
Machine Learning Approaches for Discriminating Bacterial and Viral Targeted Human Proteins


Abstract
:1. Introduction
2. Material and Methods
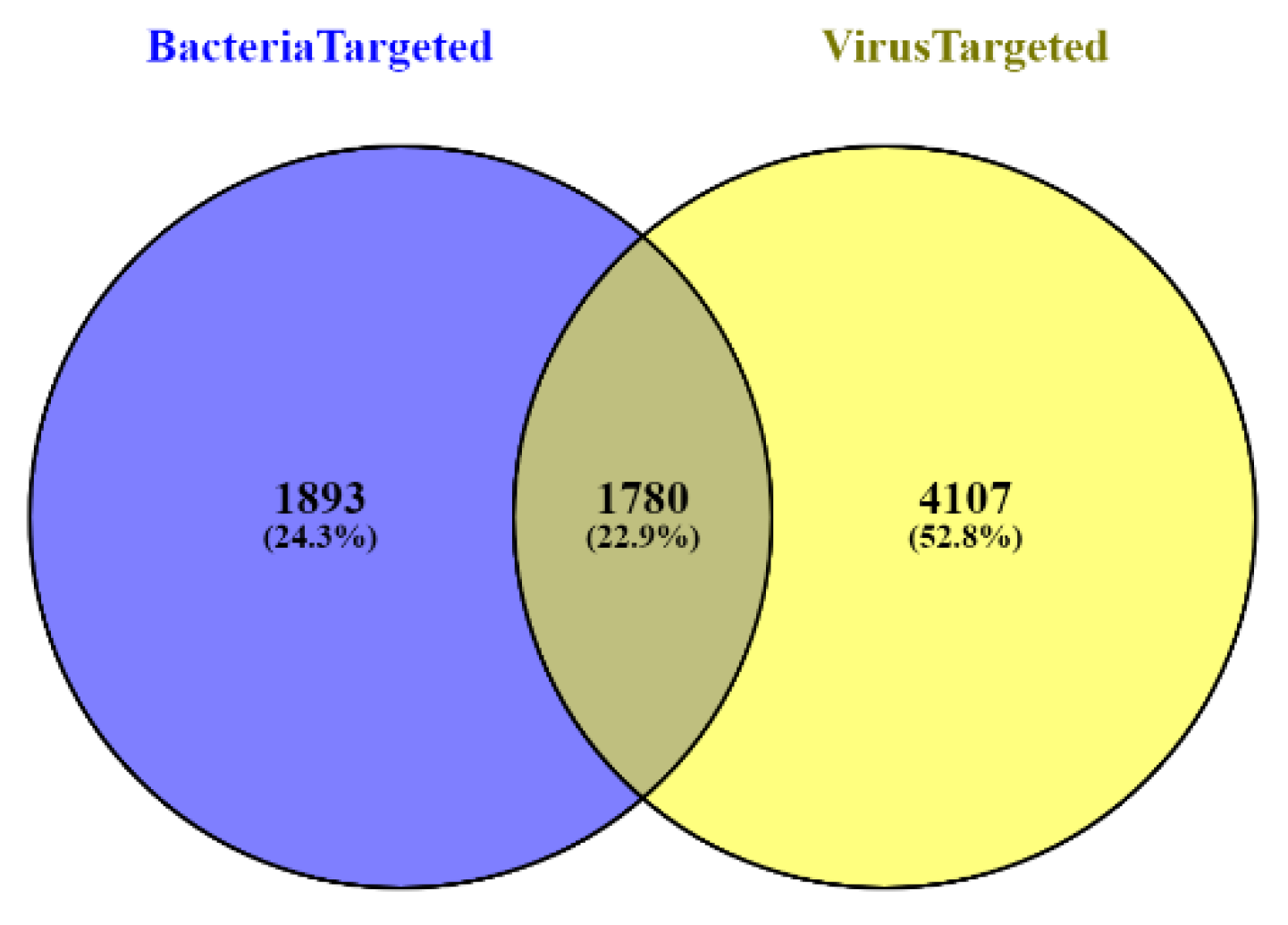
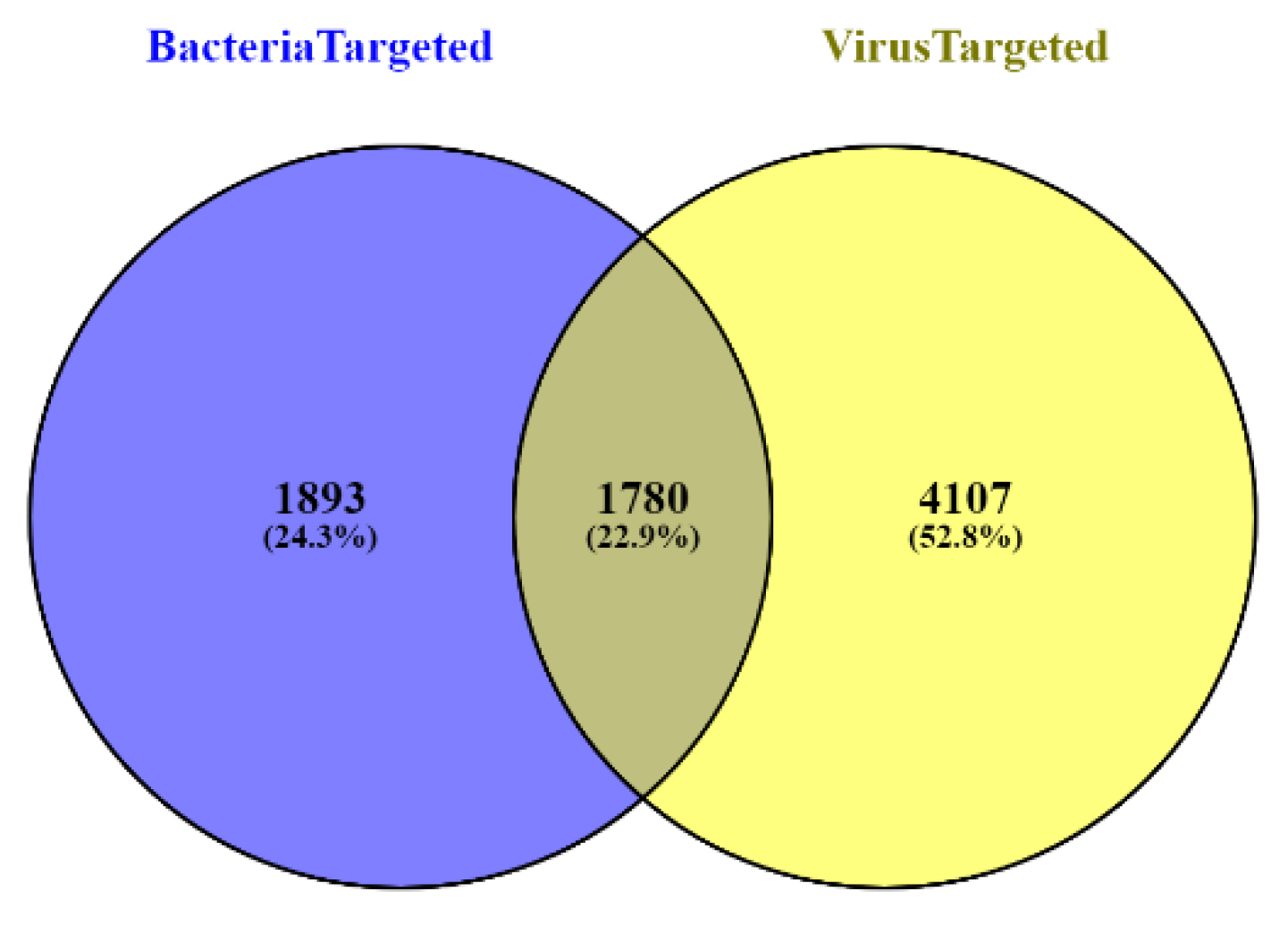
2.1. Data Collection
2.2. Sequence Features
2.3. Network Features
2.4. Gene Ontology (GO) Features
2.5. Classification
2.5.1. Support Vector Machines (SVM)
2.5.2. Random Forest (RF)
2.5.3. Deep Neural Networks (DNN)
2.6. 10-Fold Cross-Validation
2.7. Feature Selection
2.8. Performance Measures
2.9. GO Enrichment Analysis
2.10. Pathway Enrichment Analysis
3. Results
3.1. Selection of Features
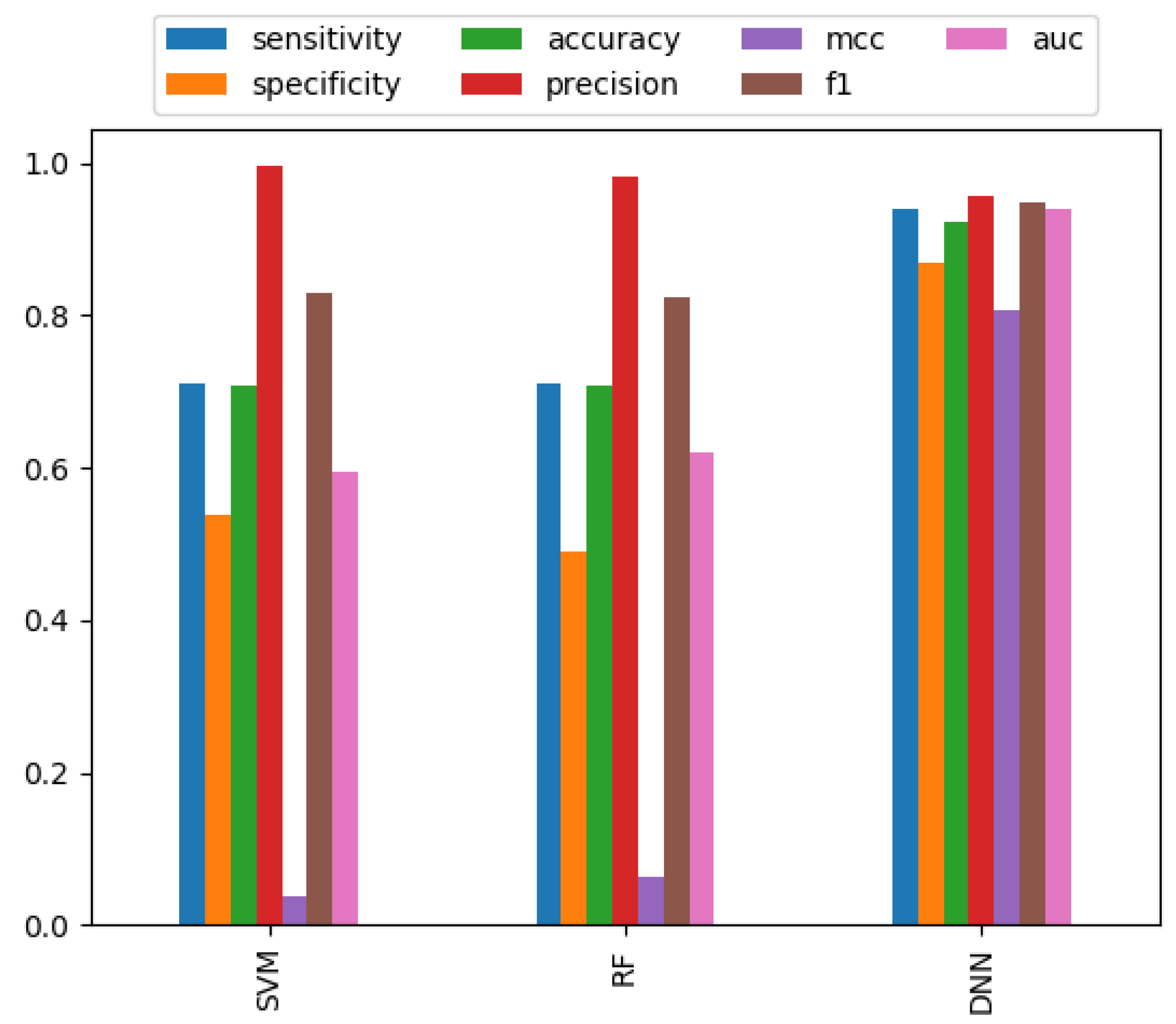
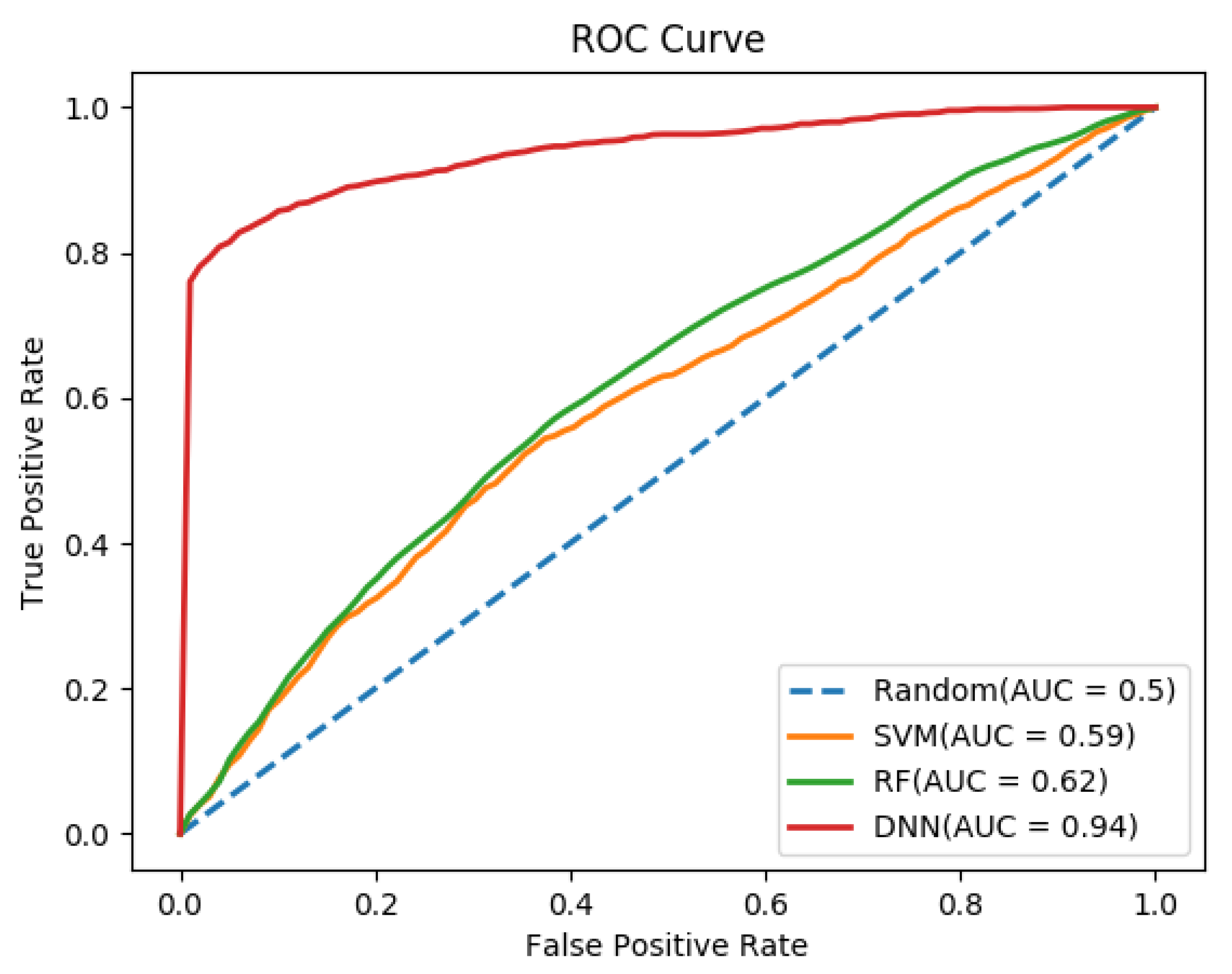
3.2. Performance Comparison of Different Classifiers
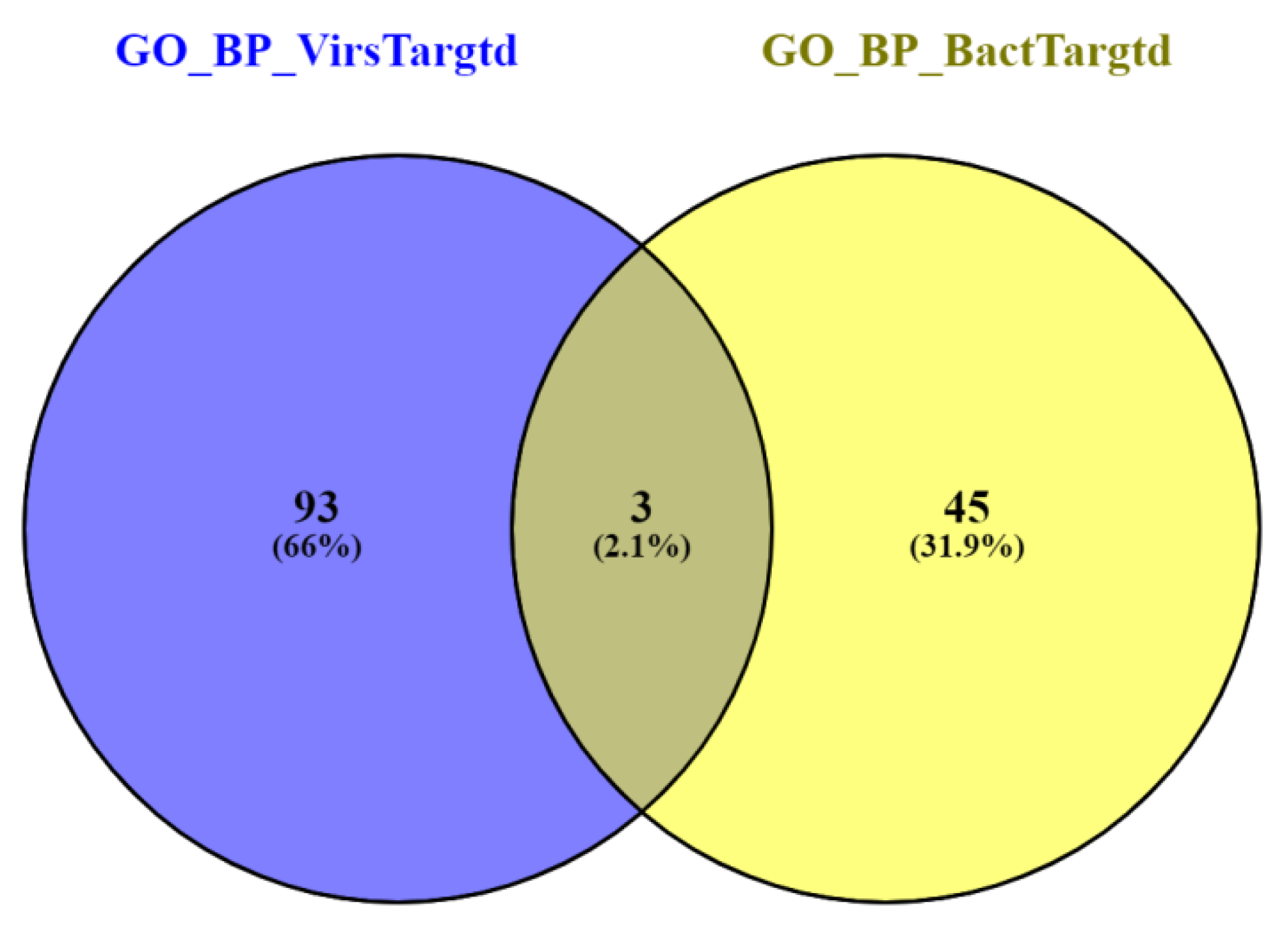
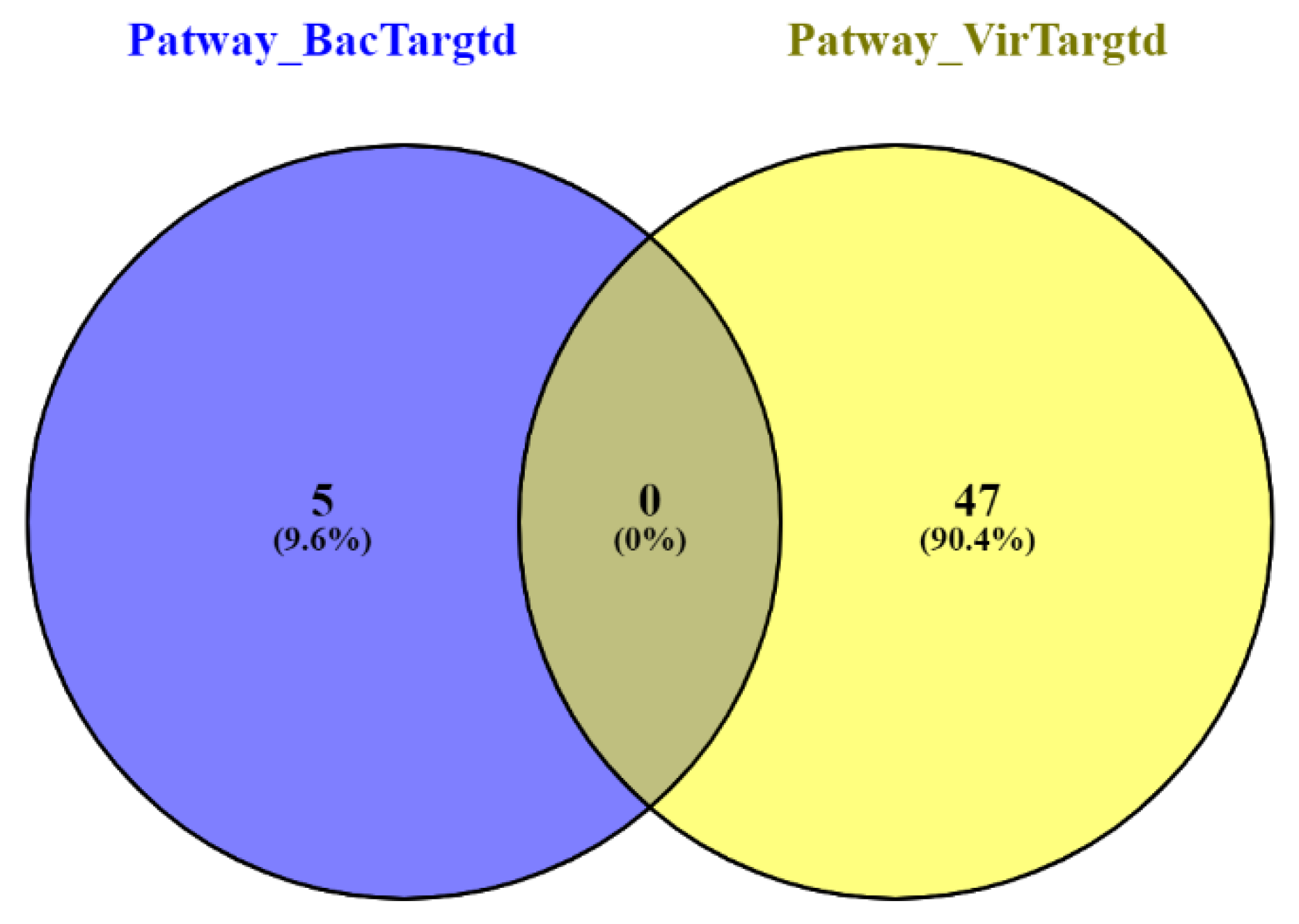
3.3. Gene Ontology Enrichment Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- WHO. Health in 2015: From MDGs to SDGs; WHO Press: Geneva, Switzerland, 2015; pp. 101–130. [Google Scholar]
- Nicholson, L.B. The immune system. Essays Biochem. 2016, 60, 275–301. [Google Scholar] [CrossRef] [Green Version]
- Nicod, C.; Banaei-Esfahani, A.; Collins, B.C. Elucidation of host-pathogen protein-protein interactions to uncover mechanisms of host cell rewiring. Curr. Opin. Microbiol. 2017, 39, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Gao, S.; Nguyen, N.N.; Fan, M.; Jin, J.; Liu, B.; Zhao, L.; Xiong, G.; Tan, M.; Li, S.; et al. Stringent homology-based prediction of H. sapiens-M. tuberculosis H37Rv protein-protein interactions. Biol. Direct 2014, 9, 5. [Google Scholar] [CrossRef] [Green Version]
- Kosesoy, I.; Gok, M.; Oz, C. A new sequence based encoding for prediction of host-pathogen protein interactions. Comput. Biol. Chem. 2019, 78, 170–177. [Google Scholar] [CrossRef]
- Alguwaizani, S.; Park, B.; Zhou, X.; Huang, D.S.; Han, K. Predicting Interactions between Virus and Host Proteins Using Repeat Patterns and Composition of Amino Acids. J. Healthc. Eng. 2018, 2018, 1391265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lian, X.; Yang, S.; Li, H.; Fu, C.; Zhang, Z. Machine-Learning-Based Predictor of Human-Bacteria Protein-Protein Interactions by Incorporating Comprehensive Host-Network Properties. J. Proteome Res. 2019, 18, 2195–2205. [Google Scholar] [CrossRef]
- Tyagi, N.; Krishnadev, O.; Srinivasan, N. Prediction of protein-protein interactions between Helicobacter pylori and a human host. Mol. Biosyst. 2009, 5, 1630–1635. [Google Scholar] [CrossRef]
- Penn, B.H.; Netter, Z.; Johnson, J.R.; Von Dollen, J.; Jang, G.M.; Johnson, T.; Ohol, Y.M.; Maher, C.; Bell, S.L.; Geiger, K.; et al. An Mtb-Human Protein-Protein Interaction Map Identifies a Switch between Host Antiviral and Antibacterial Responses. Mol. Cell 2018, 71, 637–648.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barman, R.K.; Saha, S.; Das, S. Prediction of interactions between viral and host proteins using supervised machine learning methods. PLoS ONE 2014, 9, e112034. [Google Scholar] [CrossRef]
- Wuchty, S. Computational prediction of host-parasite protein interactions between P. falciparum and H. sapiens. PLoS ONE 2011, 6, e26960. [Google Scholar] [CrossRef] [Green Version]
- Dyer, M.D.; Murali, T.M.; Sobral, B.W. The landscape of human proteins interacting with viruses and other pathogens. PLoS Pathog. 2008, 4, e32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uetz, P.; Dong, Y.A.; Zeretzke, C.; Atzler, C.; Baiker, A.; Berger, B.; Rajagopala, S.V.; Roupelieva, M.; Rose, D.; Fossum, E.; et al. Herpesviral protein networks and their interaction with the human proteome. Science 2006, 311, 239–242. [Google Scholar] [CrossRef] [Green Version]
- Farooq, Q.U.A.; Khan, F.F. Construction and analysis of a comprehensive protein interaction network of HCV with its host Homo sapiens. BMC Infect. Dis. 2019, 19, 367. [Google Scholar] [CrossRef]
- Li, Y.; Liu, G.; Zhang, J.; Zhong, X.; He, Z. Identification of key genes in human airway epithelial cells in response to respiratory pathogens using microarray analysis. BMC Microbiol. 2018, 18, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Zhang, Y.; Li, Y.H.; Wang, S.; Zhang, J.J.; Zhang, C.X.; Zhang, Z.S. Investigating dysregulated pathways in Staphylococcus aureus (SA) exposed macrophages based on pathway interaction network. Comput. Biol. Chem. 2017, 66, 21–25. [Google Scholar] [CrossRef]
- Ehsani Ardakani, M.J.; Safaei, A.; Arefi Oskouie, A.; Haghparast, H.; Haghazali, M.; Mohaghegh Shalmani, H.; Peyvandi, H.; Naderi, N.; Zali, M.R. Evaluation of liver cirrhosis and hepatocellular carcinoma using Protein-Protein Interaction Networks. Gastroenterol. Hepatol. Bed Bench 2016, 9, S14–S22. [Google Scholar] [PubMed]
- Simos, T.; Georgopoulou, U.; Thyphronitis, G.; Koskinas, J.; Papaloukas, C. Analysis of protein interaction networks for the detection of candidate hepatitis B and C biomarkers. IEEE J. Biomed. Health Inform. 2015, 19, 181–189. [Google Scholar] [CrossRef]
- Wang, Q.; Lou, Z.; Zhai, L.; Zhao, H. Detection of Significant Pneumococcal Meningitis Biomarkers by Ego Network. Indian J. Pediatrics 2017, 84, 430–436. [Google Scholar] [CrossRef]
- Liu, J.; Ma, Z.; Liu, Y.; Wu, L.; Hou, Z.; Li, W. Screening of potential biomarkers in hepatitis C virus-induced hepatocellular carcinoma using bioinformatic analysis. Oncol. Lett. 2019, 18, 2500–2508. [Google Scholar] [CrossRef] [PubMed]
- Durmus Tekir, S.; Cakir, T.; Ulgen, K.O. Infection Strategies of Bacterial and Viral Pathogens through Pathogen-Human Protein-Protein Interactions. Front. Microbiol. 2012, 3, 46. [Google Scholar] [CrossRef] [Green Version]
- Durmus Tekir, S.; Cakir, T.; Ardic, E.; Sayilirbas, A.S.; Konuk, G.; Konuk, M.; Sariyer, H.; Ugurlu, A.; Karadeniz, I.; Ozgur, A.; et al. PHISTO: Pathogen-host interaction search tool. Bioinformatics 2013, 29, 1357–1358. [Google Scholar] [CrossRef] [Green Version]
- UniProt, C. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [Green Version]
- Meher, P.K.; Sahu, T.K.; Banchariya, A.; Rao, A.R. DIRProt: A computational approach for discriminating insecticide resistant proteins from non-resistant proteins. BMC Bioinform. 2017, 18, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meher, P.K.; Sahu, T.K.; Mohanty, J.; Gahoi, S.; Purru, S.; Grover, M.; Rao, A.R. nifPred: Proteome-Wide Identification and Categorization of Nitrogen-Fixation Proteins of Diaztrophs Based on Composition-Transition-Distribution Features Using Support Vector Machine. Front. Microbiol. 2018, 9, 1100. [Google Scholar] [CrossRef] [PubMed]
- Bhadra, P.; Yan, J.; Li, J.; Fong, S.; Siu, S.W.I. AmPEP: Sequence-based prediction of antimicrobial peptides using distribution patterns of amino acid properties and random forest. Sci. Rep. 2018, 8, 1697. [Google Scholar] [CrossRef] [Green Version]
- Cao, D.S.; Liang, Y.Z.; Yan, J.; Tan, G.S.; Xu, Q.S.; Liu, S. PyDPI: Freely available python package for chemoinformatics, bioinformatics, and chemogenomics studies. J. Chem. Inf. Model. 2013, 53, 3086–3096. [Google Scholar] [CrossRef] [PubMed]
- Keshava Prasad, T.S.; Goel, R.; Kandasamy, K.; Keerthikumar, S.; Kumar, S.; Mathivanan, S.; Telikicherla, D.; Raju, R.; Shafreen, B.; Venugopal, A.; et al. Human Protein Reference Database--2009 update. Nucleic Acids Res. 2009, 37, D767–D772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assenov, Y.; Ramirez, F.; Schelhorn, S.E.; Lengauer, T.; Albrecht, M. Computing topological parameters of biological networks. Bioinformatics 2008, 24, 282–284. [Google Scholar] [CrossRef] [Green Version]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Abadi, M.; Barham, P.; Chen, J.; Chen, Z.; Davis, A.; Dean, J.; Devin, M.; Ghemawat, S.; Irving, G.; Isard, M.; et al. TensorFlow: A system for large-scale machine learning. In Proceedings of the 12th USENIX Symposium on Operating Systems Design and Implementation (OSDI 16), Savannah, GA, USA, 2–4 November 2016; pp. 265–283. [Google Scholar]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [Green Version]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef] [PubMed]
- Grijalva, C.G.; Nuorti, J.P.; Griffin, M.R. Antibiotic prescription rates for acute respiratory tract infections in US ambulatory settings. JAMA 2009, 302, 758–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drijkoningen, J.J.; Rohde, G.G. Pneumococcal infection in adults: Burden of disease. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2014, 20 (Suppl. S5), 45–51. [Google Scholar] [CrossRef] [Green Version]
- Mathew, B.; Roy, D.D.; Kumar, T.V. The use of procalcitonin as a marker of sepsis in children. J. Clin. Diagn. Res. JCDR 2013, 7, 305–307. [Google Scholar] [CrossRef]
- Yusa, T.; Tateda, K.; Ohara, A.; Miyazaki, S. New possible biomarkers for diagnosis of infections and diagnostic distinction between bacterial and viral infections in children. J. Infect. Chemother. Off. J. Jpn. Soc. Chemother. 2017, 23, 96–100. [Google Scholar] [CrossRef] [Green Version]
- Zav’yalov, V.P.; Hamalainen-Laanaya, H.; Korpela, T.K.; Wahlroos, T. Interferon-Inducible Myxovirus Resistance Proteins: Potential Biomarkers for Differentiating Viral from Bacterial Infections. Clin. Chem. 2019, 65, 739–750. [Google Scholar] [CrossRef] [Green Version]
- Srugo, I.; Klein, A.; Stein, M.; Golan-Shany, O.; Kerem, N.; Chistyakov, I.; Genizi, J.; Glazer, O.; Yaniv, L.; German, A.; et al. Validation of a Novel Assay to Distinguish Bacterial and Viral Infections. Pediatrics 2017, 140. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.; Zhu, J.; Song, L.; Cai, W.; Wang, J. Combined use of biomarkers for distinguishing between bacterial and viral etiologies in pediatric lower respiratory tract infections. Infect. Dis. 2015, 47, 289–293. [Google Scholar] [CrossRef] [PubMed]
- ten Oever, J.; Tromp, M.; Bleeker-Rovers, C.P.; Joosten, L.A.; Netea, M.G.; Pickkers, P.; van de Veerdonk, F.L. Combination of biomarkers for the discrimination between bacterial and viral lower respiratory tract infections. J. Infect. 2012, 65, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Suarez, N.M.; Bunsow, E.; Falsey, A.R.; Walsh, E.E.; Mejias, A.; Ramilo, O. Superiority of transcriptional profiling over procalcitonin for distinguishing bacterial from viral lower respiratory tract infections in hospitalized adults. J. Infect. Dis. 2015, 212, 213–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Yu, J.; Crosby, S.D.; Storch, G.A. Gene expression profiles in febrile children with defined viral and bacterial infection. Proc. Natl. Acad. Sci. USA 2013, 110, 12792–12797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsalik, E.L.; Henao, R.; Nichols, M.; Burke, T.; Ko, E.R.; McClain, M.T.; Hudson, L.L.; Mazur, A.; Freeman, D.H.; Veldman, T.; et al. Host gene expression classifiers diagnose acute respiratory illness etiology. Sci. Transl. Med. 2016, 8, 322ra11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweeney, T.E.; Wong, H.R.; Khatri, P. Robust classification of bacterial and viral infections via integrated host gene expression diagnostics. Sci. Transl. Med. 2016, 8, 346ra91. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence Features | ||||||
|---|---|---|---|---|---|---|
| Features Set | Vector Length | Method | Accuracy (%) | MCC | F1-Score (%) | AUC |
| Amino acid composition (AAC) | 20 | SVM | 69.20 | 0.10 | 80.60 | 0.580 |
| Amino acid composition (AAC) | 20 | RF | 70.20 | 0.05 | 82.30 | 0.629 |
| Amino acid composition (AAC) | 20 | DNN | 71.90 | 0.21 | 82.30 | 0.699 |
| Dipeptide composition (DC) | 400 | SVM | 70.10 | 0.09 | 81.90 | 0.598 |
| Dipeptide composition (DC) | 400 | RF | 70.70 | 0.06 | 82.50 | 0.614 |
| Dipeptide composition (DC) | 400 | DNN | 86.40 | 0.67 | 90.30 | 0.931 |
| Pseudo-amino acid composition (PAAC) | 25 | SVM | 65.40 | 0.09 | 76.80 | 0.582 |
| Pseudo-amino acid composition (PAAC) | 25 | RF | 70.70 | 0.09 | 82.30 | 0.628 |
| Pseudo-amino acid composition (PAAC) | 25 | DNN | 71.00 | 0.19 | 81.30 | 0.708 |
| Composition, Transition, and Distribution (CTD) | 147 | SVM | 71.00 | 0.01 | 83.00 | 0.525 |
| Composition, Transition, and Distribution (CTD) | 147 | RF | 70.90 | 0.09 | 82.50 | 0.622 |
| Composition, Transition, and Distribution (CTD) | 147 | DNN | 70.70 | 0.02 | 82.80 | 0.603 |
| AAC_DC | 420 | SVM | 70.60 | 0.05 | 82.80 | 0.602 |
| AAC_DC | 420 | RF | 70.50 | 0.06 | 82.50 | 0.620 |
| AAC_DC | 420 | DNN | 86.00 | 0.66 | 90.00 | 0.924 |
| AAC_DC_PAAC | 445 | SVM | 70.70 | 0.04 | 82.90 | 0.594 |
| AAC_DC_PAAC | 445 | RF | 70.70 | 0.06 | 82.50 | 0.621 |
| AAC_DC_PAAC | 445 | DNN | 92.40 | 0.81 | 94.90 | 0.939 |
| AAC_DC_PAAC_CTD | 592 | SVM | 71.00 | 0.07 | 83.00 | 0.566 |
| AAC_DC_PAAC_CTD | 592 | RF | 70.40 | 0.04 | 823.00 | 0.627 |
| AAC_DC_PAAC_CTD | 592 | DNN | 70.10 | 0.03 | 83.00 | 0.588 |
| Gene Ontology Features | ||||||
| Gene Ontology (GO) | 282 | SVM | 52.60 | 0.03 | 61.40 | 0.283 |
| Gene Ontology (GO) | 282 | RF | 66.70 | 0.13 | 77.90 | 0.613 |
| Gene Ontology (GO) | 282 | DNN | 80.20 | 0.51 | 86.40 | 0.886 |
| Network Features | ||||||
| Network | 9 | SVM | 54.20 | 0.06 | 62.70 | 0.538 |
| Network | 9 | RF | 53.90 | 0.06 | 62.60 | 0.527 |
| Network | 9 | DNN | 53.30 | 0.05 | 61.90 | 0.512 |
| Mixed features | ||||||
| AAC_DC_PAAC_GO | 727 | SVM | 70.90 | 0.02 | 83.00 | 0.609 |
| AAC_DC_PAAC_GO | 727 | RF | 70.50 | 0.05 | 82.30 | 0.635 |
| AAC_DC_PAAC_GO | 727 | DNN | 83.40 | 0.60 | 88.30 | 0.914 |
| AAC_DC_PAAC_CTD_GO | 874 | SVM | 71.00 | 0.06 | 83.00 | 0.567 |
| AAC_DC_PAAC_CTD_GO | 874 | RF | 70.40 | 0.06 | 82.30 | 0.635 |
| AAC_DC_PAAC_CTD_GO | 874 | DNN | 70.12 | 0.04 | 83.00 | 0.563 |
| AAC_DC_PAAC_CTD_GO_Network | 883 | SVM | 70.30 | 0.06 | 81.50 | 0.595 |
| AAC_DC_PAAC_CTD_GO_Network | 883 | RF | 70.50 | 0.07 | 82.60 | 0.642 |
| AAC_DC_PAAC_CTD_GO_Network | 883 | DNN | 72.10 | 0.18 | 83.10 | 0.725 |
| Features with Feature Selection Methods | Vector Length | Method | Accuracy (%) | MCC | F1-Score (%) | AUC |
|---|---|---|---|---|---|---|
| AAC_DC_PAAC_UFS_chi2 | 44 | SVM | 65.70 | 0.09 | 77.60 | 0.568 |
| AAC_DC_PAAC_UFS_chi2 | 44 | RF | 70.40 | 0.08 | 82.30 | 0.634 |
| AAC_DC_PAAC_UFS_chi2 | 44 | DNN | 71.90 | 0.21 | 82.10 | 0.704 |
| AAC_DC_PAAC_UFS_f_classif | 44 | SVM | 64.80 | 0.08 | 76.40 | 0.550 |
| AAC_DC_PAAC_UFS_f_classif | 44 | RF | 70.30 | 0.07 | 82.20 | 0.631 |
| AAC_DC_PAAC_UFS_f_classif | 44 | DNN | 72.60 | 0.22 | 82.60 | 0.705 |
| AAC_DC_PAAC_UFS_mutual_info_classif | 44 | SVM | 62.20 | 0.14 | 71.90 | 0.604 |
| AAC_DC_PAAC_UFS_mutual_info_classif | 44 | RF | 70.30 | 0.06 | 82.40 | 0.622 |
| AAC_DC_PAAC_UFS_mutual_info_classif | 44 | DNN | 72.10 | 0.23 | 82.00 | 0.714 |
| AAC_DC_PAAC_RFE | 44 | SVM | 69.90 | 0.08 | 81.50 | 0.584 |
| AAC_DC_PAAC_RFE | 44 | RF | 70.20 | 0.06 | 82.30 | 0.633 |
| AAC_DC_PAAC_RFE | 44 | DNN | 73.10 | 0.25 | 83.00 | 0.716 |
| AAC_DC_PAAC_SFM | 376 | SVM | 70.60 | 0.04 | 82.80 | 0.595 |
| AAC_DC_PAAC_SFM | 376 | RF | 70.60 | 0.06 | 82.40 | 0.627 |
| AAC_DC_PAAC_SFM | 376 | DNN | 75.10 | 0.39 | 82.60 | 0.796 |
| AAC_DC_PAAC_TBFS | 227 | SVM | 70.30 | 0.07 | 82.30 | 0.604 |
| AAC_DC_PAAC_TBFS | 227 | RF | 70.60 | 0.06 | 82.40 | 0.628 |
| AAC_DC_PAAC_TBFS | 227 | DNN | 77.00 | 0.44 | 84.00 | 0.805 |
| Term | p-Value |
|---|---|
| regulation of nucleic acid-templated transcription (GO:1903506) | 0.003414 |
| regulation of cellular macromolecule biosynthetic process (GO:2000112) | 0.004395 |
| negative regulation of catalytic activity (GO:0043086) | 0.008008 |
| glomerulus vasculature development (GO:0072012) | 0.029631 |
| regulation of relaxation of cardiac muscle (GO:1901897) | 0.029631 |
| dosage compensation by inactivation of X chromosome (GO:0009048) | 0.029631 |
| negative regulation of cellular response to hypoxia (GO:1900038) | 0.029631 |
| pronephros development (GO:0048793) | 0.029631 |
| negative regulation of cellular catabolic process (GO:0031330) | 0.033057 |
| negative regulation of nitric oxide biosynthetic process (GO:0045019) | 0.034484 |
| negative regulation of nitric oxide metabolic process (GO:1904406) | 0.034484 |
| negative regulation of calcium ion import (GO:0090281) | 0.034484 |
| negative regulation of RIG-I signaling pathway (GO:0039536) | 0.034484 |
| glycosphingolipid catabolic process (GO:0046479) | 0.034484 |
| thiamine-containing compound metabolic process (GO:0042723) | 0.034484 |
| regulation of cardiac muscle cell membrane potential (GO:0086036) | 0.034484 |
| negative regulation of cell adhesion mediated by integrin (GO:0033629) | 0.034484 |
| positive regulation of histone H4 acetylation (GO:0090240) | 0.034484 |
| negative regulation of heart rate (GO:0010459) | 0.034484 |
| regulation of relaxation of muscle (GO:1901077) | 0.034484 |
| Term | p-Value |
|---|---|
| peptide biosynthetic process (GO:0043043) | 8.36 × 10−11 |
| translation (GO:0006412) | 2.25 × 10−9 |
| mitochondrial ATP synthesis-coupled electron transport (GO:0042775) | 2.63 × 10−7 |
| mitochondrial translational elongation (GO:0070125) | 3.08 × 10−7 |
| cellular macromolecule biosynthetic process (GO:0034645) | 3.45 × 10−7 |
| mitochondrial translational termination (GO:0070126) | 3.60 × 10−7 |
| respiratory electron transport chain (GO:0022904) | 5.21 × 10−7 |
| translational termination (GO:0006415) | 6.01 × 10−7 |
| translational elongation (GO:0006414) | 1.10 × 10−6 |
| gene expression (GO:0010467) | 1.14 × 10−6 |
| mitochondrial translation (GO:0032543) | 1.25 × 10−6 |
| mitochondrial electron transport, cytochrome c to oxygen (GO:0006123) | 3.43 × 10−6 |
| epidermis development (GO:0008544) | 3.67 × 10−6 |
| cellular protein metabolic process (GO:0044267) | 6.09 × 10−6 |
| protein targeting to ER (GO:0045047) | 1.03 × 10−5 |
| intermediate filament organization (GO:0045109) | 4.37 × 10−5 |
| SRP-dependent cotranslational protein targeting to membrane (GO:0006614) | 9.30 × 10−5 |
| peptide cross-linking (GO:0018149) | 1.01 × 10−4 |
| cotranslational protein targeting to membrane (GO:0006613) | 1.14 × 10−4 |
| skin development (GO:0043588) | 1.20 × 10−4 |
| Pathway Name | Entities p Value |
|---|---|
| Uptake and function of anthrax toxins | 0.009407594 |
| ARL13B-mediated ciliary trafficking of INPP5E | 0.02672975 |
| Defective NEU1 causes sialidosis | 0.02672975 |
| Vitamin B1 (thiamin) metabolism | 0.044155321 |
| RUNX1 interacts with cofactors whose precise effect on RUNX1 targets is not known | 0.044545497 |
| Pathway Name | Entities p Value |
|---|---|
| Formation of the cornified envelope | 1.78 × 10−10 |
| Keratinization | 9.88 × 10−9 |
| Translation | 3.97 × 10−7 |
| Respiratory electron transport, ATP synthesis by chemiosmotic coupling, and heat production by uncoupling proteins. | 1.94 × 10−6 |
| Mitochondrial translation termination | 1.58 × 10−5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barman, R.K.; Mukhopadhyay, A.; Maulik, U.; Das, S. Machine Learning Approaches for Discriminating Bacterial and Viral Targeted Human Proteins. Processes 2022, 10, 291. https://doi.org/10.3390/pr10020291
Barman RK, Mukhopadhyay A, Maulik U, Das S. Machine Learning Approaches for Discriminating Bacterial and Viral Targeted Human Proteins. Processes. 2022; 10(2):291. https://doi.org/10.3390/pr10020291
Chicago/Turabian StyleBarman, Ranjan Kumar, Anirban Mukhopadhyay, Ujjwal Maulik, and Santasabuj Das. 2022. "Machine Learning Approaches for Discriminating Bacterial and Viral Targeted Human Proteins" Processes 10, no. 2: 291. https://doi.org/10.3390/pr10020291
APA StyleBarman, R. K., Mukhopadhyay, A., Maulik, U., & Das, S. (2022). Machine Learning Approaches for Discriminating Bacterial and Viral Targeted Human Proteins. Processes, 10(2), 291. https://doi.org/10.3390/pr10020291