1. Introduction
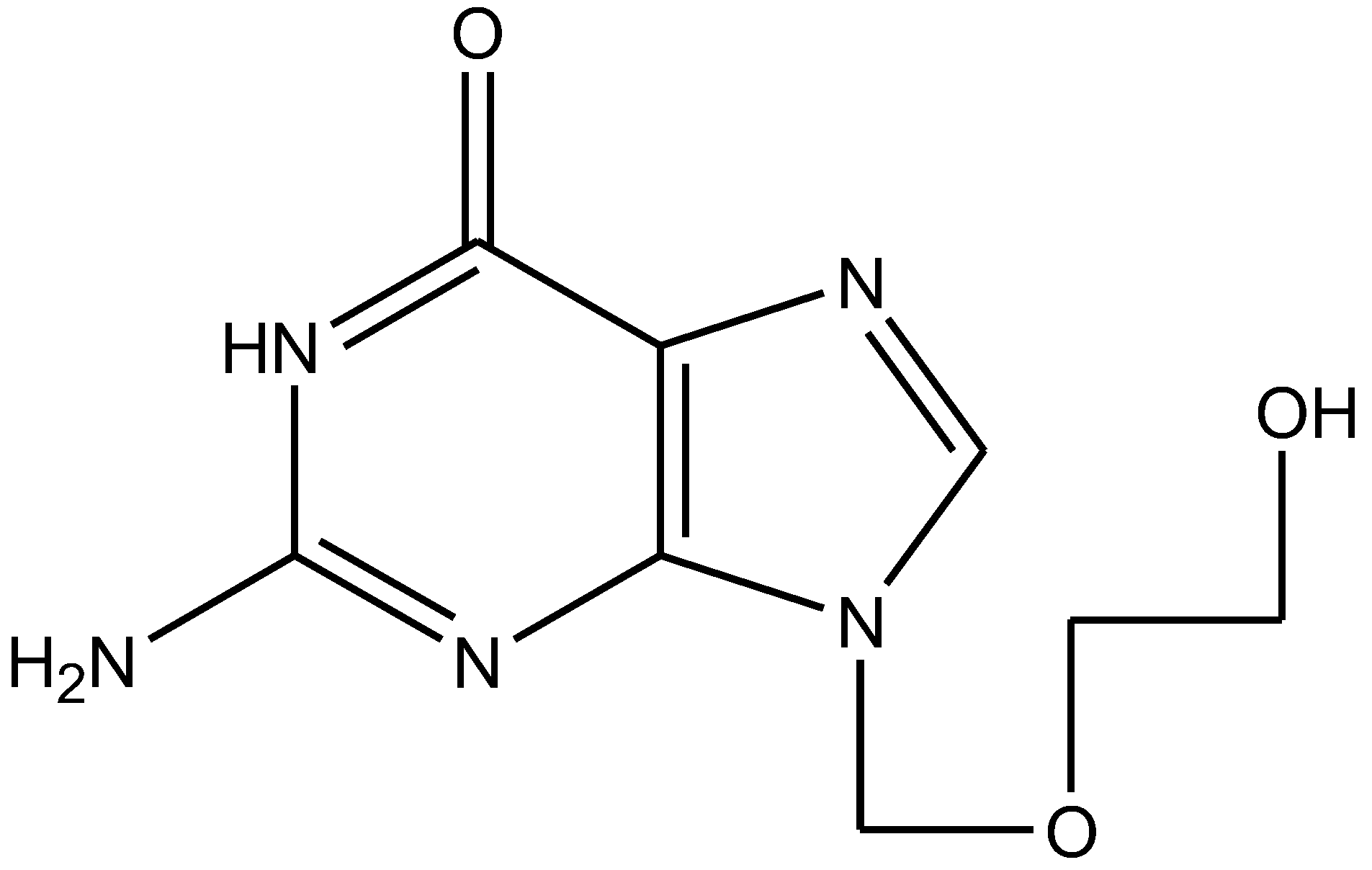
Acyclovir (ACV) 9-[(2-hydroxyethoxy)-methyl]-guanosine, is an acyclic guanosine derivative (
Figure 1), which exhibits a selective inhibition of herpes virus replication with potent clinical anti-viral activity against the herpes simplex virus (HSV) and varicella-zoster viruses [
1]. The Food and Drug Administration (FDA) approved treatment of genital herpes and HSV encephalitis [
2]. ACV is the first-line treatment for HSV encephalitis. Currently, there are no other medications indicated for the treatment of this condition [
3]. ACV incorporates itself into viral deoxyribonucleic acid (DNA), preventing further synthesis as it inhibits DNA synthesis and viral replication after it is converted to acyclovir triphosphate by viral and cellular enzymes and is available in both oral and intravenous doses [
4].
The analysis method is a quantitative measurement of ACV in biological fluids, such as plasma, serum, urine, or tissue extracts [
5]. Chromatography is an analytical method, wherein mainly high-performance liquid chromatography (HPLC) is widely used for quantification of pharmaceutical ACV in biological samples. The HPLC method is considered as an advanced analytical technique of liquid chromatography (LC), a technique of high precision, sensitivity, and specificity in the quantification of components [
6]. HPLC allows the quantification of the targeted drug independently from blood and its metabolites in a very short span of time. Different HPLC methods were published for determination of acyclovir in human serum using a UV detector [
7,
8]. Development of the bioanalytical method is to provide an assessment and evaluation of ACV bioavailability (in plasma). All components (parameters) were selected for the analysis of a considerably high concentration (C
max) of the ACV in plasma. Furthermore, this bioanalytical method can be useful in tissue distribution, toxicity (toxicokinetics), and drug therapeutic monitoring. The chromatography technique involves steps such as (i) plasma separation, (ii) preparation, (iii) extraction, (iv) selection of suitable organic/inorganic solvents, and (v) analyte separation and its validation [
9].
Over the years, several methods were reported with the quantification of ACV in biological systems, employing RP-HPLC, ultra-filtration HPLC, fluorescent techniques, Raman spectroscopy, micellar electrokinetic chromatography, and UV spectroscopy [
10,
11,
12,
13,
14,
15,
16,
17]. However, these methods are inefficient and uneconomical due to the use of high-cost solvents or complicated buffers with low active ACV detection limits. This manuscript addressed the quantification of ACV using HPLC with photo diode array (PDA) and integrating the quality by design (QbD) tool for high robustness, ruggedness, and flexibility. A QbD approach is a systemic step for the method development with predefined objectives and an understanding of product and process variables (process controls) based on the sound science and quality risk management, in order to achieve a high quality product, flexible process, regulatory flexibility, and improvement [
18]. Analytical science is an integral part of pharmaceutical product development and is defined as a science and risk-based paradigm to understand predefined objectives and critical process variables. This results in the intended outputs over the entire product life cycle [
19]. Thus, a QbD approach is used to reduce time and energy to revalidate, redevelop, and retransfer while tech transfers.
This method was optimized and validated in accordance with the FDA guidelines. The developed method gives better recovery from plasma (>90.0%), and a very low detection limit (LLOD; 50 ng). The study aimed to apply QbD analytical science to understand the critical process variables and set low-risk analysis with high robustness and analysis flexibility during method development of ACV throughout the product development life cycle. To optimize process parameters for simplified application and cost reduction, it was interesting to apply the quality by design tool (software) in the method development and validation not reported so far for the drug (ACV in biological matrix) [
20,
21]. This will avoid unnecessary use of organic solvents in method development, suitable to scale up for large-scale industrial analysis, easy revalidation, high robustness, and rapid tech transfer at low energy and in a short time.
Furthermore, LLOD from earlier published reports were 1.06 µg/mL and 156 ng/mL [
22,
23]. The novelty and uniqueness of the current study employs an integrated method of experimental designs and simulations to determine the appropriate chromatographic method for the drug analysis in biological samples. Moreover, in silico solubility analysis was carried out of ACV in various solvents using Hansen solubility parameters (HSP) for the confirmation of components of the mobile phase. In this study, we addressed the HPLC method development for quantification of ACV in rat plasma and subsequently, the pharmacokinetic study of transdermally applied vesicular systems (elastic liposomes and ethosomes) (previously published) (ELP3, ETHO2, ELP3 gel, and ETHO2 gel) [
22]. Bioanalytical method validation, along with design expert and in silico model of solubility tools, were extensively employed in the development of an accurate, sensitive, specific, and precise HPLC method with a PDA detector.
2. Materials
ACV (99.8 % pure) was obtained as a kind gift sample from Cipla Pharmaceuticals (Mumbai, Maharashtra, India). Analytical grade potassium dihydrogen phosphate was purchased from S.D. Fine Chemicals Ltd. Mumbai, Maharashtra, India. Acetonitrile, ethyl acetate and methanol HPLC grade were obtained from Sigma-Aldrich, Mumbai, India. HPLC-grade triple distilled water was prepared by using Milli-Q system, Millipore, Chandigarh, Punjab, India.
3. Methods
3.1. Estimation of Hansen Solubility Parameters (HSP) for Determination of Maximum Solubility for Mobile Phase Selection
The drug is poorly soluble in water due to lipophilic nature and nucleoside analogue. The HSPiP program (version 5.02.6, UK) was used to predict the most suitable solvent for analytical purposes. In general, Hansen solubility parameters are based on the physicochemical nature of solute and solvent in terms of dispersion, polarity, and hydrogen bonding formation capacity. These properties are innate in a molecule due to functional groups (number of hydrogen bonding acceptor counts and hydrogen bonding acceptor counts). The idea of solubility parameters was first discovered by Hildebrand and Scott by considering the solute behavior in a specific solvent [
24,
25]. Hansen expressed these three interactive forces of dispersion, polarity, and hydrogen bonding capacity as δ
d, δ
p, and δ
h, respectively. The total cohesive force (δ
t) of solute or solvent was the sum of the square of these forces, as in Equation (1).
Experimental solubility of ACV was carried out using a shaker water bath at 30 °C. In brief, a weighed quantity of the drug was added to a 5 mL of solvent contained in a clear glass vial. The vial was tightly closed and placed in the water bath at a constant temperature. Saturated solubility was conducted for 72 h to attain equilibrium in each solvent. After achieving equilibrium, the mixture was filtered and the filtrate was used for the drug assay using a UV Vis spectrophotometer (Shimadzu U-1800, Tokyo, Japan) at 256 nm. Experiments were replicated to get mean and standard deviation.
3.2. Instrumentation and Chromatographic Conditions
The HPLC system consisted of a separation module (Alliance e2695, Waters, Waters Corporation, 34 Maple street, Milford, MA, USA), an auto-sampler, a thermostat, and a 2998 photodiode array (PDA) detector. The HPLC method development and validation was carried out using a C18 column (130 × 3.6 mm, 5 µm; Waters, India). The composition of the mobile phase was potassium dihydrogen phosphate (pH 6.8): acetonitrile (ACN) in a 90:10 ratio operating in isocratic mode. The drug was detected in the wavelength range of 200–400 nm using a PDA detector and maximum detection was obtained at 256 nm. Furthermore, analytical data were processed employing Empower 2 (Waters) software (Waters Corporation, Milford, MA, USA).
3.3. Sample Preparation (Extraction Method)
Wistar rats of either sex weighing around 200–220 g were used for pharmacokinetic and validation purposes. The study protocol was approved (PCTE/LDH/1370/2013) by the Institute Animal Ethics Committee of the PCTE Institute of Pharmacy, Ludhiana, Punjab, India. The HPLC method development and validation experiments were started after taking blood from the retro-orbital plexus of eye, and the plasma was separated by cold centrifuge at 2000 rpm for 10 min. After centrifugation supernatant (plasma) was separated out and methanol was added for protein precipitation (settled down) and the remaining (clear supernatant) blank plasma was stored at −20 °C. A stock solution of ACV was prepared by dissolving the drug in methanol and ACV samples in plasma were prepared by spiking with 10 μL of each of the appropriate working dilutions of ACV, followed by vortexing it, and resulting plasma sample concentrations were in the range of 100–1000 ng/mL. These samples were subjected to HPLC analysis by injecting 10 µL of sample into the injection port. The AUC for each peak obtained was plotted against concentration to make the standard curve of ACV in plasma [
26]. Furthermore, quality control (QC) samples at various (three) concentration levels (lower QC, middle QC, and higher QC for ACV (200, 500, and 1000 ng/mL)) were prepared by diluting the stock solutions in an appropriate volume of mobile phase.
3.4. Screening Studies (Pre-Optimization)
Seven factors at two levels of the Taguchi design (
Table 1) were tried, and finally, two factors (at three levels) were employed in Design Expert (Stat-Ease Inc., 1300 Godward St NE #6400, Minneapolis, MN, USA). Several variables of Taguchi design (injection volume, flow rate, pH of buffer, ACN concentration, buffer concentration, and sampler temperature) at different levels viz. (−1) low and (+1) high were used to investigate the prime factor affecting analysis resolution in HPLC peak. Half normal and pareto charts were obtained, which revealed factors that most significantly influenced the method validation [
27]. Acyclovir is a chemically purine nucleotide (weak acid and basic groups) with slight aqueous solubility (insoluble in alcohol). The drug is associated with two pka values (pka = 2.52 and pka = 9.35) and classified as an ampholytic drug due to the dependency of solubility at ionization constant and medium pH [
28]. Therefore, we considered several deciding factors, such as (a) injection volume, (b) flow rate, (c) pH of buffer, (d) ACN concentration, (d) buffer concentration, and (e) sampler temperature (temperature dependent solubility), which were responsible for having an impact on process development for analysis. In order to minimize the variability around the analysis process, the Taguchi model (a robust design) was used as a tool to determine sets of controllable factors among these. To achieve a range of major factors, these were selected and each factor was assessed by keeping constant in other trial runs. This provided us buffer concentration and flow as prime factors affecting peak intensity for remarkable resolution and flow rate to obtain at an optimized time point. Other factors, such as column temperature, ACN content, and injection volume, had no impact on the resolution of the characteristic peak. Notably, increase in injection volume proportionally increased peak height and broad base. These were used as input factor parameters for central composite design (CCD) design.
3.5. Optimization Studies
Two factors (flow rate and buffer concentration) at three levels (low, medium, and high) in the central composite design (CCD) were finally selected to study the effect of significant factors/variables, which directly influence the responses/attributes. Thirteen runs with three levels were carried out at low (−1), medium (0), and high (+1) levels employed during each experimental run of the CCD matrix (
Table 2).
3.6. HPLC Method Validation
The quantitative method validation was in accordance with ICH Q2 (R1) guidelines. The optimized chromatographic method was validated for the parameters, such as linearity, accuracy, precision, sensitivity, and system suitability with lower limit of detection (LLOD) and lower limit of quantification (LLOQ) [
29].
3.7. Linearity and Range
Linearity was established in rat plasma by spiking the amount of ACV and appropriate dilutions were made with the mobile phase, ranging from 100 to 1000 ng/mL as working concentrations. Dilutions were passed through syringe filters (0.2 mm) before injecting into HPLC. The calibration curve was plotted using peak area (area under curve: AUC) vs nominal concentration. Data analysis was carried out using the linear regression method.
3.8. System Suitability
The suitability of the developed HPLC method of ACV was performed by determining the peak area after injecting MQC of medium QC (500 ng/mL) (without spiking in to rat plasma). AUC value was compared with mean value obtained for the MQC injections (n = 3) [
30].
3.9. Method Specificity
Blank plasma (without ACV) samples were prepared with a suitable volume of mobile phase and the samples were directly filtered into HPLC vials. The vials were kept in an auto sampler tray and were injected (10 µL) into HPLC and screened for any interfering peaks corresponding near the retention time exhibited by ACV.
3.10. Plasma Recovery
Recovery of ACV from spiked rat plasma was determined at three different concentration levels (400, 600 and 800 ng/mL). HPLC analysis was performed in triplicate (n = 3) for each of the drug concentrations and the extraction efficiency was determined (mean ± SD) [
30,
31,
32].
3.11. Method Sensitivity
The lower limit of quantification (LLOQ) and lower limit of detection (LLOD) were determined by injecting series of known concentrations of ACV to observe the signal-to-noise ratio (S/N).
3.12. Precision and Accuracy
Precision and accuracy were evaluated by inter-day and intra-day spiking of a predetermined concentration of ACV in a plasma sample. Relative standard deviation (%RSD) of the observed values was used as parameter of precision. Samples were prepared at different concentrations (LQC: 200 ng/mL, MQC: 500 ng/mL and HQC: 800 ng/mL). Precision and accuracy were evaluated over a period of three days (inter-day) using replicate (n = 3) determinations for the spiked plasma samples, whereas precision and accuracy were assessed on three separate occasions (intra-day) on the same day (n = 3) for each concentration. Relative standard deviation (%RSD) of the observed values was used as an index of precision [
32].
3.13. Robustness
The robustness of the bioanalytical method was evaluated by altering the chromatographic conditions, such as flow rate and composition of mobile phase, and their effects were observed on each of the responses (peak area and peak retention time) at different quality control samples (200, 500, and 800 ng/mL).
3.14. Stability in Plasma
The stability of ACV in plasma was evaluated at two different concentrations (500 ng and 1000 ng/mL). Briefly short-term stability at ambient temperature (30 °C) for 2 h, freeze-thaw cycles (4 °C) for 24 h, and long-term stability (30 days) at −20 °C were carried out. The stability of ACV in human plasma was also investigated for three freeze–thaw cycles. A stability experiment was carried out in triplicate (n = 3) to estimate mean ± SD.
3.15. Vesicular Formulations (Ethosomes and Elastic Liposomes) for Transdermal Applications
In our previous report, we prepared, optimized, and explored mechanistic perspective of ACV-loaded elastic liposomes and ethosomes [
22]. These reported formulations were ETHO3 (ethosomal formulation), ELP3 (elastic liposomes), ETHO3-gel (respective gel), ELP3 gel (respective gel), and ACV-solution (ACV-sol). A summary of composition and respective characteristic parameters is presented in
Table 3. It is noteworthy that elastic liposomes were composed of PC (phosphatidylcholine) and span 80 (low HLB = 4.3), whereas ethosomes were composed of PC and cholesterol. Propylene glycol and ethanol in a suitable ratio served as a good plasticizer of vesicular membranes of both vesicular systems. Carbopol gel served as carrier for colloidal suspensions (both vesicular systems). ACV was solubilized in 5% dimethyl sulfoxide (DMSO) solution to use as control for comparative assessment.
3.16. In Vivo Quantification of ACV (Pharmacokinetic Study)
The purpose of the present method validation was to carry out a bioavailability study in rat plasma using developed HPLC-PDA method. The animal study was carried out in Wistar albino rats weighing about 200–220 g of both sexes and experimental protocol was approved (approval number PCTE/LDH/1370/2013) by the Institute Ethics Committee of Punjab (PCTE Institute, PTU, Punjab), dated 7 February 2013, India (continuous work of the previous project) [
22]. Animals were randomly distributed and divided into four different groups (n = 3 per group). The rats were inspected properly to check any possible abnormality at the site of application. The dorsal part was used to apply ACV formulations over a circle (2 cm
2) on each rat and the area was properly trimmed for removing hairs. After 24 h of trimming, formulations (ELP3, ETHO2, ELP3 gel, and ETHO2 gel) containing ACV (0.5%
w/
v) were applied with an equal concentration and dose strength (2.5 mg/0.5 mL) [
22]. Blood (1 mL) was withdrawn at varied time intervals (0, 1, 2, 4, 8, 12, and 24 h) followed by cold centrifugation and extraction, and samples were subjected to HPLC analysis for quantification of ACV. Several pharmacokinetic parameters, such as area under the curve (AUC), the maximum drug concentration (C
max), and the time required to attain C
max as T
max, were determined.
3.17. Statistical Analysis
All experiments were replicated to get mean and standard deviation (n = 3). In statistical analysis, significance was expressed at p ˂ 0.05 and ANOVA (analysis of variance) was applied wherever was required to apply for data analysis in the calculation. For optimization of process variables, Design of Expert software (version 6.0.0, Sate-Ease Inc., Minneapolis, MN, USA) was used. Moreover, graphical presentation was performed using the GraphPad Prism software trial version. HSPiP software (version 5.2.06, UK) was used estimate various Hansen solubility parameters.

 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}