
Small Extracellular Vesicle Enrichment of a Retrotransposon-Derived Double-Stranded RNA: A Means to Avoid Autoinflammation?


 and
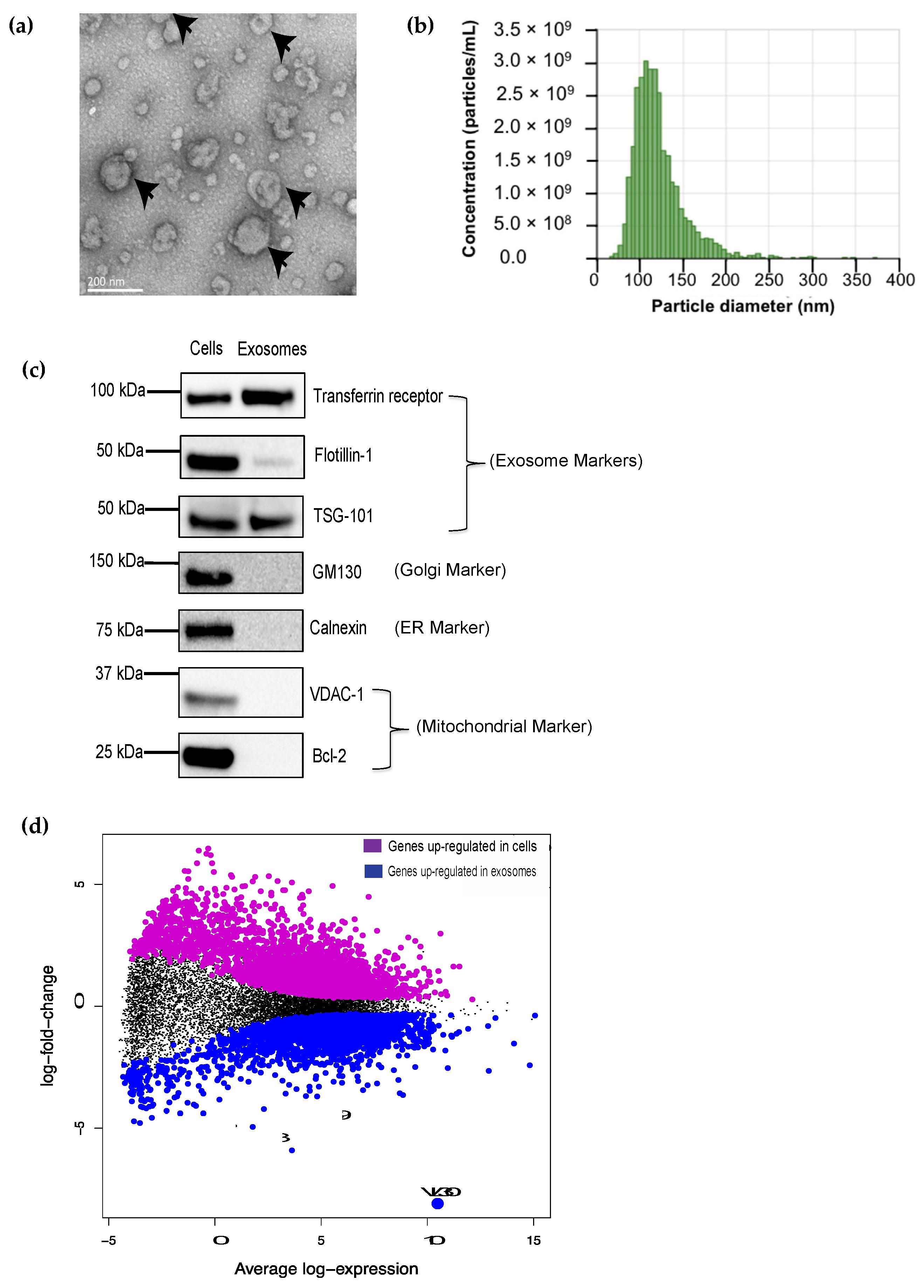
and 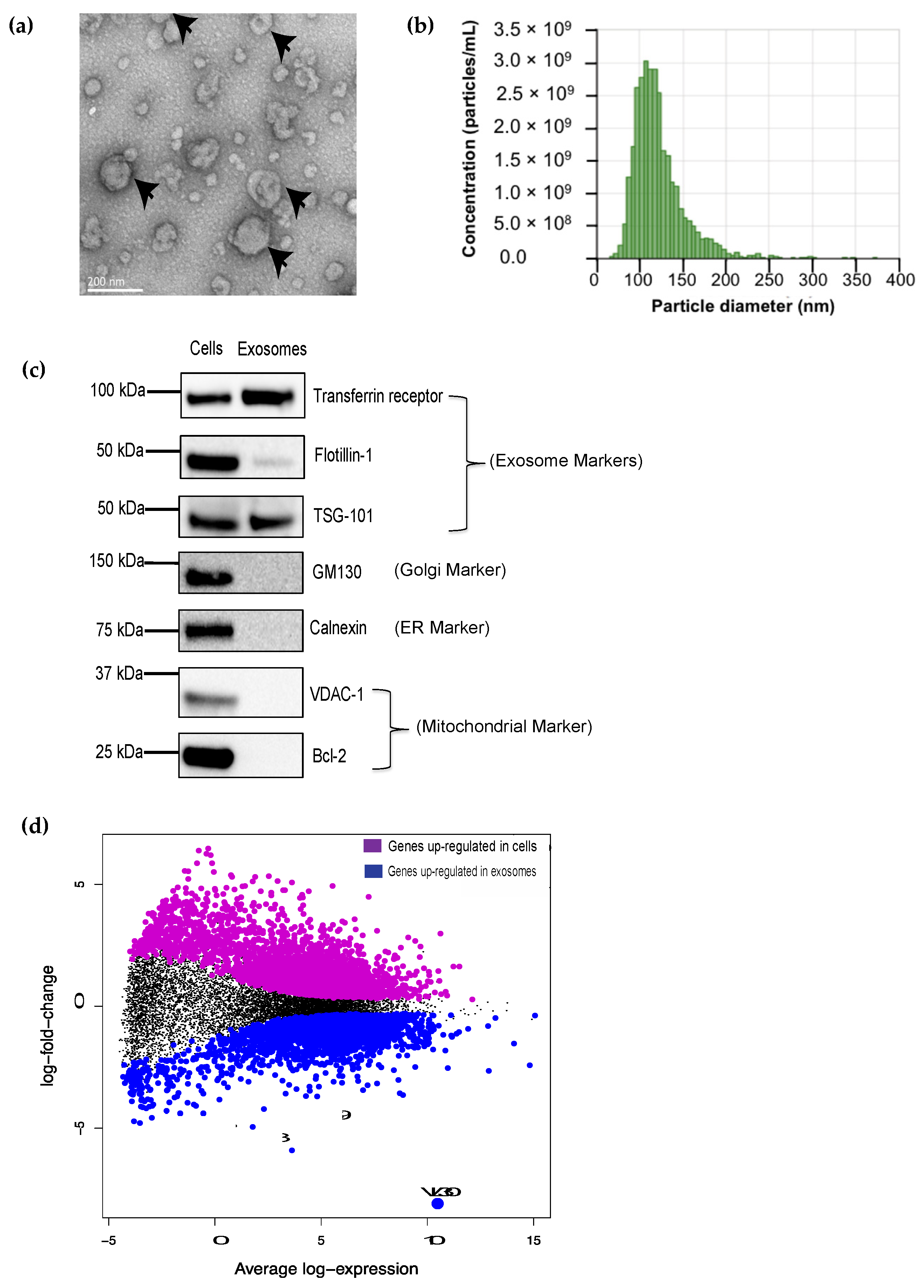{kind=link}
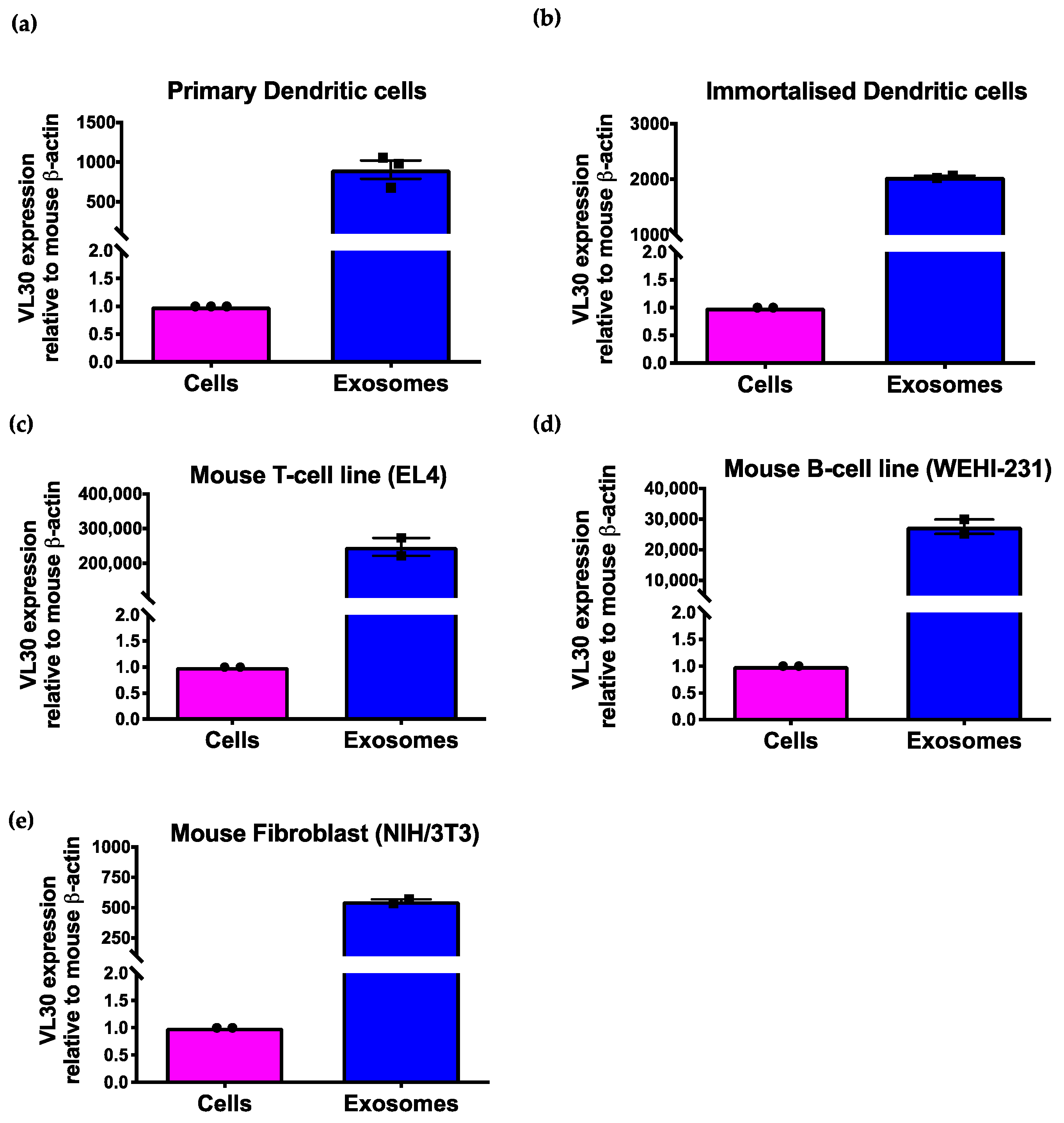{kind=link}
{kind=link}
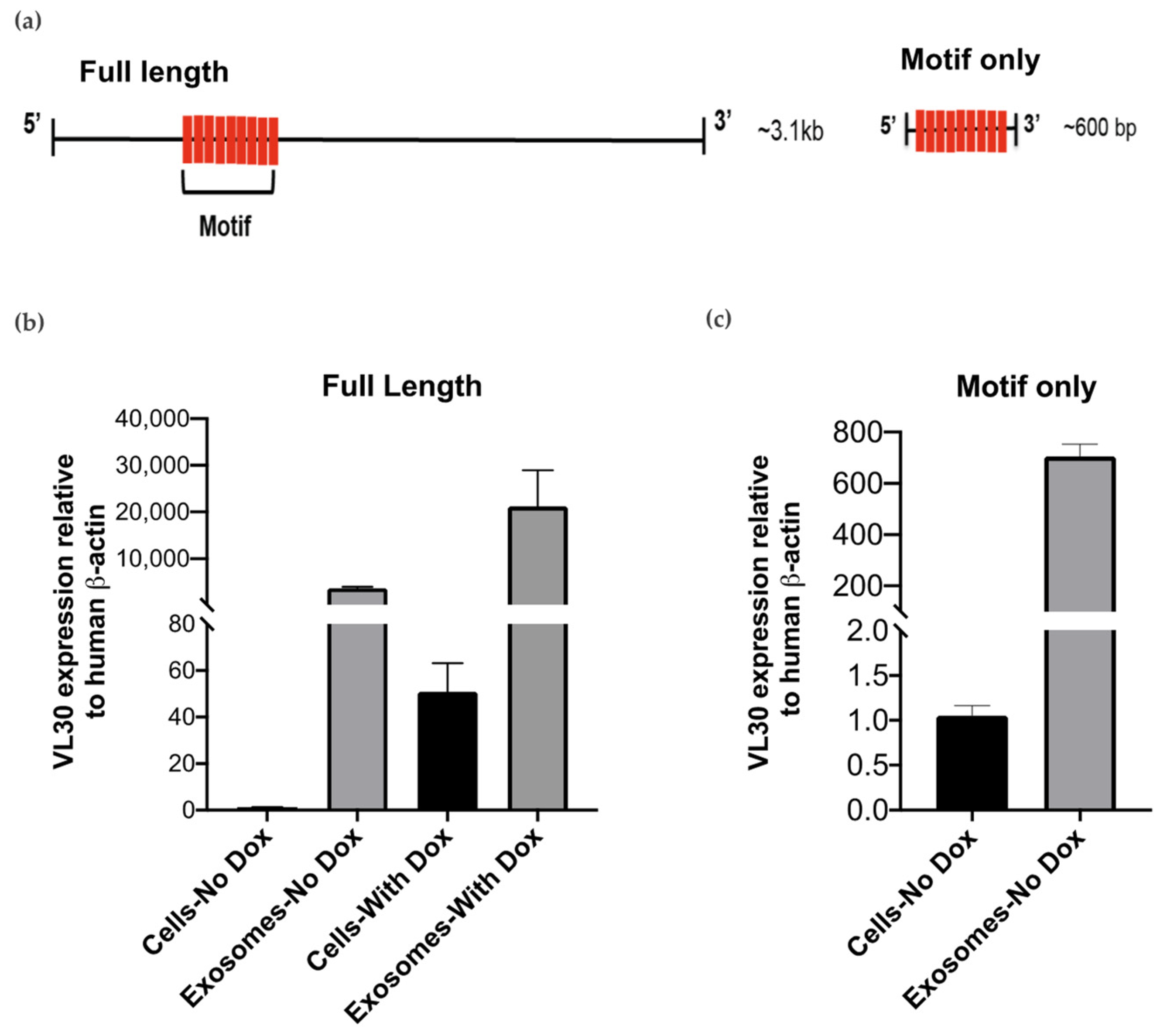{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice and Ethics
2.2. Cell Culture
2.3. SEV Isolation
2.4. SEV Size Assessment
2.5. RNA Isolation
2.6. RNA-Seq
2.7. QRT-PCR
2.8. Transmission Electron Microscopy (TEM)
2.9. VL30 Cloning
2.10. Motif Discovery Search and RNA Secondary Structure Prediction
2.11. Western Blots
2.12. Statistics
3. Results
3.1. The VL30 lncRNA Is Enriched in DC SEVs
3.2. The VL30 lncRNA Is Enriched in SEVs from Multiple Cell Types
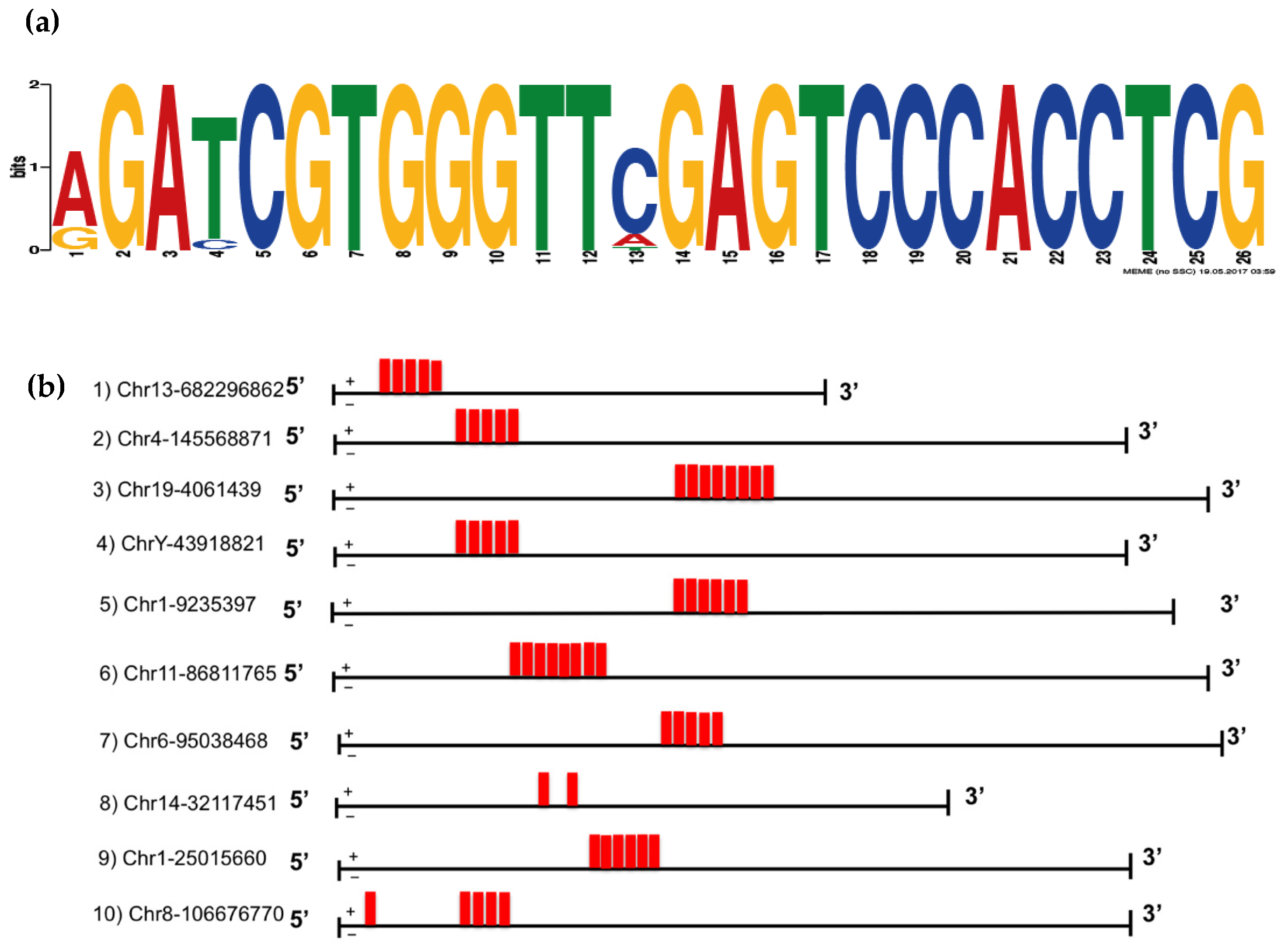
3.3. VL30 RNA Isoforms Enriched in SEVs Contain a Repeated Sequence Motif
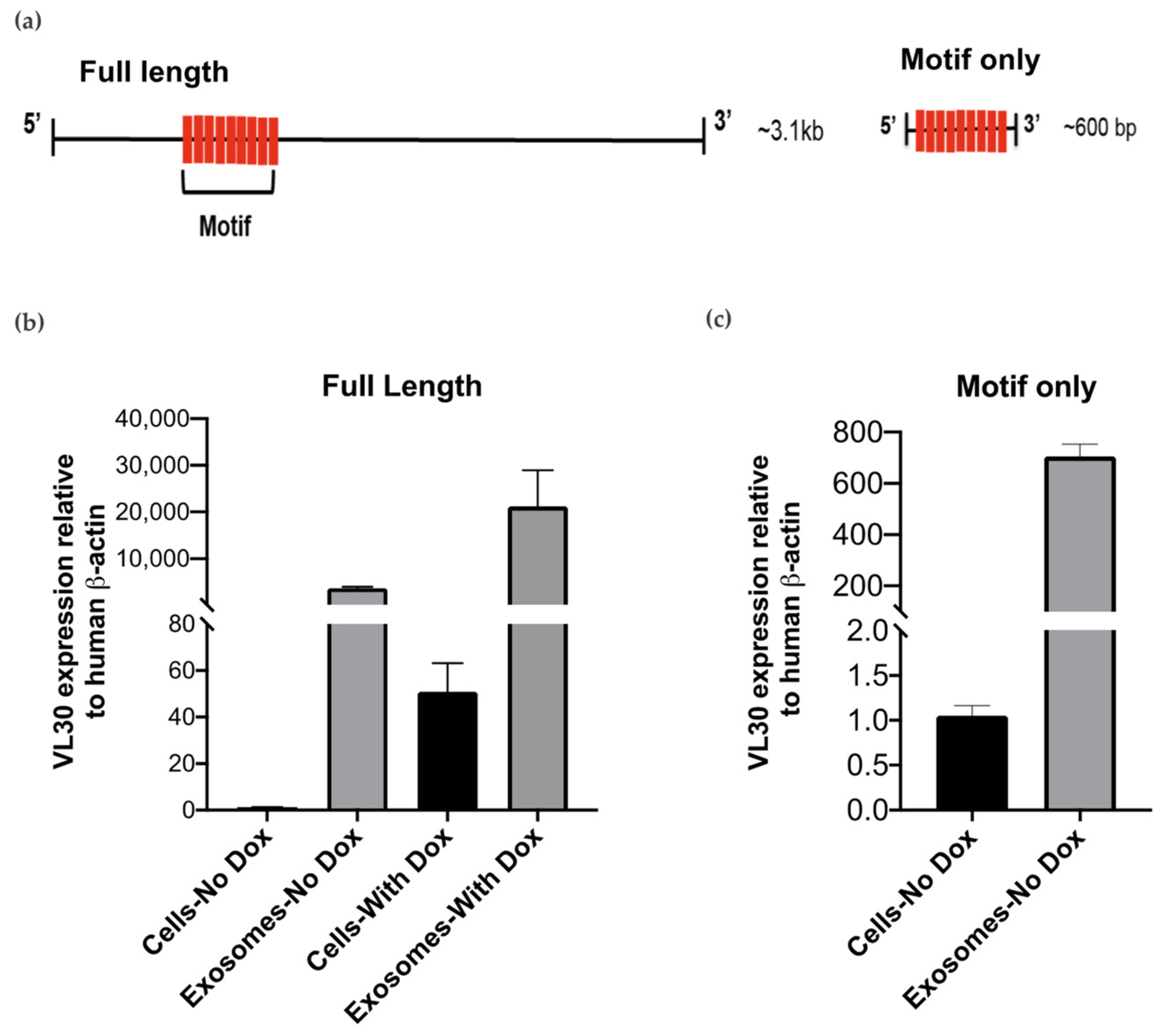
3.4. A VL30 Sequence Containing the Repeated Motif Alone Is Efficiently Incorporated into SEVs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Johnstone, R.M. Maturation of reticulocytes: Formation of exosomes as a mechanism for shedding membrane proteins. Biochem. Cell Biol. 1992, 70, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.M.; Sawant, S.S.; Kunda, N.K. Exosomes as drug delivery systems: A brief overview and progress update. Eur. J. Pharm. Biopharm. 2020, 154, 259–269. [Google Scholar] [CrossRef]
- Bunggulawa, E.J.; Wang, W.; Yin, T.; Wang, N.; Durkan, C.; Wang, Y.; Wang, G. Recent advancements in the use of exosomes as drug delivery systems. J. Nanobiotechnol. 2018, 16, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Aslan, C.; Kiaie, S.H.; Zolbanin, N.M.; Lotfinejad, P.; Ramezani, R.; Kashanchi, F.; Jafari, R. Exosomes for mRNA delivery: A novel biotherapeutic strategy with hurdles and hope. BMC Biotechnol. 2021, 21, 1–12. [Google Scholar] [CrossRef]
- O’Brien, K.; Breyne, K.; Ughetto, S.; Laurent, L.C.; Breakefield, X.O. RNA delivery by extracellular vesicles in mammalian cells and its applications. Nat. Rev. Mol. Cell Biol. 2020, 21, 585–606. [Google Scholar] [CrossRef]
- Koppers-Lalic, D.; Hackenberg, M.; Bijnsdorp, I.V.; van Eijndhoven, M.A.; Sadek, P.; Sie, D.; Zini, N.; Middeldorp, J.; Ylstra, B.; de Menezes, R.X.; et al. Nontemplated nucleotide additions distinguish the small RNA composition in cells from exosomes. Cell Rep. 2014, 8, 1649–1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villarroya-Beltri, C.; Baixauli, F.; Gutiérrez-Vázquez, C.; Sánchez-Madrid, F.; Mittelbrunn, M. Sorting it out: Regulation of exosome loading. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2014; Volume 28, pp. 3–13. [Google Scholar] [CrossRef] [Green Version]
- Villarroya-Beltri, C.; Gutierrez-Vazquez, C.; Sanchez-Cabo, F.; Pérez-Hernández, D.; Vázquez, J.; Martin-Cofreces, N.; Martinez-Herrera, D.J.; Pascual-Montano, A.; Mittelbrunn, M.; Sánchez-Madrid, F. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat. Commun. 2013, 4, 2980. [Google Scholar] [CrossRef] [Green Version]
- Groot, M.; Lee, H. Sorting mechanisms for MicroRNAs into extracellular vesicles and their associated diseases. Cells 2020, 9, 1044. [Google Scholar] [CrossRef] [PubMed]
- Bolukbasi, M.F.; Mizrak, A.; Ozdener, G.B.; Madlener, S.; Ströbel, T.; Erkan, E.P.; Fan, J.B.; Breakefield, X.O.; Saydam, O. miR-1289 and “Zipcode”-like Sequence Enrich mRNAs in Microvesicles. Mol. Ther.-Nucleic Acids 2012, 1, e10. [Google Scholar] [CrossRef]
- Batagov, A.O.; Kuznetsov, V.A.; Kurochkin, I.V. Identification of nucleotide patterns enriched in secreted RNAs as putative cis-acting elements targeting them to exosome nano-vesicles. BMC Genom. 2011, 12, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Kossinova, O.A.; Gopanenko, A.V.; Tamkovich, S.; Krasheninina, O.; Tupikin, A.E.; Kiseleva, E.; Yanshina, D.D.; Malygin, A.; Ven’Yaminova, A.G.; Kabilov, M.; et al. Cytosolic YB-1 and NSUN2 are the only proteins recognizing specific motifs present in mRNAs enriched in exosomes. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2017, 1865, 664–673. [Google Scholar] [CrossRef]
- Pitt, J.M.; André, F.; Amigorena, S.; Soria, J.-C.; Eggermont, A.; Kroemer, G.; Zitvogel, L. Dendritic cell–derived exosomes for cancer therapy. J. Clin. Investig. 2016, 126, 1224–1232. [Google Scholar] [CrossRef]
- Maas, S.L.N.; De Vrij, J.; Broekman, M.L.D. Quantification and size-Profiling of extracellular vesicles using tunable resistive pulse sensing. J. Vis. Exp. 2014, 51623, e51623. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. Feature counts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [Green Version]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef] [Green Version]
- Phipson, B.; Lee, S.; Majewski, I.; Alexander, W.S.; Smyth, G.K. Robust hyperparameter estimation protects against hypervariable genes and improves power to detect differential expression. Ann. Appl. Stat. 2016, 10, 946–963. [Google Scholar] [CrossRef]
- Bellingham, S.A.; Coleman, B.M.; Hill, A.F. Small RNA deep sequencing reveals a distinct miRNA signature released in exosomes from prion-infected neuronal cells. Nucleic Acids Res. 2012, 40, 10937–10949. [Google Scholar] [CrossRef] [Green Version]
- Bailey, T.L.; Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in bipolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994, 2, 28–36. [Google Scholar]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Boussac, M.; Véron, P.; Ricciardi-Castagnoli, P.; Raposo, G.; Garin, J.; Amigorena, S. Proteomic analysis of dendritic cell-derived exosomes: A secreted subcellular compartment distinct from apoptotic vesicles. J. Immunol. 2001, 166, 7309–7318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Sui, A.; Garen, A. Binding of mouse VL30 retrotransposon RNA to PSF protein induces genes repressed by PSF: Effects on steroidogenesis and oncogenesis. Proc. Natl. Acad. Sci. USA 2004, 101, 621–626. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Wang, B.; Bromberg, M.; Hu, Z.; Konigsberg, W.; Garen, A. Retroviral-mediated transmission of a mouse VL30 RNA to human melanoma cells promotes metastasis in an immunodeficient mouse model. Proc. Natl. Acad. Sci. USA 2002, 99, 6269–6273. [Google Scholar] [CrossRef] [Green Version]
- Markopoulos, G.; Noutsopoulos, D.; Mantziou, S.; Gerogiannis, D.; Thrasyvoulou, S.; Vartholomatos, G.; Kolettas, E.; Tzavaras, T. Genomic analysis of mouse VL30 retrotransposons. Mob. DNA 2016, 7, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreux, M.; Garaigorta, U.; Boyd, B.; Décembre, E.; Chung, J.; Whitten-Bauer, C.; Wieland, S.; Chisari, F.V. Short-Range exosomal transfer of viral rna from infected cells to plasmacytoid dendritic cells triggers innate immunity. Cell Host Microbe 2012, 12, 558–570. [Google Scholar] [CrossRef] [Green Version]
- Skog, J.; Würdinger, T.; Van Rijn, S.; Meijer, D.H.; Gainche, L.; Curry, W.T., Jr.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nature 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- Darlix, J.-L.; Torrent, C. Retroviral Vectors Comprising a VL30-Derived psi Region. U.S. Patent 5,747,323, 5 May 1998. [Google Scholar]
- Torrent, C.; Gabus, C.; Darlix, J.L. A small and efficient dimerization/packaging signal of rat VL30 RNA and its use in murine leukemia virus-VL30-derived vectors for gene transfer. J. Virol. 1994, 68, 661–667. [Google Scholar] [CrossRef] [Green Version]
- Gould, S.J.; Booth, A.M.; Hildreth, J.E.K. The Trojan exosome hypothesis. Proc. Natl. Acad. Sci. USA 2003, 100, 10592–10597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelchen-Matthews, A.; Raposo, G.; Marsh, M. Endosomes, exosomes and Trojan viruses. Trends Microbiol. 2004, 12, 310–316. [Google Scholar] [CrossRef]
- Dzananovic, E.; Patel, T.R.; Deo, S.; McEleney, K.; Stetefeld, J.; McKenna, S.A. Recognition of viral RNA stem-loops by the tandem double-stranded RNA binding domains of PKR. RNA 2013, 19, 333–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayanan, A.; Iordanskiy, S.; Das, R.; Van Duyne, R.; Santos, S.; Jaworski, E.; Guendel, I.; Sampey, G.; Dalby, E.; Iglesias-Ussel, M.; et al. Exosomes derived from HIV-1-infected cells contain trans-Activation response element RNA. J. Biol. Chem. 2013, 288, 20014–20033. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.; Smith, B.R.; Tate, M.; Belz, G.; Barrios, M.H.; Elgass, K.D.; Weisman, A.S.; Baker, P.J.; Preston, S.; Whitehead, L.; et al. SIDT2 Transports extracellular dsRNA into the cytoplasm for innate immune recognition. Immunity 2017, 47, 498–509. [Google Scholar] [CrossRef] [Green Version]
- Sampey, G.C.; Saifuddin, M.; Schwab, A.; Barclay, R.; Punya, S.; Chung, M.-C.; Hakami, R.M.; Zadeh, M.A.; Lepene, B.; Klase, Z.A.; et al. Exosomes from HIV-1-infected cells stimulate production of pro-inflammatory cytokines through trans-activating response (TAR) RNA. J. Biol. Chem. 2016, 291, 1251–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlee, M.; Hartmann, G. Discriminating self from non-self in nucleic acid sensing. Nat. Rev. Immunol. 2016, 16, 566–580. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barrios, M.H.; Garnham, A.L.; Foers, A.D.; Cheng-Sim, L.; Masters, S.L.; Pang, K.C. Small Extracellular Vesicle Enrichment of a Retrotransposon-Derived Double-Stranded RNA: A Means to Avoid Autoinflammation? Biomedicines 2021, 9, 1136. https://doi.org/10.3390/biomedicines9091136
Barrios MH, Garnham AL, Foers AD, Cheng-Sim L, Masters SL, Pang KC. Small Extracellular Vesicle Enrichment of a Retrotransposon-Derived Double-Stranded RNA: A Means to Avoid Autoinflammation? Biomedicines. 2021; 9(9):1136. https://doi.org/10.3390/biomedicines9091136
Chicago/Turabian StyleBarrios, Marilou H., Alexandra L. Garnham, Andrew D. Foers, Lesley Cheng-Sim, Seth L. Masters, and Ken C. Pang. 2021. "Small Extracellular Vesicle Enrichment of a Retrotransposon-Derived Double-Stranded RNA: A Means to Avoid Autoinflammation?" Biomedicines 9, no. 9: 1136. https://doi.org/10.3390/biomedicines9091136
APA StyleBarrios, M. H., Garnham, A. L., Foers, A. D., Cheng-Sim, L., Masters, S. L., & Pang, K. C. (2021). Small Extracellular Vesicle Enrichment of a Retrotransposon-Derived Double-Stranded RNA: A Means to Avoid Autoinflammation? Biomedicines, 9(9), 1136. https://doi.org/10.3390/biomedicines9091136