneja, M.; Allen, J.; Kurrasch, R.H.; Chu, M.E.; Quattrocchi, E.; Manson, S.C.; Chang, D.J. Epidemiology and Treatment of New-Onset and Established Rheumatoid Arthritis in an Insured US Population. Arthritis Care Res. 2015, 67, 1646–1655. [Google Scholar] [CrossRef]
- Lawrence, R.C.; Felson, D.T.; Helmick, C.G.; Arnold, L.M.; Choi, H.; Deyo, R.A.; Gabriel, S.; Hirsch, R.; Hochberg, M.C.; Hunder, G.G.; et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part II. Arthritis Rheum. 2008, 58, 26–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhari, K.; Rizvi, S.; Syed, B.A. Rheumatoid arthritis: Current and future trends. Nat. Rev. Drug Discov. 2016, 15, 305–306. [Google Scholar] [CrossRef] [PubMed]
- Hua, C.; Buttgereit, F.; Combe, B. Glucocorticoids in rheumatoid arthritis: Current status and future studies. RMD Open 2020, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briot, K.; Roux, C. Glucocorticoid-induced osteoporosis. RMD Open 2015, 1, e000014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schakman, O.; Kalista, S.; Barbe, C.; Loumaye, A.; Thissen, J.P. Glucocorticoid-induced skeletal muscle atrophy. Int. J. Biochem. Cell Biol. 2013, 45, 2163–2172. [Google Scholar] [CrossRef]
- van Vollenhoven, R. Treat-to-target in rheumatoid arthritis—Are we there yet? Nat. Rev. Rheumatol. 2019, 15, 180–186. [Google Scholar] [CrossRef]
- Pokharel, G.; Deardon, R.; Johnson, S.R.; Tomlinson, G.; Hull, P.M.; Hazlewood, G.S. Effectiveness of initial methotrexate-based treatment approaches in early rheumatoid arthritis: An elicitation of rheumatologists’ beliefs. Rheumatology 2020. [Google Scholar] [CrossRef]
- Friedman, B.; Cronstein, B. Methotrexate mechanism in treatment of rheumatoid arthritis. Jt. Bone Spine 2019, 86, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.M.; Pratt, A.G.; Isaacs, J.D. Mechanism of action of methotrexate in rheumatoid arthritis, and the search for biomarkers. Nat. Rev. Rheumatol. 2016, 12, 731–742. [Google Scholar] [CrossRef]
- Fox, R.I.; Herrmann, M.L.; Frangou, C.G.; Wahl, G.M.; Morris, R.E.; Strand, V.; Kirschbaum, B.J. Mechanism of action for leflunomide in rheumatoid arthritis. Clin. Immunol. 1999, 93, 198–208. [Google Scholar] [CrossRef]
- Hua, L.; Du, H.; Ying, M.; Wu, H.; Fan, J.; Shi, X. Efficacy and safety of low-dose glucocorticoids combined with methotrexate and hydroxychloroquine in the treatment of early rheumatoid arthritis: A single-center, randomized, double-blind clinical trial. Medicine 2020, 99, e20824. [Google Scholar] [CrossRef] [PubMed]
- Schrezenmeier, E.; Dorner, T. Mechanisms of action of hydroxychloroquine and chloroquine: Implications for rheumatology. Nat. Rev. Rheumatol. 2020, 16, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Smedegard, G.; Bjork, J. Sulphasalazine: Mechanism of action in rheumatoid arthritis. Br. J. Rheumatol. 1995, 34 (Suppl. 2), 7–15. [Google Scholar] [CrossRef]
- Wahl, C.; Liptay, S.; Adler, G.; Schmid, R.M. Sulfasalazine: A potent and specific inhibitor of nuclear factor kappa B. J. Clin. Investig. 1998, 101, 1163–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edrees, A.F.; Misra, S.N.; Abdou, N.I. Anti-tumor necrosis factor (TNF) therapy in rheumatoid arthritis: Correlation of TNF-alpha serum level with clinical response and benefit from changing dose or frequency of infliximab infusions. Clin. Exp. Rheumatol. 2005, 23, 469–474. [Google Scholar]
- Wu, C.; Wang, S.; Xian, P.; Yang, L.; Chen, Y.; Mo, X. Effect of Anti-TNF Antibodies on Clinical Response in Rheumatoid Arthritis Patients: A Meta-Analysis. Biomed. Res. Int. 2016, 2016, 7185708. [Google Scholar] [CrossRef]
- Mathieu, S.; Couderc, M.; Pereira, B.; Soubrier, M. The effects of TNF-alpha inhibitor therapy on arterial stiffness and endothelial dysfunction in rheumatoid arthritis: A meta-analysis. Semin. Arthritis Rheum. 2013, 43, e1–e2. [Google Scholar] [CrossRef]
- Ma, X.; Xu, S. TNF inhibitor therapy for rheumatoid arthritis. Biomed. Rep. 2013, 1, 177–184. [Google Scholar] [CrossRef] [Green Version]
- Biggioggero, M.; Crotti, C.; Becciolini, A.; Favalli, E.G. Tocilizumab in the treatment of rheumatoid arthritis: An evidence-based review and patient selection. Drug Des. Devel. Ther. 2019, 13, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Cohen, M.D.; Keystone, E. Rituximab for Rheumatoid Arthritis. Rheumatol. Ther. 2015, 2, 99–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vital, E.M.; Emery, P. Abatacept in the treatment of rheumatoid arthritis. Ther. Clin. Risk Manag. 2006, 2, 365–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Zhou, H.; Liu, L. Side effects of methotrexate therapy for rheumatoid arthritis: A systematic review. Eur. J. Med. Chem. 2018, 158, 502–516. [Google Scholar] [CrossRef]
- Aletaha, D.; Kapral, T.; Smolen, J.S. Toxicity profiles of traditional disease modifying antirheumatic drugs for rheumatoid arthritis. Ann. Rheum. Dis. 2003, 62, 482–486. [Google Scholar] [CrossRef] [Green Version]
- Moots, R.J.; Xavier, R.M.; Mok, C.C.; Rahman, M.U.; Tsai, W.C.; Al-Maini, M.H.; Pavelka, K.; Mahgoub, E.; Kotak, S.; Korth-Bradley, J.; et al. The impact of anti-drug antibodies on drug concentrations and clinical outcomes in rheumatoid arthritis patients treated with adalimumab, etanercept, or infliximab: Results from a multinational, real-world clinical practice, non-interventional study. PLoS ONE 2017, 12, e0175207. [Google Scholar] [CrossRef] [Green Version]
- Kloppenburg, M.; Berenbaum, F. Osteoarthritis year in review 2019: Epidemiology and therapy. Osteoarthr. Cartil. 2020, 28, 242–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philp, A.M.; Collier, R.L.; Grover, L.M.; Davis, E.T.; Jones, S.W. Resistin promotes the abnormal Type I collagen phenotype of subchondral bone in obese patients with end stage hip osteoarthritis. Sci. Rep. 2017, 7, 4042. [Google Scholar] [CrossRef] [Green Version]
- Pearson, M.J.; Herndler-Brandstetter, D.; Tariq, M.A.; Nicholson, T.A.; Philp, A.M.; Smith, H.L.; Davis, E.T.; Jones, S.W.; Lord, J.M. IL-6 secretion in osteoarthritis patients is mediated by chondrocyte-synovial fibroblast cross-talk and is enhanced by obesity. Sci. Rep. 2017, 7, 3451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loeser, R.F.; Goldring, S.R.; Scanzello, C.R.; Goldring, M.B. Osteoarthritis: A disease of the joint as an organ. Arthritis Rheum. 2012, 64, 1697–1707. [Google Scholar] [CrossRef] [Green Version]
- Belluzzi, E.; Macchi, V.; Fontanella, C.G.; Carniel, E.L.; Olivotto, E.; Filardo, G.; Sarasin, G.; Porzionato, A.; Granzotto, M.; Pozzuoli, A.; et al. Infrapatellar Fat Pad Gene Expression and Protein Production in Patients with and without Osteoarthritis. Int. J. Mol. Sci. 2020, 21, 6016. [Google Scholar] [CrossRef] [PubMed]
- Safiri, S.; Kolahi, A.A.; Smith, E.; Hill, C.; Bettampadi, D.; Mansournia, M.A.; Hoy, D.; Ashrafi-Asgarabad, A.; Sepidarkish, M.; Almasi-Hashiani, A.; et al. Global, regional and national burden of osteoarthritis 1990–2017: A systematic analysis of the Global Burden of Disease Study 2017. Ann. Rheum. Dis. 2020, 79, 819–828. [Google Scholar] [CrossRef]
- Philp, A.M.; Davis, E.T.; Jones, S.W. Developing anti-inflammatory therapeutics for patients with osteoarthritis. Rheumatology 2017, 56, 869–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonge, D.P.; Pearson, M.J.; Jones, S.W. The hallmarks of osteoarthritis and the potential to develop personalised disease-modifying pharmacological therapeutics. Osteoarthr. Cartil. 2014, 22, 609–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gato-Calvo, L.; Magalhaes, J.; Ruiz-Romero, C.; Blanco, F.J.; Burguera, E.F. Platelet-rich plasma in osteoarthritis treatment: Review of current evidence. Ther. Adv. Chronic Dis. 2019, 10, 2040622319825567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogata, T.; Ideno, Y.; Akai, M.; Seichi, A.; Hagino, H.; Iwaya, T.; Doi, T.; Yamada, K.; Chen, A.Z.; Li, Y.; et al. Effects of glucosamine in patients with osteoarthritis of the knee: A systematic review and meta-analysis. Clin. Rheumatol. 2018, 37, 2479–2487. [Google Scholar] [CrossRef] [Green Version]
- Maricar, N.; Parkes, M.J.; Callaghan, M.J.; Hutchinson, C.E.; Gait, A.D.; Hodgson, R.; Felson, D.T.; O’Neill, T.W. Structural predictors of response to intra-articular steroid injection in symptomatic knee osteoarthritis. Arthritis Res. Ther. 2017, 19, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, C.H. Drug delivery to chondrocytes. Osteoarthr. Cartil. 2016, 24, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshi, H.; Akagi, R.; Yamaguchi, S.; Muramatsu, Y.; Akatsu, Y.; Yamamoto, Y.; Sasaki, T.; Takahashi, K.; Sasho, T. Effect of inhibiting MMP13 and ADAMTS5 by intra-articular injection of small interfering RNA in a surgically induced osteoarthritis model of mice. Cell Tissue Res. 2017, 368, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.C.; Yang, R.S.; Hsieh, K.H.; Fong, Y.C.; Way, T.D.; Lee, T.S.; Wu, H.C.; Fu, W.M.; Tang, C.H. Stromal cell-derived factor-1 induces matrix metalloprotease-13 expression in human chondrocytes. Mol. Pharmacol. 2007, 72, 695–703. [Google Scholar] [CrossRef]
- Dai, L.; Zhang, X.; Hu, X.; Liu, Q.; Man, Z.; Huang, H.; Meng, Q.; Zhou, C.; Ao, Y. Silencing of miR-101 Prevents Cartilage Degradation by Regulating Extracellular Matrix-related Genes in a Rat Model of Osteoarthritis. Mol. Ther. 2015, 23, 1331–1340. [Google Scholar] [CrossRef] [Green Version]
- Lian, W.S.; Ko, J.Y.; Wu, R.W.; Sun, Y.C.; Chen, Y.S.; Wu, S.L.; Weng, L.H.; Jahr, H.; Wang, F.S. MicroRNA-128a represses chondrocyte autophagy and exacerbates knee osteoarthritis by disrupting Atg12. Cell Death Dis. 2018, 9, 919. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Zhao, X.; Wang, C.; Geng, Y.; Zhao, J.; Xu, J.; Zuo, B.; Zhao, C.; Wang, C.; Zhang, X. MicroRNA-145 attenuates TNF-alpha-driven cartilage matrix degradation in osteoarthritis via direct suppression of MKK4. Cell Death Dis. 2017, 8, e3140. [Google Scholar] [CrossRef]
- Jones, S.W.; Watkins, G.; Le Good, N.; Roberts, S.; Murphy, C.L.; Brockbank, S.M.; Needham, M.R.; Read, S.J.; Newham, P. The identification of differentially expressed microRNA in osteoarthritic tissue that modulate the production of TNF-alpha and MMP13. Osteoarthr. Cartil. 2009, 17, 464–472. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, A.; Rampersaud, Y.R.; Nakamura, S.; Sharma, A.; Zeng, F.; Rossomacha, E.; Ali, S.A.; Krawetz, R.; Haroon, N.; Perruccio, A.V.; et al. microRNA-181a-5p antisense oligonucleotides attenuate osteoarthritis in facet and knee joints. Ann. Rheum. Dis. 2019, 78, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.B.; Zhao, F.C.; Yi, L.H.; Tang, J.L.; Zhu, Z.Y.; Pang, Y.; Chen, Y.S.; Li, D.Y.; Guo, K.J.; Zheng, X. MicroRNA-21-5p as a novel therapeutic target for osteoarthritis. Rheumatology 2019. [Google Scholar] [CrossRef]
- Hu, J.; Wang, Z.; Pan, Y.; Ma, J.; Miao, X.; Qi, X.; Zhou, H.; Jia, L. MiR-26a and miR-26b mediate osteoarthritis progression by targeting FUT4 via NF-kappaB signaling pathway. Int. J. Biochem. Cell Biol. 2018, 94, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Chen, X.; Gao, Z.Y.; Liu, K.; Hou, Y.; Zheng, J. The role of miR-320a and IL-1beta in human chondrocyte degradation. Bone Jt. Res. 2017, 6, 196–203. [Google Scholar] [CrossRef] [Green Version]
- Endisha, H.; Datta, P.; Sharma, A.; Nakamura, S.; Rossomacha, E.; Younan, C.; Ali, S.A.; Tavallaee, G.; Lively, S.; Potla, P.; et al. MicroRNA-34a-5p Promotes Joint Destruction During Osteoarthritis. Arthritis Rheumatol. 2021, 73, 426–439. [Google Scholar] [CrossRef]
- Baek, D.; Lee, K.M.; Park, K.W.; Suh, J.W.; Choi, S.M.; Park, K.H.; Lee, J.W.; Kim, S.H. Inhibition of miR-449a Promotes Cartilage Regeneration and Prevents Progression of Osteoarthritis in In Vivo Rat Models. Mol. Ther. Nucleic Acids 2018, 13, 322–333. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.W.; Brockbank, S.M.; Clements, K.M.; Le Good, N.; Campbell, D.; Read, S.J.; Needham, M.R.; Newham, P. Mitogen-activated protein kinase-activated protein kinase 2 (MK2) modulates key biological pathways associated with OA disease pathology. Osteoarthr. Cartil. 2009, 17, 124–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.W.; Brockbank, S.M.; Mobbs, M.L.; Le Good, N.J.; Soma-Haddrick, S.; Heuze, A.J.; Langham, C.J.; Timms, D.; Newham, P.; Needham, M.R. The orphan G-protein coupled receptor RDC1: Evidence for a role in chondrocyte hypertrophy and articular cartilage matrix turnover. Osteoarthr. Cartil. 2006, 14, 597–608. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.; Li, S.J.; Liu, R.; Zhan, J.F.; Tan, C.; Fang, Y.F.; Chen, Y.; Yu, B. Inhibition of YAP with siRNA prevents cartilage degradation and ameliorates osteoarthritis development. J. Mol. Med. 2019, 97, 103–114. [Google Scholar] [CrossRef]
- Weng, L.H.; Wang, C.J.; Ko, J.Y.; Sun, Y.C.; Wang, F.S. Control of Dkk-1 ameliorates chondrocyte apoptosis, cartilage destruction, and subchondral bone deterioration in osteoarthritic knees. Arthritis Rheum. 2010, 62, 1393–1402. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.H.; Tsai, C.H.; Fong, Y.C.; Huang, Y.L.; Wang, S.J.; Chang, Y.S.; Tang, C.H. Leptin induces oncostatin M production in osteoblasts by downregulating miR-93 through the Akt signaling pathway. Int. J. Mol. Sci. 2014, 15, 15778–15790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lian, W.S.; Wu, R.W.; Lee, M.S.; Chen, Y.S.; Sun, Y.C.; Wu, S.L.; Ke, H.J.; Ko, J.Y.; Wang, F.S. Subchondral mesenchymal stem cells from osteoarthritic knees display high osteogenic differentiation capacity through microRNA-29a regulation of HDAC4. J. Mol. Med. 2017, 95, 1327–1340. [Google Scholar] [CrossRef]
- Li, L.; Li, M.; Pang, Y.; Wang, J.; Wan, Y.; Zhu, C.; Yin, Z. Abnormal thyroid hormone receptor signaling in osteoarthritic osteoblasts regulates microangiogenesis in subchondral bone. Life Sci. 2019, 239, 116975. [Google Scholar] [CrossRef] [PubMed]
- Weng, L.H.; Ko, J.Y.; Wang, C.J.; Sun, Y.C.; Wang, F.S. Dkk-1 promotes angiogenic responses and cartilage matrix proteinase secretion in synovial fibroblasts from osteoarthritic joints. Arthritis Rheum. 2012, 64, 3267–3277. [Google Scholar] [CrossRef] [Green Version]
- Palao, G.; Santiago, B.; Galindo, M.; Paya, M.; Ramirez, J.C.; Pablos, J.L. Down-regulation of FLIP sensitizes rheumatoid synovial fibroblasts to Fas-mediated apoptosis. Arthritis Rheum. 2004, 50, 2803–2810. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, Y.; Zhang, H.; Gao, W.; Lu, M.; Liu, W.; Li, Y.; Yin, Z. Forkhead box C1 promotes the pathology of osteoarthritis by upregulating beta-catenin in synovial fibroblasts. FEBS J. 2020, 287, 3065–3087. [Google Scholar] [CrossRef]
- Pearson, M.J.; Bik, M.A.; Ospelt, C.; Naylor, A.J.; Wehmeyer, C.; Jones, S.W.; Buckley, C.D.; Gay, S.; Filer, A.; Lord, J.M. Endogenous Galectin-9 Suppresses Apoptosis in Human Rheumatoid Arthritis Synovial Fibroblasts. Sci. Rep. 2018, 8, 12887. [Google Scholar] [CrossRef]
- Tong, K.M.; Shieh, D.C.; Chen, C.P.; Tzeng, C.Y.; Wang, S.P.; Huang, K.C.; Chiu, Y.C.; Fong, Y.C.; Tang, C.H. Leptin induces IL-8 expression via leptin receptor, IRS-1, PI3K, Akt cascade and promotion of NF-kappaB/p300 binding in human synovial fibroblasts. Cell Signal. 2008, 20, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.H.; Liu, S.C.; Tsai, C.H.; Fong, Y.C.; Wang, S.J.; Chang, Y.S.; Tang, C.H. Leptin induces IL-6 expression through OBRl receptor signaling pathway in human synovial fibroblasts. PLoS ONE 2013, 8, e75551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanus, D.E.; Wijesinghe, S.N.; Pearson, M.J.; Hadjicharalambous, M.R.; Rosser, A.; Davis, E.T.; Lindsay, M.A.; Jones, S.W. Regulation of the Inflammatory Synovial Fibroblast Phenotype by Metastasis-Associated Lung Adenocarcinoma Transcript 1 Long Noncoding RNA in Obese Patients With Osteoarthritis. Arthritis Rheumatol. 2020, 72, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, M.; Ishii, H.; Aono, H.; Takai, M.; Honda, T.; Aratani, S.; Fukamizu, A.; Nakamura, H.; Yoshino, S.; Kobata, T.; et al. Role of Notch-1 intracellular domain in activation of rheumatoid synoviocytes. Arthritis Rheum. 2001, 44, 1545–1554. [Google Scholar] [CrossRef]
- Stanford, S.M.; Maestre, M.F.; Campbell, A.M.; Bartok, B.; Kiosses, W.B.; Boyle, D.L.; Arnett, H.A.; Mustelin, T.; Firestein, G.S.; Bottini, N. Protein tyrosine phosphatase expression profile of rheumatoid arthritis fibroblast-like synoviocytes: A novel role of SH2 domain-containing phosphatase 2 as a modulator of invasion and survival. Arthritis Rheum. 2013, 65, 1171–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Y.; Lopez-Ruiz, E.; Wengel, J.; Creemers, L.B.; Howard, K.A. A hyaluronic acid-based hydrogel enabling CD44-mediated chondrocyte binding and gapmer oligonucleotide release for modulation of gene expression in osteoarthritis. J. Control. Release 2017, 253, 153–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, J.P.; Stein, J.; Cai, Y.; Riemers, F.; Wexselblatt, E.; Wengel, J.; Tryfonidou, M.; Yayon, A.; Howard, K.A.; Creemers, L.B. Fibrin-hyaluronic acid hydrogel-based delivery of antisense oligonucleotides for ADAMTS5 inhibition in co-delivered and resident joint cells in osteoarthritis. J. Control. Release 2019, 294, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Moschos, S.A.; Jones, S.W.; Perry, M.M.; Williams, A.E.; Erjefalt, J.S.; Turner, J.J.; Barnes, P.J.; Sproat, B.S.; Gait, M.J.; Lindsay, M.A. Lung delivery studies using siRNA conjugated to TAT(48-60) and penetratin reveal peptide induced reduction in gene expression and induction of innate immunity. Bioconjug. Chem. 2007, 18, 1450–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, T.; Weber, H.; DiMuzio, J.; Matter, A.; Dogdas, B.; Shah, T.; Thankappan, A.; Disa, J.; Jadhav, V.; Lubbers, L.; et al. Silencing Myostatin Using Cholesterol-conjugated siRNAs Induces Muscle Growth. Mol. Ther. Nucleic Acids 2016, 5, e342. [Google Scholar] [CrossRef] [Green Version]
- Sacchetti, C.; Liu-Bryan, R.; Magrini, A.; Rosato, N.; Bottini, N.; Bottini, M. Polyethylene-glycol-modified single-walled carbon nanotubes for intra-articular delivery to chondrocytes. ACS Nano 2014, 8, 12280–12291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, M.L.; Jiang, H.; Wu, F.; Geng, R.; Ya, L.K.; Lin, Y.C.; Xu, J.H.; Wu, X.T.; Lu, J. Precise targeting of miR-141/200c cluster in chondrocytes attenuates osteoarthritis development. Ann. Rheum. Dis. 2020. [Google Scholar] [CrossRef]
- Ospelt, C. Synovial fibroblasts in 2017. RMD Open 2017, 3, e000471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farah, H.; Young, S.P.; Mauro, C.; Jones, S.W. Metabolic dysfunction and inflammatory disease: The role of stromal fibroblasts. FEBS J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kung, C.C.; Dai, S.P.; Chiang, H.; Huang, H.S.; Sun, W.H. Temporal expression patterns of distinct cytokines and M1/M2 macrophage polarization regulate rheumatoid arthritis progression. Mol. Biol. Rep. 2020, 47, 3423–3437. [Google Scholar] [CrossRef]
- Ostergaard, M.; Peterfy, C.; Conaghan, P.; McQueen, F.; Bird, P.; Ejbjerg, B.; Shnier, R.; O’Connor, P.; Klarlund, M.; Emery, P.; et al. OMERACT Rheumatoid Arthritis Magnetic Resonance Imaging Studies. Core set of MRI acquisitions, joint pathology definitions, and the OMERACT RA-MRI scoring system. J. Rheumatol. 2003, 30, 1385–1386. [Google Scholar]
- Myers, S.L.; Brandt, K.D.; Ehlich, J.W.; Braunstein, E.M.; Shelbourne, K.D.; Heck, D.A.; Kalasinski, L.A. Synovial inflammation in patients with early osteoarthritis of the knee. J. Rheumatol. 1990, 17, 1662–1669. [Google Scholar]
- Rhodes, L.A.; Conaghan, P.G.; Radjenovic, A.; Grainger, A.J.; Emery, P.; McGonagle, D. Further evidence that a cartilage-pannus junction synovitis predilection is not a specific feature of rheumatoid arthritis. Ann. Rheum. Dis. 2005, 64, 1347–1349. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Madrid, F.; Karvonen, R.L.; Teitge, R.A.; Miller, P.R.; An, T.; Negendank, W.G. Synovial thickening detected by MR imaging in osteoarthritis of the knee confirmed by biopsy as synovitis. Magn. Reson. Imaging 1995, 13, 177–183. [Google Scholar] [CrossRef]
- Oehler, S.; Neureiter, D.; Meyer-Scholten, C.; Aigner, T. Subtyping of osteoarthritic synoviopathy. Clin. Exp. Rheumatol. 2002, 20, 633–640. [Google Scholar] [PubMed]
- Toussirot, E.; Streit, G.; Wendling, D. The contribution of adipose tissue and adipokines to inflammation in joint diseases. Curr. Med. Chem. 2007, 14, 1095–1100. [Google Scholar] [CrossRef]
- Huang, M.; Wang, H.; Hu, X.; Cao, X. lncRNA MALAT1 binds chromatin remodeling subunit BRG1 to epigenetically promote inflammation-related hepatocellular carcinoma progression. Oncoimmunology 2019, 8, e1518628. [Google Scholar] [CrossRef]
- Hamann, P.D.; Roux, B.T.; Heward, J.A.; Love, S.; McHugh, N.J.; Jones, S.W.; Lindsay, M.A. Transcriptional profiling identifies differential expression of long non-coding RNAs in Jo-1 associated and inclusion body myositis. Sci. Rep. 2017, 7, 8024. [Google Scholar] [CrossRef] [PubMed]
- Heward, J.A.; Lindsay, M.A. Long non-coding RNAs in the regulation of the immune response. Trends Immunol. 2014, 35, 408–419. [Google Scholar] [CrossRef] [Green Version]
- Ilott, N.E.; Heward, J.A.; Roux, B.; Tsitsiou, E.; Fenwick, P.S.; Lenzi, L.; Goodhead, I.; Hertz-Fowler, C.; Heger, A.; Hall, N.; et al. Corrigendum: Long non-coding RNAs and enhancer RNAs regulate the lipopolysaccharide-induced inflammatory response in human monocytes. Nat. Commun. 2015, 6, 6814. [Google Scholar] [CrossRef] [Green Version]
- Pearson, M.J.; Jones, S.W. Review: Long Noncoding RNAs in the Regulation of Inflammatory Pathways in Rheumatoid Arthritis and Osteoarthritis. Arthritis Rheumatol. 2016, 68, 2575–2583. [Google Scholar] [CrossRef] [PubMed]
- Roux, B.T.; Heward, J.A.; Donnelly, L.E.; Jones, S.W.; Lindsay, M.A. Catalog of Differentially Expressed Long Non-Coding RNA following Activation of Human and Mouse Innate Immune Response. Front. Immunol. 2017, 8, 1038. [Google Scholar] [CrossRef] [Green Version]
- Pearson, M.J.; Philp, A.M.; Heward, J.A.; Roux, B.T.; Walsh, D.A.; Davis, E.T.; Lindsay, M.A.; Jones, S.W. Long Intergenic Noncoding RNAs Mediate the Human Chondrocyte Inflammatory Response and Are Differentially Expressed in Osteoarthritis Cartilage. Arthritis Rheumatol. 2016, 68, 845–856. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, Y.; Zhang, H.; Chang, J.; Lu, M.; Gao, W.; Liu, W.; Li, Y.; Yin, L.; Wang, X.; et al. Identification of a novel microRNA-141-3p/Forkhead box C1/β-catenin axis associated with rheumatoid arthritis synovial fibroblast function in vivo and in vitro. Theranostics 2020, 10, 5412–5434. [Google Scholar] [CrossRef]
- Jia, W.; Wu, W.; Yang, D.; Xiao, C.; Huang, M.; Long, F.; Su, Z.; Qin, M.; Liu, X.; Zhu, Y.Z. GATA4 regulates angiogenesis and persistence of inflammation in rheumatoid arthritis. Cell Death Dis. 2018, 9, 503. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Xiao, Z.; Wang, G.; Song, X. Knockdown of ADAM10 inhibits migration and invasion of fibroblast-like synoviocytes in rheumatoid arthritis. Mol. Med. Rep. 2015, 12, 5517–5523. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Chen, W.; Wang, L.; Li, F.; Zhang, C.; Xu, L. Tumor necrosis factor receptor-associated factor 6 promotes migration of rheumatoid arthritis fibroblast-like synoviocytes. Mol. Med. Rep. 2015, 11, 2761–2766. [Google Scholar] [CrossRef] [Green Version]
- Burr, D.B.; Gallant, M.A. Bone remodelling in osteoarthritis. Nat. Rev. Rheumatol. 2012, 8, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, N. Signaling Pathways in Osteoclast Differentiation. Chonnam. Med. J. 2016, 52, 12–17. [Google Scholar] [CrossRef] [Green Version]
- Hunter, D.J.; Gerstenfeld, L.; Bishop, G.; Davis, A.D.; Mason, Z.D.; Einhorn, T.A.; Maciewicz, R.A.; Newham, P.; Foster, M.; Jackson, S.; et al. Bone marrow lesions from osteoarthritis knees are characterized by sclerotic bone that is less well mineralized. Arthritis Res. Ther. 2009, 11, R11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, A.J. Changes in bone collagen with age and disease. J. Musculoskelet Neuronal Interact. 2002, 2, 529–531. [Google Scholar] [PubMed]
- Bailey, A.J.; Sims, T.J.; Knott, L. Phenotypic expression of osteoblast collagen in osteoarthritic bone: Production of type I homotrimer. Int. J. Biochem. Cell Biol. 2002, 34, 176–182. [Google Scholar] [CrossRef]
- Quasnichka, H.L.; Anderson-MacKenzie, J.M.; Bailey, A.J. Subchondral bone and ligament changes precede cartilage degradation in guinea pig osteoarthritis. Biorheology 2006, 43, 389–397. [Google Scholar] [PubMed]
- Radin, E.L.; Rose, R.M. Role of subchondral bone in the initiation and progression of cartilage damage. Clin. Orthop. Relat. Res. 1986, 213, 34–40. [Google Scholar] [CrossRef]
- Chang, J.; Jackson, S.G.; Wardale, J.; Jones, S.W. Hypoxia modulates the phenotype of osteoblasts isolated from knee osteoarthritis patients, leading to undermineralized bone nodule formation. Arthritis Rheumatol. 2014, 66, 1789–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisignoli, G.; Toneguzzi, S.; Pozzi, C.; Piacentini, A.; Riccio, M.; Ferruzzi, A.; Gualtieri, G.; Facchini, A. Proinflammatory cytokines and chemokine production and expression by human osteoblasts isolated from patients with rheumatoid arthritis and osteoarthritis. J. Rheumatol. 1999, 26, 791–799. [Google Scholar] [PubMed]
- Berry, P.A.; Jones, S.W.; Cicuttini, F.M.; Wluka, A.E.; Maciewicz, R.A. Temporal relationship between serum adipokines, biomarkers of bone and cartilage turnover, and cartilage volume loss in a population with clinical knee osteoarthritis. Arthritis Rheum. 2011, 63, 700–707. [Google Scholar] [CrossRef]
- Qiang, Y.W.; Barlogie, B.; Rudikoff, S.; Shaughnessy, J.D., Jr. Dkk1-induced inhibition of Wnt signaling in osteoblast differentiation is an underlying mechanism of bone loss in multiple myeloma. Bone 2008, 42, 669–680. [Google Scholar] [CrossRef]
- Hurson, C.J.; Butler, J.S.; Keating, D.T.; Murray, D.W.; Sadlier, D.M.; O’Byrne, J.M.; Doran, P.P. Gene expression analysis in human osteoblasts exposed to dexamethasone identifies altered developmental pathways as putative drivers of osteoporosis. BMC Musculoskelet Disord. 2007, 8, 12. [Google Scholar] [CrossRef] [Green Version]
- Butler, J.S.; Murray, D.W.; Hurson, C.J.; O’Brien, J.; Doran, P.P.; O’Byrne, J.M. The role of Dkk1 in bone mass regulation: Correlating serum Dkk1 expression with bone mineral density. J. Orthop. Res. 2011, 29, 414–418. [Google Scholar] [CrossRef]
- Akkiraju, H.; Nohe, A. Role of Chondrocytes in Cartilage Formation, Progression of Osteoarthritis and Cartilage Regeneration. J. Dev. Biol. 2015, 3, 177–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldring, M.B.; Marcu, K.B. Epigenomic and microRNA-mediated regulation in cartilage development, homeostasis, and osteoarthritis. Trends Mol. Med. 2012, 18, 109–118. [Google Scholar] [CrossRef] [Green Version]
- Kung, L.H.W.; Ravi, V.; Rowley, L.; Angelucci, C.; Fosang, A.J.; Bell, K.M.; Little, C.B.; Bateman, J.F. Cartilage MicroRNA Dysregulation During the Onset and Progression of Mouse Osteoarthritis Is Independent of Aggrecanolysis and Overlaps With Candidates From End-Stage Human Disease. Arthritis Rheumatol. 2018, 70, 383–395. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, A.; Ali, S.A.; Kapoor, M. Antisense oligonucleotide-based therapies for the treatment of osteoarthritis: Opportunities and roadblocks. Bone 2020, 138, 115461. [Google Scholar] [CrossRef]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.W.; Lindsay, M.A. Overview of target validation and the impact of oligonucleotides. Curr. Opin. Mol. Ther. 2004, 6, 546–550. [Google Scholar] [PubMed]
- Jones, S.W.; Christison, R.; Bundell, K.; Voyce, C.J.; Brockbank, S.M.; Newham, P.; Lindsay, M.A. Characterisation of cell-penetrating peptide-mediated peptide delivery. Br. J. Pharmacol. 2005, 145, 1093–1102. [Google Scholar] [CrossRef]
- Takakura, H.; Sato, H.; Nakajima, K.; Suzuki, M.; Ogawa, M. In Vitro and In Vivo Cell Uptake of a Cell-Penetrating Peptide Conjugated with Fluorescent Dyes Having Different Chemical Properties. Cancers 2021, 13, 2245. [Google Scholar] [CrossRef] [PubMed]
- Nair, J.K.; Willoughby, J.L.; Chan, A.; Charisse, K.; Alam, M.R.; Wang, Q.; Hoekstra, M.; Kandasamy, P.; Kel’in, A.V.; Milstein, S.; et al. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J. Am. Chem. Soc. 2014, 136, 16958–16961. [Google Scholar] [CrossRef] [Green Version]
- Pasi, K.J.; Rangarajan, S.; Georgiev, P.; Mant, T.; Creagh, M.D.; Lissitchkov, T.; Bevan, D.; Austin, S.; Hay, C.R.; Hegemann, I.; et al. Targeting of Antithrombin in Hemophilia A or B with RNAi Therapy. N. Engl. J. Med. 2017, 377, 819–828. [Google Scholar] [CrossRef]
- Huang, Y. Preclinical and Clinical Advances of GalNAc-Decorated Nucleic Acid Therapeutics. Mol. Ther. Nucleic Acids 2017, 6, 116–132. [Google Scholar] [CrossRef] [Green Version]
- Pallares, R.M.; Choo, P.; Cole, L.E.; Mirkin, C.A.; Lee, A.; Odom, T.W. Manipulating Immune Activation of Macrophages by Tuning the Oligonucleotide Composition of Gold Nanoparticles. Bioconjug. Chem. 2019, 30, 2032–2037. [Google Scholar] [CrossRef]
- Wong, C.Y.; Martinez, J.; Zhao, J.; Al-Salami, H.; Dass, C.R. Development of orally administered insulin-loaded polymeric-oligonucleotide nanoparticles: Statistical optimization and physicochemical characterization. Drug Dev. Ind. Pharm. 2020, 46, 1238–1252. [Google Scholar] [CrossRef]
- Suzuki, Y.; Onuma, H.; Sato, R.; Sato, Y.; Hashiba, A.; Maeki, M.; Tokeshi, M.; Kayesh, M.E.H.; Kohara, M.; Tsukiyama-Kohara, K.; et al. Lipid nanoparticles loaded with ribonucleoprotein-oligonucleotide complexes synthesized using a microfluidic device exhibit robust genome editing and hepatitis B virus inhibition. J. Control. Release 2021, 330, 61–71. [Google Scholar] [CrossRef]
- Geiger, B.C.; Wang, S.; Padera, R.F., Jr.; Grodzinsky, A.J.; Hammond, P.T. Cartilage-penetrating nanocarriers improve delivery and efficacy of growth factor treatment of osteoarthritis. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.; Hou, J.; Zhong, Z.; Hao, N.; Lin, Y.; Li, C. Targeted delivery of hyaluronic acid-coated solid lipid nanoparticles for rheumatoid arthritis therapy. Drug Deliv. 2018, 25, 716–722. [Google Scholar] [CrossRef]
- Bajpayee, A.G.; Scheu, M.; Grodzinsky, A.J.; Porter, R.M. Electrostatic interactions enable rapid penetration, enhanced uptake and retention of intra-articular injected avidin in rat knee joints. J. Orthop. Res. 2014, 32, 1044–1051. [Google Scholar] [CrossRef]
- Hu, H.Y.; Lim, N.H.; Juretschke, H.P.; Ding-Pfennigdorff, D.; Florian, P.; Kohlmann, M.; Kandira, A.; Peter von Kries, J.; Saas, J.; Rudolphi, K.A.; et al. In vivo visualization of osteoarthritic hypertrophic lesions. Chem. Sci. 2015, 6, 6256–6261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajpayee, A.G.; Wong, C.R.; Bawendi, M.G.; Frank, E.H.; Grodzinsky, A.J. Avidin as a model for charge driven transport into cartilage and drug delivery for treating early stage post-traumatic osteoarthritis. Biomaterials 2014, 35, 538–549. [Google Scholar] [CrossRef] [Green Version]
- Freedman, J.D.; Lusic, H.; Wiewiorski, M.; Farley, M.; Snyder, B.D.; Grinstaff, M.W. A cationic gadolinium contrast agent for magnetic resonance imaging of cartilage. Chem. Commun. 2015, 51, 11166–11169. [Google Scholar] [CrossRef] [PubMed] [Green Version]


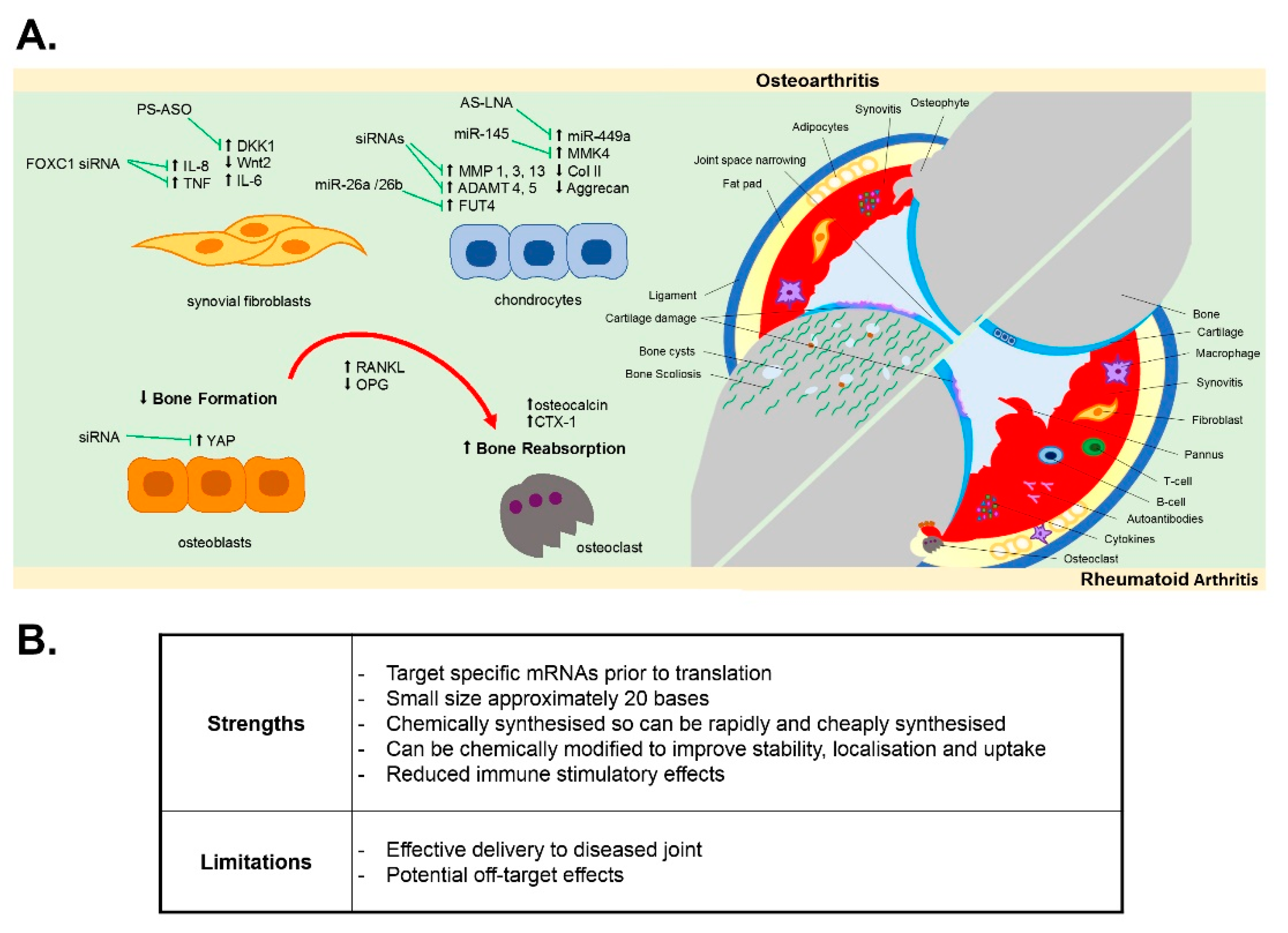{kind=link}
| Cartilage | |||||
|---|---|---|---|---|---|
| Target | Type of Oligonucleotide | Target Cell/Tissue | Study Model | Function | Ref. |
| ADAMTS5 | siRNA | cartilage | in vivo—MM mouse | Silencing alone or in combination with MMP13 siRNA improved histological scores of OA severity. | [40] |
| c-Fos | antisense oligonucleotide | chondrocytes | in vitro | Knockdown acted to inhibit the potentiating action of SDF-1α on MMP-13 promoter activity. | [41] |
| c-Jun | antisense oligonucleotide | chondrocytes | in vitro | Knockdown acted to inhibit the potentiating action of SDF-1α on MMP-13 promoter activity. | [41] |
| miR-101 | adenovirus-miRNA | cartilage | in vivo—MIA rat | Silencing by intra-articular injection reduced cartilage degeneration in an OA model. Microarray analysis found downregulation of several cartilage-related genes. | [42] |
| miR-128 | antisense oligonucleotides | chondrocyte | in vivo—ACLT rat | Intra-articular injections silencing miR-128a reduced cartilage degradation, synovitis and subchondral bone damage in the ACLT rat model via Atg12. | [43] |
| miR-145 | miRNA mimics | cartilage | in vivo—DMM rat | Intra-articular injection of miRNA mimics reduced cartilage degradation via suppression of MKK4, which negatively regulates TNFα-mediated JNK, p38, p-c-Jun and p-ATF2, thus repressing MMP3, MMP13 and Adamts-5. | [44] |
| mir-146 | pre-miRNA mimics | chondrocytes | in vitro | Overexpression significantly attenuated IL-1β-induced reduced TNFα production. | [45] |
| miR-181a-5p | locked nucleic acid | chondrocyte | in vivo –FJD rat and DMM mouse | Silencing by intra-articular injection attenuated cartilage destruction, reducing catabolic, hypertrophic and apoptotic marker expression. | [46] |
| mir-21-5p | antagomir | chondrocytes | in vivo—DMM mouse | Intra-articular injections to knockdown mir-21-5p significantly attenuated the severity of OA by modulating expression of FGF18. | [47] |
| miR-26a /26b | miRNA mimics | chondrocyte | in vivo—ACLT rat | Intra-articular over-expression attenuated development of OA in vivo and over-expression also promoted chondrocyte proliferation in vitro through the FUT4/NF-κB axis. | [48] |
| miR-320a | antisense oligonucleotides | chondrocytes | in vitro | Silencing reduced the IL-1β-mediated release of MMP13 and sGAG whilst enhancing expression of Col2a1 and ACAN. | [49] |
| miR-34a-5p | locked nucleic acid | chondrocytes and synovial fibroblasts | in vivo—DMM and high-fat diet/DMM mouse | Chondroprotective effects imparted following silencing in vivo by intra-articular injection. In vitro ASO treatment increased COL2A1 and ACAN in OA chondrocytes whilst reducing COL1A1 and TNFα in OA fibroblasts. | [50] |
| miR-449a | locked nucleic acid | cartilage and subchondral bone | in vivo—DMM rat | Intra-articular silencing promoted cartilage regeneration and expression of type II collagen and aggrecan in cartilage. | [51] |
| miR-98 | pre-miRNA mimics | chondrocytes | in vitro | Overexpression significantly attenuated IL-1β-induced reduced TNFα production. | [45] |
| MK2 | siRNA | chondrocytes | in vitro | MK2 silencing inhibited IL-1β-induced production of MMP3, MMP13 and PGE2. | [34,52] |
| MMP-13 | siRNA | cartilage | in vivo—DMM mouse | Silencing alone or in combination with ADAMTS5 siRNA improved histological scores of OA severity in vivo by intra-articular injection. | [40] |
| p38 | siRNA | chondrocytes | in vitro | P38 silencing inhibited IL-1β-induced production of MMP3, MMP13 and PGE2. | [34,52] |
| RDC1 | siRNA | chondrocytes | in vitro | Silencing RDC1 in OA chondrocytes reduced expression of MMPs and hypertrophic markers. | [53] |
| YAP | siRNA | chondrocytes | in vivo—ACLT mouse | Silencing of YAP reduced chondrocyte apoptosis and inhibited IL-1β-induced catabolic factors. Intra-articular injection ameliorated OA development and reduced aberrant subchondral bone formation in vivo. | [54] |
| Subchondral Bone | |||||
| Target | Type of Oligonucleotide | Target Cell/Tissue | Study Model | Function | Ref. |
| DKK1 | end-capped phosphorothioate antisense oligonucleotide | cartilage and subchondral bone | in vivo—ACLT and CIA rat | Oligonucleotides delivered intraperitoneally reduced disease severity, bone mineral density loss and reduced serum levels of bone resorption markers osteocalcin and CTX-1 and expression of TNFα, IL-1β, MMP3 and RANKL. | [55] |
| Leptin receptor long isoform (OBRl) | phosphorothioate double-stranded decoy oligonucleotide | osteoblasts | in vitro | Silencing abolished the leptin-mediated production of oncostatin M via miR-93/Akt signalling axis. | [56] |
| miR-29a | antisense oligonucleotides | subchondral mesenchymal stem cells | in vitro | Knockdown inhibits Wnt3a expression and impaired Wnt-mediated osteogenic differentiation via HDAC4. | [57] |
| Thyroid hormone receptor (THR) | siRNA | osteoblasts | in vivo—DMM mouse | THR knockdown downregulated THR regulatory genes including HIF-1α, VEGF and IGI-1. Intra-articular injection improved sclerotic subchondral bone formation, as well as an overall reduction in OA severity score. | [58] |
| Synovium | |||||
| Target | Type of Oligonucleotide | Target Cell/Tissue | Study Model | Function | Ref. |
| Dickkopf-1 (DKK1) | 21-mer end-capped phosphorothioate antisense oligonucleotide | synovium | in vivo—ACLT rat | Intraperitoneally administered silencing reduced proteinases and angiogenic factors, reduced vessel distribution and formation and reduced cartilage injury. | [59] |
| FLIP | antisense oligo-deoxynucleotide | synovial fibroblasts | in vitro | FLIP knockdown increased fas-mediated apoptosis by 3-fold. | [60] |
| FoxC1 | siRNA | synovial fibroblasts | in vivo—DMM mouse | Silencing inhibited IL-6, IL-8, TNF, ADAMTS-5, fibronectin, MMP3 and MMP13 and proliferation of OA synovial fibroblast, whilst intra-articular injection of FoxC1 siRNA prevented OA development in vivo. | [61] |
| Galectin-9 | siRNA | synovial fibroblasts | in vitro | Galectin-9 knockdown increased apoptosis of human RA synovial fibroblasts. | [62] |
| Leptin receptor long isoform (OBRl) | antisense oligonucleotide | synovial fibroblasts | in vitro | Inflammatory OA fibroblast phenotype mediated by leptin was inhibited, thus reducing leptin-mediated IL-8 secretion via JAK2/STAT3 pathway. | [63] |
| Leptin receptor long isoform (OBRl) | phosphorothioate double-stranded decoy oligonucleotide | synovial fibroblasts | in vitro | Inflammatory OA fibroblast phenotype mediated by leptin was inhibited resulting in reduced IL-6 via IRS-1/PI3K/Akt/AP-1 pathway. | [64] |
| MALAT1 | locked nucleic acid | synovial fibroblasts | in vitro | MALAT1 knockdown inhibited both the proliferative and inflammatory phenotype of obese OA synovial fibroblasts, resulting in reduced CXCL8 expression and secretion and increased expression of TRIM6, IL7R, HIST1H1C and MAML3. | [65] |
| Notch-1 | antisense oligonucleotide | synovial fibroblasts | in vitro | Antisense Notch-1 oligonucleotide abrogated Notch-1 expression in the nucleus, preventing TNFα-induced translocation of Notch-1 intracellular domain (NICD) to the nucleus. Inhibits both basal and TNFα-induced proliferation of RA and OA synovial fibroblasts in a dose-dependent manner. | [66] |
| PTPN11 (SHP-2) | antisense knockdown | synovial fibroblasts | in vitro | Loss of SHP-2 inhibits migration, invasion, adhesion and survival of RA synovial fibroblasts through reduced PDGF-induced activation of MAPKs and upstream FAK. | [67] |
| Target | Type of Oligonucleotide | Delivery Method | Study Model | Outcome | Ref. |
|---|---|---|---|---|---|
| Viral | |||||
| miR-128a/ATG12 | antisense oligonucleotide | lentiviral | ACLT rat | Intra-articular administration of miR-128a antisense oligonucleotide disrupted ATG12 repression, stabilizing chondrocyte autophagy and delaying OA progression. | [43] |
| miR-101 | antisense oligonucleotide | adenovirus | MIA rat | Antisense oligonucleotide silencing of miR-101 reduced cartilage degradation. | [42] |
| Biomaterial | |||||
| COX2 | locked nucleic acid | hyaluronic acid hydrogel | human primary OA chondrocytes | Hydrogel-encapsulated LNA silenced COX2 over 14 days. | [68] |
| ADAMTS5 | antisense oligonucleotide | fibrin and hyaluronic acid hydrogel | human primary OA chondrocytes | In a 3D two-gel cell construct, antisense oligonucleotides silenced ADAMTS5 at day 7 and 14 with concentrations of 5 and 10 µM. | [69] |
| Bioconjugation | |||||
| p38 MAPK | siRNA | cholesterol | L929 mouse fibroblast cell line and BALB/c mice | siRNA conjugated to cholesterol, TAT and pentratin silenced p38 MAPK in vitro whilst conjugation to cholesterol alone could circumvent immunostimulatory effects of TAT and pentratin in vivo. | [70] |
| myostatin | siRNA | cholesterol | CD-1 mice | A single intravenous injection of cholesterol-conjugated siRNA sustained myostatin silencing in skeletal muscle and in circulation over 21 days. | [71] |
| Nanoparticles | |||||
| GFP | morpholino antisense oligonucleotides | PEG-SWCNTs | DMM mice | Intra-articular injections of PEG-SWCNTs carrying anti-GFP oligonucleotides could silence GFP in GFP-transgenic mice | [72] |
| miR-141/200c | miRNA | PEG-PA | DMM mice | Intra-articular silencing of miR-141/200c has chondroprotective effects via downregulation of the IL-6/STAT3 pathway. | [73] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wijesinghe, S.N.; Lindsay, M.A.; Jones, S.W. Oligonucleotide Therapies in the Treatment of Arthritis: A Narrative Review. Biomedicines 2021, 9, 902. https://doi.org/10.3390/biomedicines9080902
Wijesinghe SN, Lindsay MA, Jones SW. Oligonucleotide Therapies in the Treatment of Arthritis: A Narrative Review. Biomedicines. 2021; 9(8):902. https://doi.org/10.3390/biomedicines9080902
Chicago/Turabian StyleWijesinghe, Susanne N., Mark A. Lindsay, and Simon W. Jones. 2021. "Oligonucleotide Therapies in the Treatment of Arthritis: A Narrative Review" Biomedicines 9, no. 8: 902. https://doi.org/10.3390/biomedicines9080902
APA StyleWijesinghe, S. N., Lindsay, M. A., & Jones, S. W. (2021). Oligonucleotide Therapies in the Treatment of Arthritis: A Narrative Review. Biomedicines, 9(8), 902. https://doi.org/10.3390/biomedicines9080902