
Betulinic Acid Decorated with Polar Groups and Blue Emitting BODIPY Dye: Synthesis, Cytotoxicity, Cell-Cycle Analysis and Anti-HIV Profiling

 , , , , , ,
, , , , , ,  and
and 
Abstract
: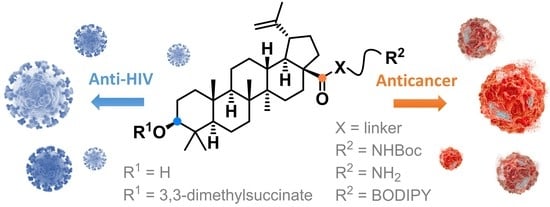
1. Introduction
2. Materials and Methods
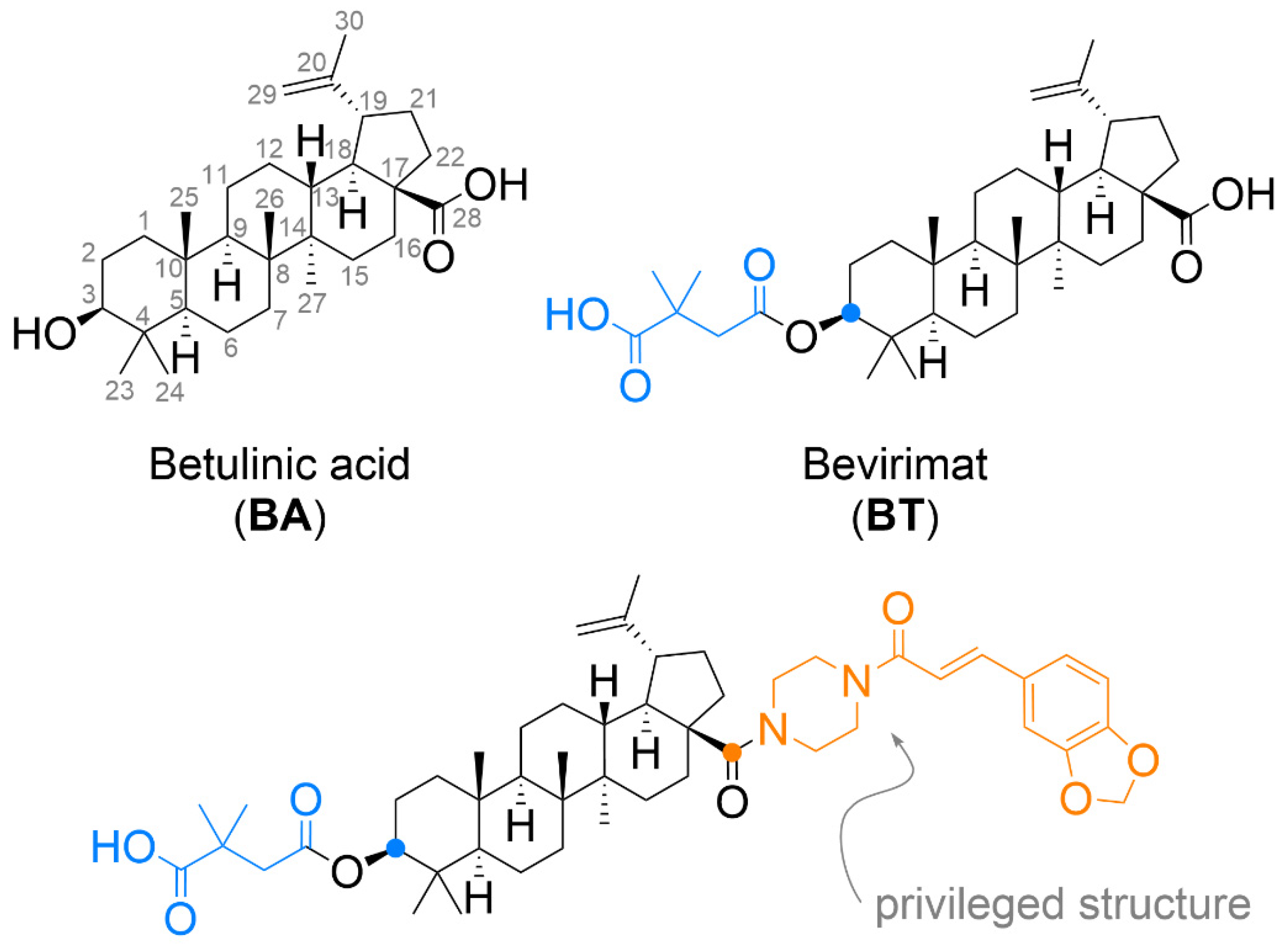
2.1. Chemical Synthesis
Compound Synthesis and Characterization
2.2. Biochemistry
2.2.1. Cell Lines
2.2.2. MTS Assay
2.2.3. Cell Cycle and Apoptosis Analysis
2.2.4. BrDU Incorporation Analysis
2.2.5. BrU Incorporation Analysis
2.2.6. Fluorescent Microscopy
2.2.7. VSV-G Pseudotyped HIV-1 Particles Production
2.2.8. Single-Round Infectivity Assay
2.2.9. Western Blot
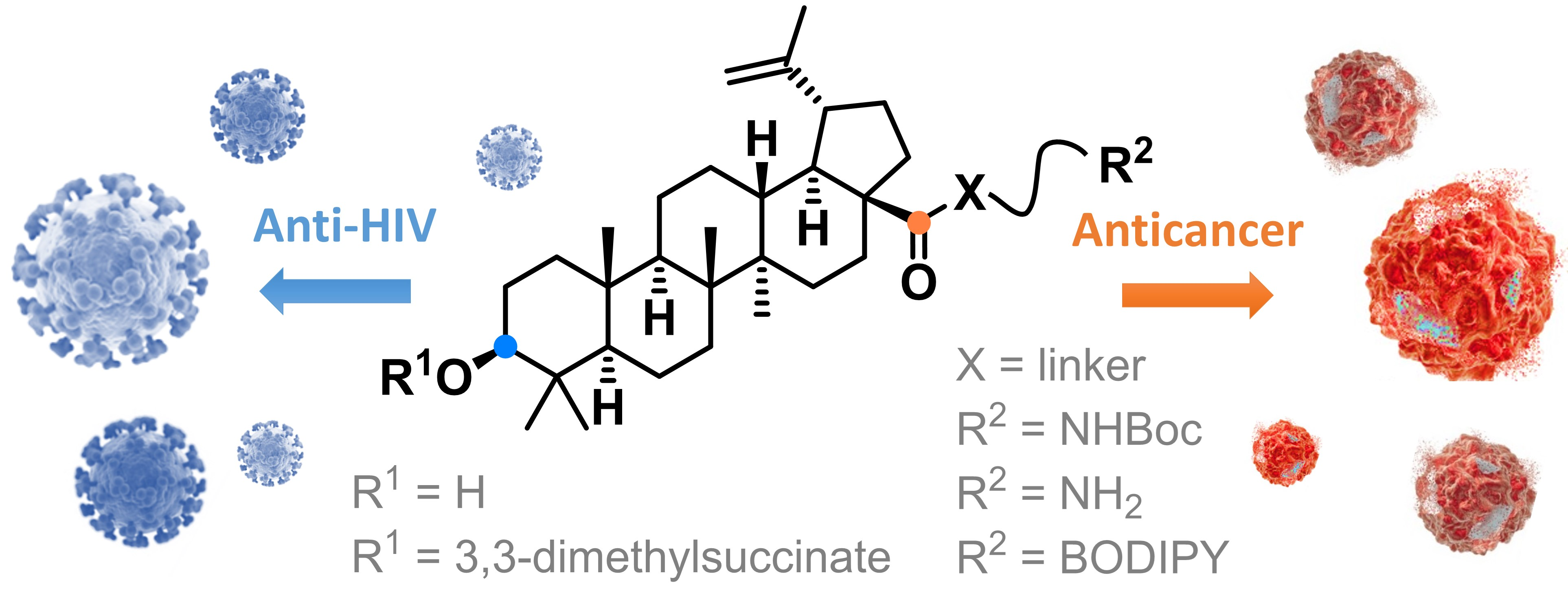
3. Results and Discussion
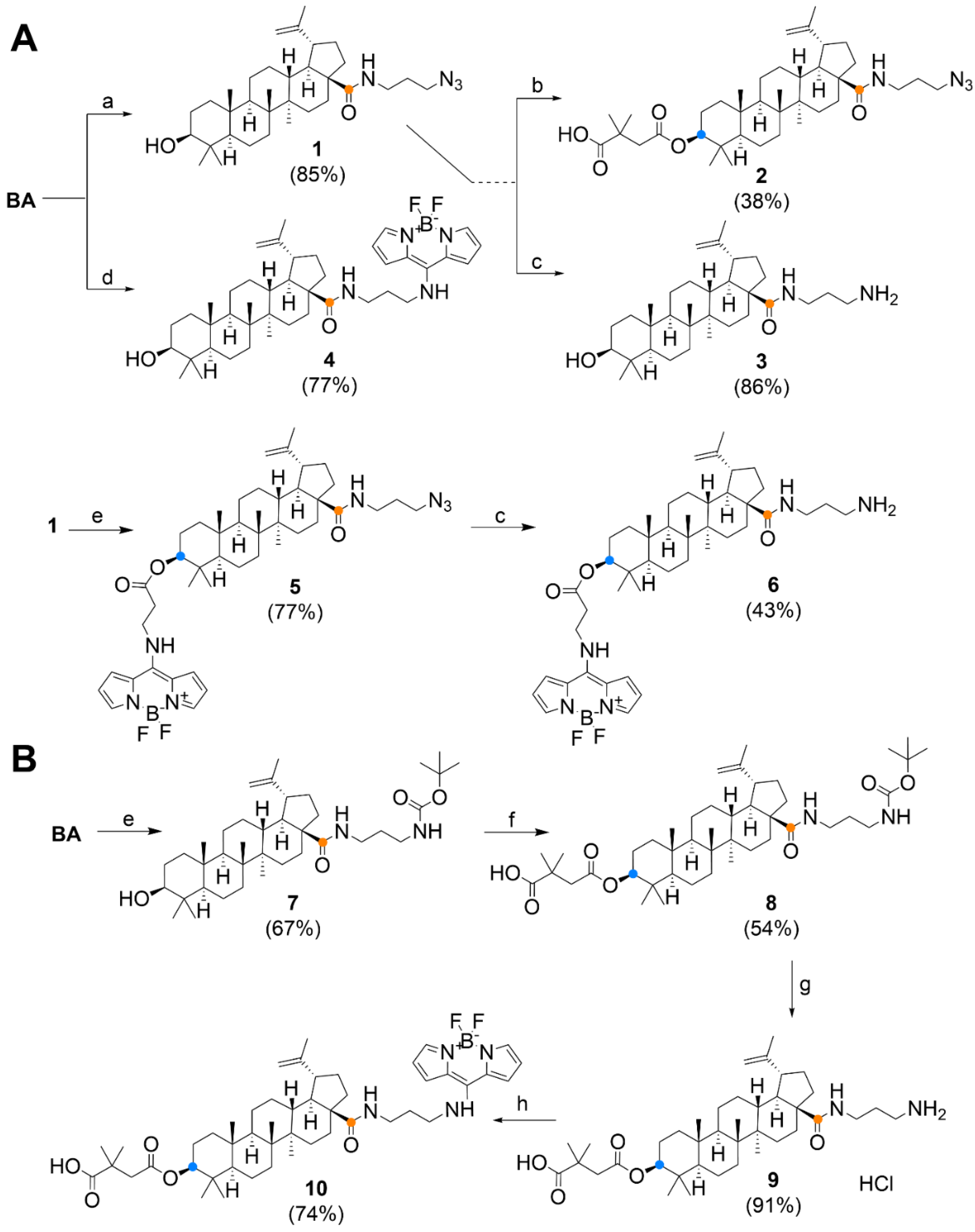
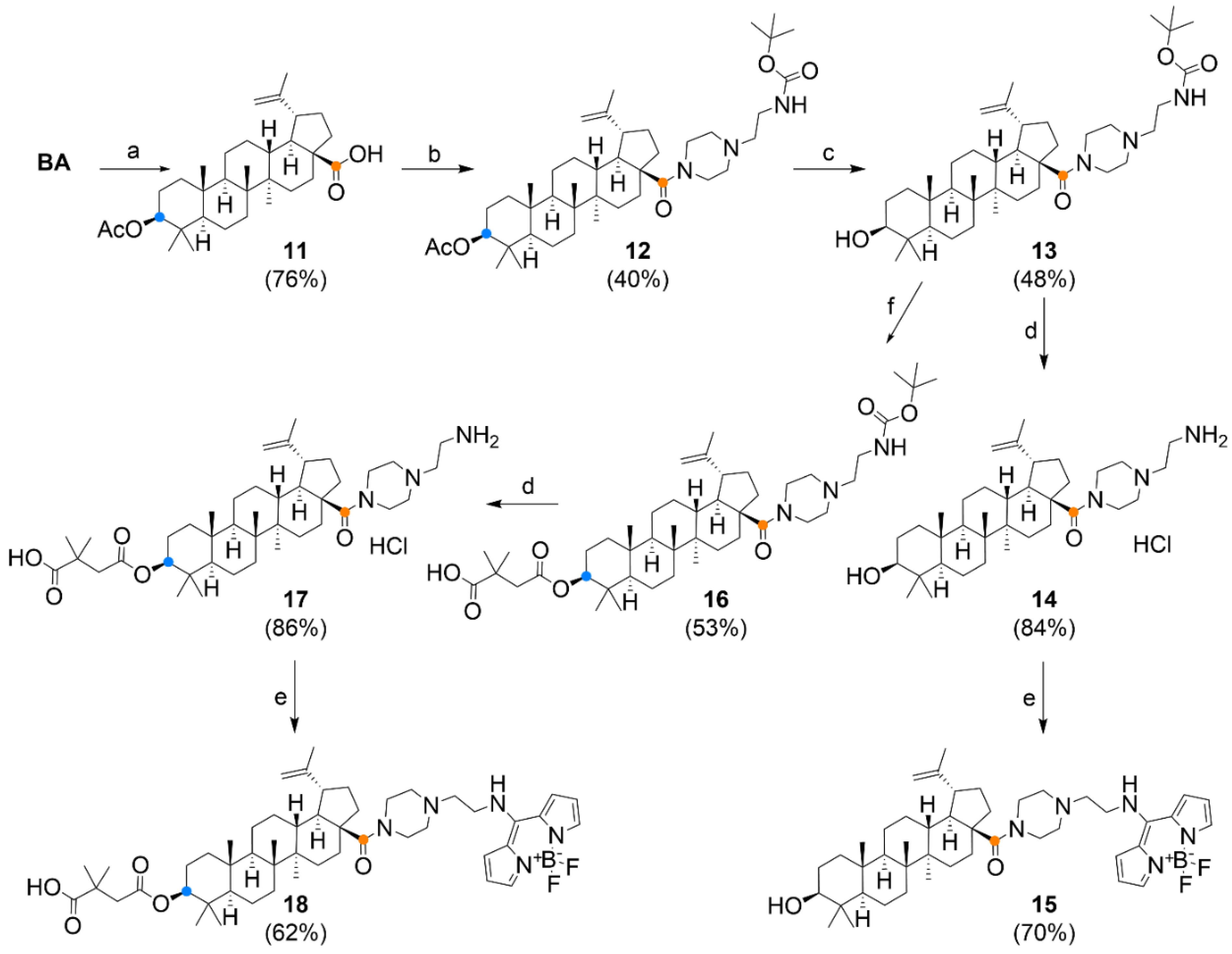
3.1. Chemistry
3.2. Cytotoxicity on a Panel of Cell Lines
3.3. Cell Cycle, Apoptosis and DNA/RNA Synthesis
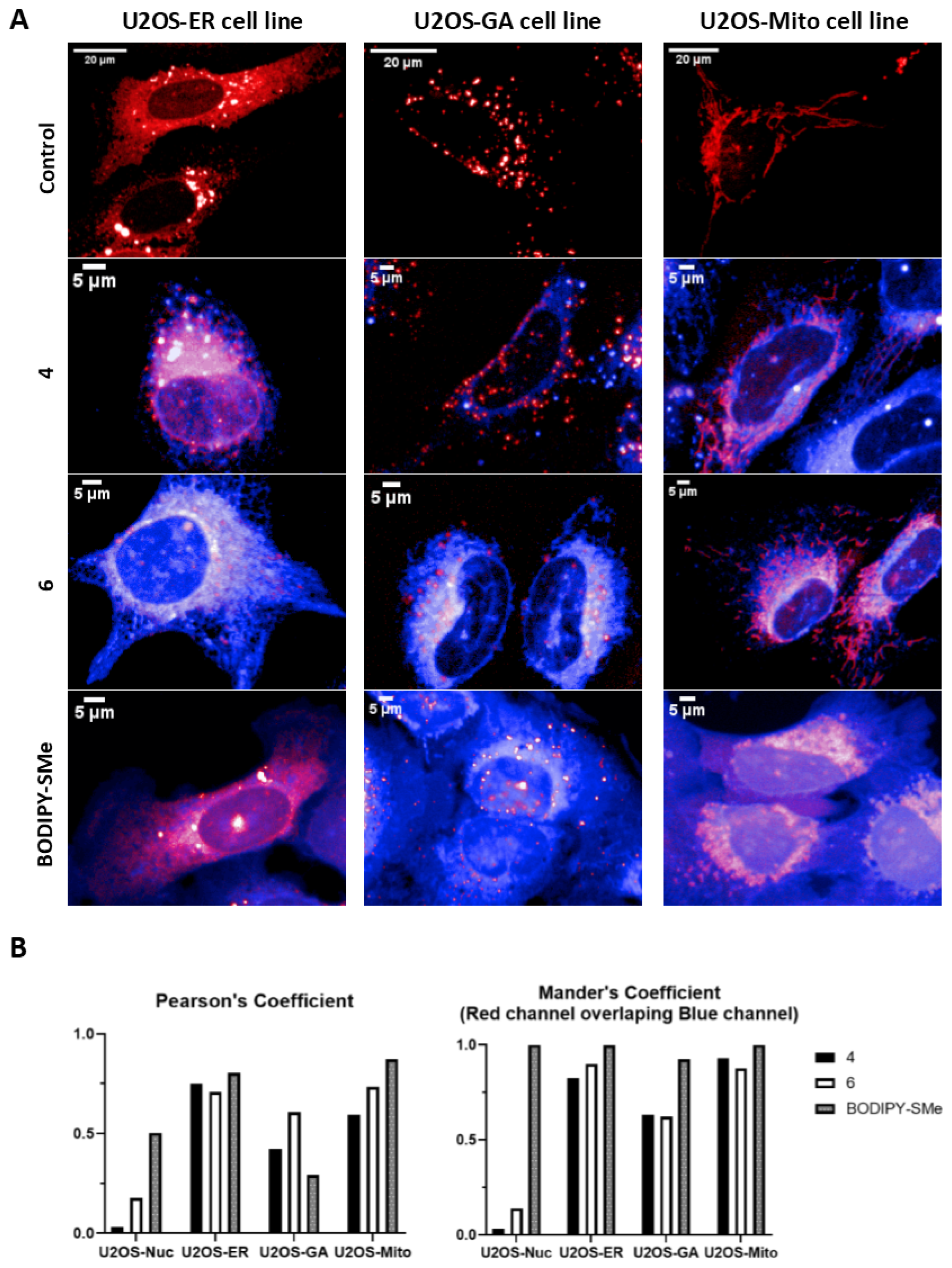
3.4. Live Cells Imaging
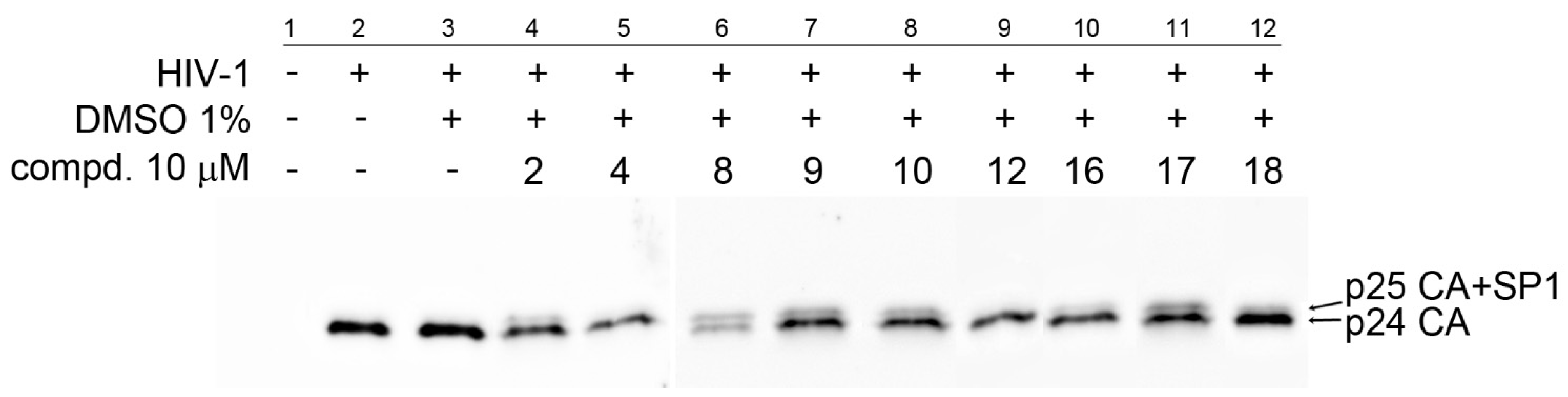
3.5. Anti-HIV Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sousa, J.L.C.; Freire, C.S.R.; Silvestre, A.J.D.; Silva, A.M.S. Recent developments in the functionalization of betulinic acid and its natural analogues: A route to new bioactive compounds. Molecules 2019, 24, 355. [Google Scholar] [CrossRef]
- Zuco, V.; Supino, R.; Righetti, S.C.; Cleris, L.; Marchesi, E.; Gambacorti-Passerini, C.; Formelli, F. Selective cytotoxicity of betulinic acid on tumor cell lines, but not on normal cells. Cancer Lett. 2002, 175, 17–25. [Google Scholar] [CrossRef]
- Pisha, E.; Chai, H.; Lee, I.S.; Chagwedera, T.E.; Farnsworth, N.R.; Cordell, G.A.; Beecher, C.W.W.; Fong, H.H.S.; Kinghorn, A.D.; Brown, D.M.; et al. Discovery of betulinic acid as a selective inhibitor of human melanoma that functions by induction of apoptosis. Nat. Med. 1995, 1, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Noda, Y.; Kaiya, T.; Kohda, K.; Kawazoe, Y. Enhanced cytotoxicity of some triterpenes toward leukemia L1210 cells cultured in low pH media: Possibility of a New Mode of Cell Killing. Chem. Pharm. Bull. 1997, 45, 1665–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujioka, T.; Kashiwada, Y.; Kilkuskie, R.E.; Cosentino, L.M.; Ballas, L.M.; Jiang, J.B.; Janzen, W.P.; Chen, I.-S.; Lee, K.-H. Anti-AIDS agents, 11. Betulinic acid and platanic acid as anti-HIV principles from Syzigium claviflorum, and the anti-HIV activity of structurally related triterpenoids. J. Nat. Prod. 1994, 57, 243–247. [Google Scholar] [CrossRef]
- Kodr, D.; Rumlová, M.; Zimmermann, T.; Džubák, P.; Drašar, P.; Jurášek, M. Antitumor and anti-HIV derivatives of betulinic acid. Chem. Listy 2020, 114, 658–667. [Google Scholar]
- Fulda, S.; Friesen, C.; Los, M.; Scaffidi, C.; Mier, W.; Benedict, M.; Nunez, G.; Krammer, P.H.; Peter, M.E.; Debatin, K.M. Betulinic acid triggers CD95 (APO-1/Fas)- and p53-independent apoptosis via activation of caspases in neuroectodermal tumors. Cancer Res. 1997, 57, 4956–4964. [Google Scholar] [PubMed]
- Fulda, S.; Scaffidi, C.; Susin, S.A.; Krammer, P.H.; Kroemer, G.; Peter, M.E.; Debatin, K.M. Activation of mitochondria and release of mitochondrial apoptogenic factors by betulinic acid. J. Biol. Chem. 1998, 273, 33942–33948. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.K.; Ho, J.C.K.; Cheung, F.W.K.; Liu, B.P.L.; Ye, W.C.; Che, C.T. Apoptotic activity of betulinic acid derivatives on murine melanoma B16 cell line. Eur. J. Pharmacol. 2004, 498, 71–78. [Google Scholar] [CrossRef]
- Fulda, S.; Jeremias, I.; Steiner, H.H.; Pietsch, T.; Debatin, K.M. Betulinic acid: A new cytotoxic agent against malignant brain-tumor cells. Int. J. Cancer 1999, 82, 435–441. [Google Scholar] [CrossRef]
- Mullauer, F.B.; van Bloois, L.; Daalhuisen, J.B.; Ten Brink, M.S.; Storm, G.; Medema, J.P.; Schiffelers, R.M.; Kessler, J.H. Betulinic acid delivered in liposomes reduces growth of human lung and colon cancers in mice without causing systemic toxicity. Anti-Cancer Drug 2011, 22, 223–233. [Google Scholar] [CrossRef]
- Takada, Y.; Aggarwal, B.B. Betulinic acid suppresses carcinogen-induced NF-kappa B activation through inhibition of I kappa B alpha kinase and p65 phosphorylation: Abrogation of cyclooxygenase-2 and matrix metalloprotease-9. J. Immunol. 2003, 171, 3278–3286. [Google Scholar] [CrossRef] [Green Version]
- Melzig, M.F.; Bormann, H. Betulinic acid inhibits aminopeptidase N activity. Planta Med. 1998, 64, 655–657. [Google Scholar] [CrossRef]
- Kwon, H.J.; Shim, J.S.; Kim, J.H.; Cho, H.Y.; Yum, Y.N.; Kim, S.H.; Yu, J. Betulinic acid inhibits growth factor-induced in vitro angiogenesis via the modulation of mitochondrial function in endothelial cells. Jpn. J. Cancer Res. 2002, 93, 417–425. [Google Scholar] [CrossRef]
- Kashiwada, Y.; Nagao, T.; Hashimoto, A.; Ikeshiro, Y.; Okabe, H.; Cosentino, L.M.; Lee, K.-H. Anti-AIDS agents 38. Anti-HIV activity of 3-O-acyl ursolic acid derivatives. J. Nat. Prod. 2000, 63, 1619–1622. [Google Scholar] [CrossRef] [PubMed]
- Sundquist, W.I.; Krausslich, H.G. HIV-1 Assembly, budding, and maturation. Cold Spring Harb. Perspect. Med. 2012, 2, a006924. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.F.; Ogundele, A.; Forrest, A.; Wilton, J.; Salzwedel, K.; Doto, J.; Allaway, G.P.; Martin, D.E. Phase I and II study of the safety, virologic effect, and pharmacokinetics/pharmacodynamics of single-dose 3-O-(3′,3′-dimethylsuccinyl)betulinic acid (bevirimat) against human immunodeficiency virus infection. Antimicrob. Agents Chemother. 2007, 51, 3574–3581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.E.; Blum, R.; Wilton, J.; Doto, J.; Galbraith, H.; Burgess, G.L.; Smith, P.C.; Ballow, C. Safety and pharmacokinetics of bevirimat (PA-457), a novel inhibitor of human immunodeficiency virus maturation, in healthy volunteers. Antimicrob. Agents Chemother. 2007, 51, 3063. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.E.; Blum, R.; Doto, J.; Galbraith, H.; Ballow, C. Multiple-Dose Pharmacokinetics and safety of bevirimat, a novel inhibitor of HIV maturation, in healthy volunteers. Clin. Pharmacokinet. 2007, 46, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Margot, N.A.; Gibbs, C.S.; Miller, M.D. Phenotypic susceptibility to bevirimat in isolates from HIV-1-infected patients without prior exposure to bevirimat. Antimicrob. Agents Chemother. 2010, 54, 2345–2353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Gu, Q.; Morris-Natschke, S.L.; Chen, C.-H.; Lee, K.-H. Incorporation of privileged structures into bevirimat can improve activity against wild-type and bevirimat-resistant HIV-1. J. Med. Chem. 2016, 59, 9262–9268. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Chen, C.-H.; Morris-Natschke, S.L.; Lee, K.-H. Design, synthesis, and structure activity relationship analysis of new betulinic acid derivatives as potent HIV inhibitors. Eur. J. Med. Chem. 2021, 215, 113287. [Google Scholar] [CrossRef]
- Mukherjee, R.; Jaggi, M.; Rajendran, P.; Siddiqui, M.J.A.; Srivastava, S.K.; Vardhan, A.; Burman, A.C. Betulinic acid and its derivatives as anti-angiogenic agents. Bioorg. Med. Chem. Lett. 2004, 14, 2181–2184. [Google Scholar] [CrossRef]
- Kim, J.Y.; Koo, H.M.; Kim, D.S.H.L. Development of C-20 modified betulinic acid derivatives as antitumor agents. Bioorg. Med. Chem. Lett. 2001, 11, 2405–2408. [Google Scholar] [CrossRef]
- Chowdhury, A.R.; Mandal, S.; Mittra, B.; Sharma, S.; Mukhopadhyay, S.; Majumder, H.K. Betulinic acid, a potent inhibitor of eukaryotic topoisomerase I: Identification of the inhibitory step, the major functional group responsible and development of more potent derivatives. Med. Sci. Monit. 2002, 8, BR254–BR265. [Google Scholar] [PubMed]
- Bildziukevich, U.; Rarova, L.; Janovska, L.; Saman, D.; Wimmer, Z. Enhancing effect of cystamine in its amides with betulinic acid as antimicrobial and antitumor agent in vitro. Steroids 2019, 148, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Bildziukevich, U.; Vida, N.; Rárová, L.; Kolář, M.; Šaman, D.; Havlíček, L.; Drašar, P.; Wimmer, Z. Polyamine derivatives of betulinic acid and beta-sitosterol: A comparative investigation. Steroids 2015, 100, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Brandes, B.; Hoenke, S.; Fischer, L.; Csuk, R. Design, synthesis and cytotoxicity of BODIPY-FL labelled triterpenoids. Eur. J. Med. Chem. 2020, 185, 111858. [Google Scholar] [CrossRef] [PubMed]
- Krajčovičová, S.; Staňková, J.; Džubák, P.; Hajdúch, M.; Soural, M.; Urban, M. A synthetic approach for the rapid preparation of BODIPY conjugates and their use in imaging of cellular drug uptake and distribution. Chem. Eur. J. 2018, 24, 4957–4966. [Google Scholar] [CrossRef] [PubMed]
- Sommerwerk, S.; Heller, L.; Kerzig, C.; Kramell, A.E.; Csuk, R. Rhodamine B conjugates of triterpenoic acids are cytotoxic mitocans even at nanomolar concentrations. Eur. J. Med. Chem. 2017, 127, 1–9. [Google Scholar] [CrossRef]
- Pal, A.; Ganguly, A.; Chowdhuri, S.; Yousuf, M.; Ghosh, A.; Barui, A.K.; Kotcherlakota, R.; Adhikari, S.; Banerjee, R. Bis-arylidene oxindole-betulinic acid conjugate: A fluorescent cancer cell detector with potent anticancer activity. ACS Med. Chem. Lett. 2015, 6, 612–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rumlová, M.; Křížová, I.; Keprová, A.; Hadravová, R.; Doležal, M.; Strohalmová, K.; Pichová, I.; Hájek, M.; Ruml, T. HIV-1 protease-induced apoptosis. Retrovirology 2014, 11, 37. [Google Scholar] [CrossRef]
- Dostálková, A.; Kaufman, F.; Křížová, I.; Kultová, A.; Strohalmová, K.; Hadravová, R.; Ruml, T.; Rumlová, M. Mutations in the basic region of the Mason-Pfizer monkey virus nucleocapsid protein affect reverse transcription, genomic RNA packaging, and the virus assembly site. J. Virol. 2018, 92, e00106-18. [Google Scholar] [CrossRef] [Green Version]
- Křížová, I.; Hadravová, R.; Štokrová, J.; Günterová, J.; Doležal, M.; Ruml, T.; Rumlová, M.; Pichová, I. The G-patch domain of Mason-Pfizer monkey virus is a part of reverse transcriptase. J. Virol. 2012, 86, 1988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strohalmová-Bohmová, K.; Spiwok, V.; Lepšík, M.; Hadravová, R.; Křížová, I.; Ulbrich, P.; Pichová, I.; Bednárová, L.; Ruml, T.; Rumlová, M. Role of Mason-Pfizer monkey virus CA-NC spacer peptide-like domain in assembly of immature particles. J. Virol. 2014, 88, 14148. [Google Scholar] [CrossRef] [Green Version]
- Goud, T.V.; Tutar, A.; Biellmann, J.-F. Synthesis of 8-heteroatom-substituted 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene dyes (BODIPY). Tetrahedron 2006, 62, 5084–5091. [Google Scholar] [CrossRef]
- Kim, D.; Ma, D.; Kim, M.; Jung, Y.; Kim, N.H.; Lee, C.; Cho, S.W.; Park, S.; Huh, Y.; Jung, J.; et al. Fluorescent labeling of protein using blue-emitting 8-amino-BODIPY derivatives. J. Fluoresc. 2017, 27, 2231–2238. [Google Scholar] [CrossRef]
- Chang, Y.-T.; Alamudi, S.H.; Satapathy, R.; Su, D. Background-free fluorescent probes for live cell imaging. US Patent WO2017078623A1, 2017. [Google Scholar]
- Qian, K.; Bori, I.D.; Chen, C.-H.; Huang, L.; Lee, K.-H. Anti-AIDS agents 90. Novel C-28 modified bevirimat analogues as potent HIV maturation inhibitors. J. Med. Chem. 2012, 55, 8128–8136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staudinger, H.; Meyer, J. Über neue organische Phosphorverbindungen III. Phosphinmethylenderivate und Phosphinimine. Helv. Chim. Acta 1919, 2, 635–646. [Google Scholar] [CrossRef] [Green Version]
- Neises, B.; Steglich, W. Simple method for the esterification of carboxylic acids. Angew. Chem. Int. Ed. 1978, 17, 522–524. [Google Scholar] [CrossRef]
- Kotsantis, P.; Silva, L.M.; Irmscher, S.; Jones, R.M.; Folkes, L.; Gromak, N.; Petermann, E. Increased global transcription activity as a mechanism of replication stress in cancer. Nat. Commun. 2016, 7, 13087. [Google Scholar] [CrossRef] [PubMed]
- Fantin, V.R.; St-Pierre, J.; Leder, P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 2006, 9, 425–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zielonka, J.; Joseph, J.; Sikora, A.; Hardy, M.; Ouari, O.; Vasquez-Vivar, J.; Cheng, G.; Lopez, M.; Kalyanaraman, B. Mitochondria-targeted triphenylphosphonium-based compounds: Syntheses, mechanisms of action, and therapeutic and diagnostic applications. Chem. Rev. 2017, 117, 10043–10120. [Google Scholar] [CrossRef] [PubMed]
- Evers, M.; Poujade, C.; Soler, F.; Ribeill, Y.; James, C.; Lelievre, Y.; Gueguen, J.C.; Reisdorf, D.; Morize, I.; Pauwels, R.; et al. Betulinic acid derivatives: A new class of human immunodeficiency virus type 1 specific inhibitors with a new mode of action. J. Med. Chem. 1996, 39, 1056–1068. [Google Scholar] [CrossRef]
- Kashiwada, Y.; Hashimoto, F.; Cosentino, L.M.; Chen, C.H.; Garrett, P.E.; Lee, K.H. Betulinic acid and dihydrobetulinic acid derivatives as potent anti-HIV agents. J. Med. Chem. 1996, 39, 1016–1017. [Google Scholar] [CrossRef]
- Soler, F.; Poujade, C.; Evers, M.; Carry, J.C.; Henin, Y.; Bousseau, A.; Huet, T.; Pauwels, R.; DeClercq, E.; Mayaux, J.F.; et al. Betulinic acid derivatives: A new class of specific inhibitors of human immunodeficiency virus type 1 entry. J. Med. Chem. 1996, 39, 1069–1083. [Google Scholar] [CrossRef]
- Li, F.; Goila-Gaur, R.; Salzwedel, K.; Kilgore, N.R.; Reddick, M.; Matallana, C.; Castillo, A.; Zoumplis, D.; Martin, D.E.; Orenstein, J.M.; et al. PA-457: A potent HIV inhibitor that disrupts core condensation by targeting a late step in Gag processing. Proc. Natl. Acad. Sci. USA 2003, 100, 13555–13560. [Google Scholar] [CrossRef] [Green Version]
- Schur, F.K.M.; Obr, M.; Hagen, W.J.H.; Wan, W.; Jakobi, A.J.; Kirkpatrick, J.M.; Sachse, C.; Krausslich, H.G.; Briggs, J.A.G. An atomic model of HIV-1 capsid-SP1 reveals structures regulating assembly and maturation. Science 2016, 353, 506–508. [Google Scholar] [CrossRef]
- Adamson, C.S.; Sakalian, M.; Salzwedel, K.; Freed, E.O. Polymorphisms in Gag spacer peptide 1 confer varying levels of resistance to the HIV-1 maturation inhibitor bevirimat. Retrovirology 2010, 7, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.X.; Salzwedel, K.; Wang, D.; Chakravarty, S.; Freed, E.O.; Wild, C.T.; Li, F. A single polymorphism in HIV-1 subtype C SP1 is sufficient to confer natural resistance to the maturation inhibitor bevirimat. Antimicrob. Agents Chemother. 2011, 55, 3324–3329. [Google Scholar] [CrossRef] [Green Version]
- Van Baelen, K.; Salzwedel, K.; Rondelez, E.; Van Eygen, V.; De Vos, S.; Verheyen, A.; Steegen, K.; Verlinden, Y.; Allaway, G.P.; Stuyver, L.J. Susceptibility of human immunodeficiency virus type 1 to the maturation inhibitor bevirimat is modulated by baseline polymorphisms in Gag spacer peptide 1. Antimicrob. Agents Chemother. 2009, 53, 2185–2188. [Google Scholar] [CrossRef] [Green Version]
- Coric, P.; Turcaud, S.; Souquet, F.; Briant, L.; Gay, B.; Royer, J.; Chazal, N.; Bouaziz, S. Synthesis and biological evaluation of a new derivative of bevirimat that targets the Gag CA-SP1 cleavage site. Eur. J. Med. Chem. 2013, 62, 453–465. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Lu, W.X.; Li, F. Pharmacological intervention of HIV-1 maturation. Acta Pharm. Sin. B 2015, 5, 493–499. [Google Scholar] [CrossRef] [Green Version]
- Gu, M.; Zhao, P.; Zhang, S.Y.; Fan, S.J.; Yang, L.; Tong, Q.C.; Ji, G.; Huan, C. Betulinic acid alleviates endoplasmic reticulum stress-mediated nonalcoholic fatty liver disease through activation of farnesoid X receptors in mice. Brit. J. Pharmacol. 2019, 176, 847–863. [Google Scholar] [CrossRef]
- Ye, Y.Q.; Zhang, T.; Yuan, H.Q.; Li, D.F.; Lou, H.X.; Fan, P.H. Mitochondria-targeted lupane triterpenoid derivatives and their selective apoptosis-inducing anticancer mechanisms. J. Med. Chem. 2017, 60, 6353–6363. [Google Scholar] [CrossRef] [PubMed]
- Dubinin, M.V.; Semenova, A.A.; Ilzorkina, A.I.; Mikheeva, I.B.; Yashin, V.A.; Penkov, N.V.; Vydrina, V.A.; Ishmuratov, G.Y.; Sharapov, V.A.; Khoroshavina, E.I.; et al. Effect of betulin and betulonic acid on isolated rat liver mitochondria and liposomes. Biochim. Biophys. Acta-Biomembr. 2020, 1862. [Google Scholar] [CrossRef] [PubMed]
- Dubinin, M.V.; Semenova, A.A.; Nedopekina, D.A.; Davletshin, E.V.; Spivak, A.Y.; Belosludtsev, K.N. Effect of F16-betulin conjugate on mitochondrial membranes and its role in cell death initiation. Membranes 2021, 11, 352. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Semenova, A.A.; Ilzorkina, A.I.; Penkov, N.V.; Nedopekina, D.A.; Sharapov, V.A.; Khoroshavina, E.I.; Davletshin, E.V.; Belosludtseva, N.V.; Spivak, A.Y.; et al. Mitochondria-targeted prooxidant effects of betulinic acid conjugated with delocalized lipophilic cation F16. Free Radic. Bio. Med. 2021, 168, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lu, X.C.; Zhu, R.L.; Zhang, K.X.; Li, S.; Chen, Z.J.; Li, L.X. Betulinic acid induces apoptosis in differentiated PC12 cells via ROS-mediated mitochondrial pathway. Neurochem. Res. 2017, 42, 1130–1140. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Cell Line a | BA | BT | 1 | 2 | 3 | 4 | 6 | 7 | 8 | 9 | 10 | 12 | 13 | 14 | 16 | 17 | 18 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CCRF-CEM | >50 | 12.82 | 8.98 | 8.14 | 0.21 | 23.65 | 1.55 | >50 | 8.18 | 22.48 | 2.92 | 9.62 | >50 | 0.29 | >50 | 5.76 | 8.61 |
| CEM-DNR | 23.05 | 22.17 | 16.84 | 10.23 | 1.22 | >50 | 11.53 | >50 | 9.24 | >50 | 7.00 | >50 | 4.76 | 0.35 | >50 | >50 | >50 |
| K562 | >50 | 23.60 | >50 | 25.99 | 0.90 | >50 | 5.25 | >50 | 19.18 | >50 | 23.19 | >50 | 5.09 | 0.40 | 47.90 | >50 | >50 |
| K562-Tax | >50 | 22.03 | 10.58 | 15.94 | 0.37 | >50 | 31.80 | >50 | 13.30 | >50 | 12.21 | >50 | 8.77 | 0.52 | >50 | >50 | >50 |
| A549 | 22.68 | 23.06 | >50 | 22.87 | 1.80 | >50 | 6.65 | 21.29 | 18.84 | >50 | 13.55 | >50 | 5.15 | 1.26 | 44.93 | >50 | 47.80 |
| HCT116 | >50 | 14.17 | >50 | 19.40 | 0.82 | >50 | 3.85 | >50 | 13.21 | >50 | 7.92 | >50 | 6.02 | 0.39 | 46.63 | >50 | 30.84 |
| HCT116p53−/− | >50 | 18.20 | >50 | 29.24 | 0.44 | >50 | 3.39 | >50 | 21.56 | >50 | 8.80 | >50 | 4.99 | 0.44 | 44.76 | >50 | 45.50 |
| U2OS | 29.69 | 27.63 | 29.04 | 22.22 | 0.89 | >50 | 5.00 | 18.39 | 17.16 | >50 | 12.38 | >50 | 4.16 | 0.42 | 44.62 | 44.58 | >50 |
| MRC-5 | >50 | >50 | >50 | 24.19 | 2.59 | >50 | 8.07 | 17.58 | 23.14 | >50 | 14.12 | >50 | 5.18 | 1.58 | 44.61 | >50 | >50 |
| BJ | >50 | >50 | >50 | 25.33 | 1.91 | >50 | 8.37 | 20.69 | 21.54 | >50 | 15.49 | >50 | 5.36 | 1.59 | 47.63 | >50 | >50 |
| Compound | <G1 | G0/G1 | S | G2/M | pH3Ser10 a | BrDU b | BrU c |
|---|---|---|---|---|---|---|---|
| control | 2.20 | 40.77 | 37.64 | 21.60 | 1.77 | 37.22 | 40.77 |
1  | 1.36 | 37.03 | 44.13 | 18.84 | 1.53 | 36.54 | 52.69 |
1  | 1.93 | 42.33 | 35.87 | 21.80 | 1.71 | 37.99 | 44.47 |
| 2 | 4.33 | 39.88 | 46.65 | 13.47 | 1.33 | 52.07 | 28.60 |
| 2 | 41.17 | 23.94 | 56.21 | 19.85 | 0.58 | 22.75 | 4.05 |
| 3 | 1.30 | 35.07 | 36.54 | 28.40 | 2.04 | 41.88 | 50.56 |
| 3 | 3.53 | 31.73 | 41.18 | 27.09 | 0.16 | 2.06 | 0.40 |
| 6 | 2.23 | 33.39 | 49.74 | 16.87 | 1.50 | 44.97 | 42.25 |
| 6 | 5.24 | 37.16 | 39.52 | 23.32 | 0.50 | 7.10 | 26.05 |
| 8 | 2.64 | 31.93 | 47.72 | 20.35 | 1.70 | 46.44 | 33.61 |
| 8 | 39.65 | 40.78 | 37.66 | 21.56 | 1.25 | 8.51 | 1.85 |
| 10 | 3.80 | 37.98 | 44.59 | 17.43 | 1.44 | 42.25 | 35.04 |
| 10 | 10.69 | 44.58 | 42.32 | 13.10 | 1.25 | 10.18 | 13.02 |
| 12 | 2.45 | 33.64 | 43.16 | 23.20 | 1.40 | 59.73 | 41.98 |
| 12 | 3.01 | 29.75 | 56.69 | 13.55 | 0.68 | 64.07 | 34.00 |
| 14 | 8.63 | 21.46 | 56.29 | 22.25 | 0.15 | 24.55 | 1.56 |
| 14 | 8.16 | 27.28 | 51.82 | 20.90 | 0.18 | 0.52 | 1.53 |
| 17 | 2.89 | 34.83 | 46.96 | 18.21 | 1.56 | 48.07 | 26.70 |
| 17 | 4.04 | 43.42 | 38.47 | 18.10 | 1.49 | 29.41 | 3.39 |
| 18 | 2.06 | 36.06 | 44.31 | 19.63 | 1.25 | 55.81 | 46.98 |
| 18 | 2.97 | 38.01 | 44.12 | 17.87 | 1.55 | 49.64 | 70.23 |
) and 5 × IC50 ( ) values. a phospho-Histone (Ser10); b 5-bromo-2-deoxyuridine; c BrU, 5-bromouridine.| Compd. | 1 | 2 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 12 | 13 | 15 | 16 | 17 | 18 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
 IC50 [μM] IC50 [μM] | >40 | 36.4 | >40 | >40 | 12.0 | >40 | 37.8 | >40 | >40 | >40 | >40 | >40 | >40 | >40 | >40 |
 IC50i [μM] IC50i [μM] | >50 | 11.7 | 44.1 | >50 | n.d. | >50 | 1.4 | 14.0 | 8.4 | 31.9 | >50 | >50 | 9.1 | 7.6 | 7.1 |
). To determine the effect of the compounds on HIV-1 infectivity ( ), HEK 293 cells were transfected with the lentiviral vectors and treated with the tested compounds. The cells producing HIV-1 particles in the presence or absence of DMSO (at a final concentration of 1%) were used as controls. At 48 h post-transfection, the content of HIV-1 capsid (CA) protein from the culture media was quantified by ELISA and normalized amounts of VSV-G pseudotyped HIV-1 viruses were used to infect fresh HEK 293 cells. HIV-1 infectivity was determined 48 h later by quantification of GFP-producing cells by flow cytometry. The 50% infection inhibition (IC50i) was defined as the concentration of the compound that reduced the HIV-1 infectivity by 50% compared to the untreated controls.Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kodr, D.; Stanková, J.; Rumlová, M.; Džubák, P.; Řehulka, J.; Zimmermann, T.; Křížová, I.; Gurská, S.; Hajdúch, M.; Drašar, P.B.; et al. Betulinic Acid Decorated with Polar Groups and Blue Emitting BODIPY Dye: Synthesis, Cytotoxicity, Cell-Cycle Analysis and Anti-HIV Profiling. Biomedicines 2021, 9, 1104. https://doi.org/10.3390/biomedicines9091104
Kodr D, Stanková J, Rumlová M, Džubák P, Řehulka J, Zimmermann T, Křížová I, Gurská S, Hajdúch M, Drašar PB, et al. Betulinic Acid Decorated with Polar Groups and Blue Emitting BODIPY Dye: Synthesis, Cytotoxicity, Cell-Cycle Analysis and Anti-HIV Profiling. Biomedicines. 2021; 9(9):1104. https://doi.org/10.3390/biomedicines9091104
Chicago/Turabian StyleKodr, David, Jarmila Stanková, Michaela Rumlová, Petr Džubák, Jiří Řehulka, Tomáš Zimmermann, Ivana Křížová, Soňa Gurská, Marián Hajdúch, Pavel B. Drašar, and et al. 2021. "Betulinic Acid Decorated with Polar Groups and Blue Emitting BODIPY Dye: Synthesis, Cytotoxicity, Cell-Cycle Analysis and Anti-HIV Profiling" Biomedicines 9, no. 9: 1104. https://doi.org/10.3390/biomedicines9091104
APA StyleKodr, D., Stanková, J., Rumlová, M., Džubák, P., Řehulka, J., Zimmermann, T., Křížová, I., Gurská, S., Hajdúch, M., Drašar, P. B., & Jurášek, M. (2021). Betulinic Acid Decorated with Polar Groups and Blue Emitting BODIPY Dye: Synthesis, Cytotoxicity, Cell-Cycle Analysis and Anti-HIV Profiling. Biomedicines, 9(9), 1104. https://doi.org/10.3390/biomedicines9091104