
Predicting the Solubility of Nonelectrolyte Solids Using a Combination of Molecular Simulation with the Solubility Parameter Method MOSCED: Application to the Wastewater Contaminants Monuron, Diuron, Atrazine and Atenolol

 ,
,
Abstract
:1. Introduction
2. Method
2.1. MOSCED
Molecular Simulation
2.2.
3. Computational Details
3.1. Molecular Simulation
3.2. Regressing MOSCED Parameters
4. Results and Discussion
4.1. Pure Component Fugacity
4.2. MOSCED Parameters
4.3. Solubility Predictions
5. Summary and Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. MOSCED Calculator
Appendix B. Alternative to Calculating
References
- Sene, L.; Converti, A.; Aparecida Ribeiro Secchi, G.; de Cássia Garcia Simão, R. New Aspects on Atrazine Biodegradation. Braz. Arch. Biol. Technol. 2010, 53, 487–496. [Google Scholar] [CrossRef] [Green Version]
- Aparecido dos Santos, E.; da Cruz, C.; Patrícia Carraschi, S.; Roberto Marques Silva, J.; Grossi Botelho, R.; Domingues Velini, E.; Antonio Pitelli, R. Atrazine levels in the Jaboticabal water stream (São Paulo State, Brazil) and its toxicological effects on the pacu fish Piaractus mesopotamicus. Arch. Hig. Rada. Toksikol. 2015, 66, 73–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hvězdová, M.; Kosubová, P.; Košíková, M.; Scherr, K.E.; Šimek, Z.; Brodský, L.; Šudoma, M.; Škulcová, L.; Sáňka, M.; Svobodová, M.; et al. Currently and recently used pesticides in Central European arable soils. Sci. Total Environ. 2018, 613–614, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Beek, T.A.D.; Weber, F.A.; Bergmann, A.; Hickmann, S.; Ebert, I.; Hein, A.; Küster, A. Pharmaceuticals in the Environment–Global Occurances and Perspectives. Environ. Toxicol. Chem. 2016, 35, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Küster, A.; Alder, A.C.; Escher, B.I.; Duis, K.; Fenner, K.; Garric, J.; Hutchinson, T.H.; Lapen, D.R.; Péry, A.; Römbke, J.; et al. Environmental risk assessment of human pharmaceuticals in the European Union: A case study with the β-blocker atenolol. Integr. Environ. Assess. Manag. 2010, 6, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Kuster, M.; de Alda, L.; Hernando, M.D.; Petrovic, M.; Martín-Alonso, J.; Barceló, M.J.D. Analysis and occurance of pharmaceuticals, progestogens and polar pesticides in sewage treatment plant effluents, river water and drinking water in Llobregat river basin (Barcelona, Spain). J. Hydrol. 2008, 358, 112–123. [Google Scholar] [CrossRef]
- Taheran, M.; Brar, S.K.; Verma, M.; Surampalli, R.Y.; Zhang, T.C.; Valero, J.R. Membrane processes for removal of pharmaceutically active compounds (PhACs) from water and wastewaters. Sci. Total Environ. 2016, 547, 60–77. [Google Scholar] [CrossRef]
- Schröder, P.; Helmreich, B.; Škrbić, B.; Carballa, M.; Papa, M.; Pastore, C.; Emre, Z.; Oehmen, A.; Langenhoff, A.; Molinos, M.; et al. Status of hormones and painkillers in wastewater effluents across several European states–considerations for the EU watch list concerning estradiols and diclofenac. Environ. Sci. Pollut. Res. 2016, 23, 12835–12866. [Google Scholar] [CrossRef]
- Chaukura, N.; Gwenzi, W.; Tavengwa, N.; Manyuchi, M.M. Biosorbents for the removal of synthetic organics and emerging pollutants: Opportunities and challenges for developing countries. Environ. Dev. 2016, 19, 84–89. [Google Scholar] [CrossRef]
- Al Qarni, H.; Collier, P.; O’Keefe, J.; Akunna, J. Investigating the removal of some pharmaceutical compounds in hospital wastewater treatment plants operating in Saudi Arabia. Environ. Sci. Pollut. Res. 2016, 23, 13003–13014. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Crafts, P.A. Correlation and Prediction of Drug Molecule Solubility in Mixed Solvent Systems with the Nonrandom Two-Liquid Segment Activity Coefficient (NRTL-SAC) Model. Ind. Eng. Chem. Res. 2006, 45, 4816–4824. [Google Scholar] [CrossRef]
- Lazzaroni, M.J.; Bush, D.; Eckert, C.A.; Frank, T.C.; Gupta, S.; Olson, J.D. Revision of MOSCED Parameters and Extension to Solid Solubility Calculations. Ind. Eng. Chem. Res. 2005, 44, 4075–4083. [Google Scholar] [CrossRef]
- Cassens, J.; Ruether, F.; Leonhard, K.; Sadowski, G. Solubility calculation of pharmaceutical compounds—A priori parameter estimation using quantum-chemistry. Fluid Phase Equilib. 2010, 299, 161–170. [Google Scholar] [CrossRef]
- Spyriouni, T.; Krokidis, X.; Economou, I.G. Thermodynamics of pharmaceuticals: Prediction of solubility in pure and mixed solvents with PC-SAFT. Fluid Phase Equilib. 2011, 302, 331–337. [Google Scholar] [CrossRef]
- Paluch, A.S.; Jayaraman, S.; Shah, J.K.; Maginn, E.J. A method for computing the solubility limit of solids: Application to sodium chloride in water and alcohols. J. Chem. Phys. 2010, 133, 124504. [Google Scholar] [CrossRef]
- Belluci, M.A.; Gobbo, G.; Wijethunga, T.K.; Ciccotti, G.; Trout, B.L. Solubility of paracetamol in ethanol by molecular dynamics using the extended Einstein crystal method and experiments. J. Chem. Phys. 2019, 150, 094107. [Google Scholar] [CrossRef]
- Li, L.; Totton, T.; Frenkel, D. Computational methodology for solubility prediction: Application to the sparingly soluble solutes. J. Chem. Phys. 2017, 146, 214110. [Google Scholar] [CrossRef] [Green Version]
- Aragones, J.L.; Sanz, E.; Vega, C. Solubility of NaCl in water by molecular simulation revisited. J. Chem. Phys. 2012, 136, 244508. [Google Scholar] [CrossRef]
- Ley, R.T.; Fuerst, G.B.; Redeker, B.N.; Paluch, A.S. Developing a Predictive Form of MOSCED for Nonelectrolyte Solids Using Molecular Simulation: Application to Acetanilide, Acetaminophen, and Phenacetin. Ind. Eng. Chem. Res. 2016, 55, 5415–5430. [Google Scholar] [CrossRef]
- Cox, C.E.; Phifer, J.R.; da Silva, L.F.; Nogueira, G.G.; Ley, R.T.; O’Loughlin, E.J.; Barbosa, A.K.P.; Rygelski, B.T.; Paluch, A.S. Combining MOSCED with molecular simulation free energy calculations or electronic structure calculations to develop an efficient tool for solvent formulation and selection. J. Comput.-Aided Mol. Des. 2017, 31, 183–199. [Google Scholar] [CrossRef]
- Phifer, J.R.; Cox, C.E.; da Silva, L.F.; Nogueira, G.G.; Barbosa, A.K.P.; Ley, R.T.; Bozada, S.M.; O’Loughlin, E.J.; Paluch, A.S. Predicting the equilibrium solubility of solid polycyclic aromatic hydrocarbons and dibenzothiophene using a combination of MOSCED plus molecular simulation or electronic structure calculations. Mol. Phys. 2017, 115, 1286–1300. [Google Scholar] [CrossRef]
- Prausnitz, J.M.; Lichtenthaler, R.N.; de Azevedo, E.G. Molecular Thermodynamics of Fluid-Phase Equilibria, 2nd ed.; Prentice-Hall, Inc.: Englewood Cliffs, NJ, USA, 1986. [Google Scholar]
- Hildebrand, J.H.; Prausnitz, J.M.; Scott, R.L. Regular and Related Solutions; Van Nostrand Reinhold Company: New York, NY, USA, 1970. [Google Scholar]
- Nordström, F.L.; Rasmuson, A.C. Determination of the activity of a molecular solute in saturated solution. J. Chem. Thermodyn. 2008, 40, 1684–1692. [Google Scholar] [CrossRef]
- Nordström, F.L.; Rasmuson, A.C. Prediction of solubility curves and melting properties of organic and pharmaceutical compounds. Eur. J. Pharm. Sci. 2009, 36, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Thati, J.; Rasmuson, A.C. Thermodynamics of molecular solids in organic solvents. J. Chem. Thermodyn. 2012, 48, 150–159. [Google Scholar] [CrossRef]
- Thomas, E.R.; Eckert, C.A. Prediction of limiting activity coefficients by a modified separation of cohesive energy density model and UNIFAC. Ind. Eng. Chem. Proc. Des. Dev. 1984, 23, 194–209. [Google Scholar] [CrossRef]
- Park, J.H.; Carr, P.W. Predictive Ability of the MOSCED and UNIFAC Activity Coefficient Estimation Methods. Anal. Chem. 1987, 59, 2596–2602. [Google Scholar] [CrossRef]
- Howell, W.J.; Karachewski, A.M.; Stephenson, K.M.; Eckert, C.A.; Park, J.H.; Carr, P.W.; Rutan, S.C. An Improved MOSCED Equation for the Prediction and Application of Infinite Dilution Activity Coefficients. Fluid Phase Equilib. 1989, 52, 151–160. [Google Scholar] [CrossRef]
- Hait, M.J.; Liotta, C.L.; Eckert, C.A.; Bergmann, D.L.; Karachewski, A.M.; Dallas, A.J.; Eikens, D.I.; Li, J.J.; Carr, P.W.; Poe, R.B.; et al. Space Predictor for Infinite Dilution Activity Coefficients. Ind. Eng. Chem. Res. 1993, 32, 2905–2914. [Google Scholar] [CrossRef]
- Castells, C.B.; Carr, P.W.; Eikens, D.I.; Bush, D.; Eckert, C.A. Comparative Study of Semitheoretical Models for Predicting Infinite Dilution Activity Coefficients of Alkanes in Organic Solvents. Ind. Eng. Chem. Res. 1999, 38, 4104–4109. [Google Scholar] [CrossRef]
- Draucker, L.C.; Janakat, M.; Lazzaroni, M.J.; Bush, D.; Eckert, C.A.; Olson, T.C.F.D. Experimental determination and model prediction of solid solubility of multifunctional compounds in pure and mixed nonelectrolyte solvents. Ind. Eng. Chem. Res. 2007, 46, 2198–2204. [Google Scholar] [CrossRef]
- Frank, T.C.; Anderson, J.J.; Olson, J.D.; Eckert, C.A. Application of MOSCED and UNIFAC to screen hydrophobic solvents for extraction of hydrogen-bonding organics from aqueous solution. Ind. Eng. Chem. Res. 2007, 46, 4621–4625. [Google Scholar] [CrossRef]
- Dhakal, P.; Roese, S.N.; Stalcup, E.M.; Paluch, A.S. Application of MOSCED to Predict Hydration Free Energies, Henry’s Constants, Octanol/Water Partition Coefficients, and Isobaric Azeotropic Vapor-Liquid Equilibrium. J. Chem. Eng. Data 2018, 63, 352–364. [Google Scholar] [CrossRef]
- Dhakal, P.; Roese, S.N.; Stalcup, E.M.; Paluch, A.S. GC-MOSCED: A Group Contribution Method for Predicting MOSCED Parameters with Application to Limiting Activity Coefficients in Water and Octanol/Water Partition Coefficients. Fluid Phase Equilib. 2018, 470, 232–240. [Google Scholar] [CrossRef]
- Dhakal, P.; Paluch, A.S. Assessment and Revision of the MOSCED Parameters for Water: Applicability to Limiting Activity Coefficients and Binary Liquid-Liquid Equilibrium. Ind. Eng. Chem. Res. 2018, 57, 1689–1695. [Google Scholar] [CrossRef]
- Dhakal, P.; Roese, S.N.; Lucas, M.A.; Paluch, A.S. Predicting Limiting Activity Coefficients and Phase Behavior from Molecular Structure: Expanding MOSCED to Alkanediols Using Group Contribution Methods and Electronic Structure Calculations. J. Chem. Eng. Data 2018, 63, 2586–2598. [Google Scholar] [CrossRef]
- Phifer, J.R.; Solomon, K.J.; Young, K.L.; Paluch, A.S. Computing MOSCED parameters of nonelectrolyte solids with electronic structure methods in SMD and SM8 continuum solvents. AIChE J. 2017, 63, 781–791. [Google Scholar] [CrossRef]
- Diaz-Rodriguez, S.; Bozada, S.M.; Phifer, J.R.; Paluch, A.S. Predicting cyclohexane/water distribution coefficients for the SAMPL5 challenge with MOSCED and the SMD solvation model. J. Comput.-Aided Mol. Des. 2016, 30, 1007–1017. [Google Scholar] [CrossRef]
- Dhakal, P.; Ouimet, J.A.; Roese, S.N.; Paluch, A.S. MOSCED parameters for 1-n-alkyl-3-methylimidazolium-based ionic liquids: Application to limiting activity coefficients and intuitive entrainer selection for extractive distillation processes. J. Mol. Liq. 2019, 293, 111552. [Google Scholar] [CrossRef]
- Dhakal, P.; Weise, A.R.; Fritsch, M.C.; O’Dell, C.M.; Paluch, A.S. Expanding the Solubility Parameter Method MOSCED to Pyridinium, Quinolinium, Pyrrolidinium, Piperidinium, Bicyclic, Morpholinium, Ammonium, Phosphonium, and Sulfonium Based Ionic Liquids. ACS Omega 2020, 5, 3863–3877. [Google Scholar] [CrossRef] [Green Version]
- Gnap, M.; Elliott, J.R. Estimation of MOSCED parameters from the COSMO-SAC database. Fluid Phase Equilib. 2018, 470, 241–248. [Google Scholar] [CrossRef]
- Brouwer, T.; Schuur, B. Model Performances Evaluated for Infinite Dilution Activity Coefficients Prediction at 298.15 K. Ind. Eng. Chem. Res. 2019, 58, 8903–8914. [Google Scholar] [CrossRef] [Green Version]
- Widenski, D.J.; Abbas, A.; Romagnoli, J.A. Use of Predictive Solubility Models for Isothermal Antisolvent Crystallization Modeling and Optimization. Ind. Eng. Chem. Res. 2011, 50, 8304–8313. [Google Scholar] [CrossRef]
- Abrams, D.S.; Prausnitz, J.M. Statistical thermodynamics of liquid mixtures: A new expression for the excess Gibbs energy or partly or completely miscible systems. AIChE J. 1975, 21, 116–128. [Google Scholar] [CrossRef]
- Schacht, C.S.; Zubeir, L.; de Loos, T.W.; Gross, J. Application of Infinite Dilution Activity Coefficients for Determining Binary Equation of State Parameters. Ind. Eng. Chem. Res. 2010, 49, 7646–7653. [Google Scholar] [CrossRef]
- Schreiber, L.B.; Eckert, C.A. Use of Infinite Dilution Activity Coefficients with Wilson’s Equation. Ind. Eng. Chem. Process Des. Dev. 1971, 10, 572–576. [Google Scholar] [CrossRef]
- Chipot, C.; Pohorille, A. (Eds.) Free Energy Calculations: Theory and Applications in Chemistry and Biology; Springer Series in Chemical Physics; Springer: New York, NY, USA, 2007; Volume 86. [Google Scholar]
- Roese, S.N.; Heintz, J.D.; Uzat, C.B.; Schmidt, A.J.; Margulis, G.V.; Sabatino, S.J.; Paluch, A.S. Assessment of the SM12, SM8, and SMD Solvation Models for Predicting Limiting Activity Coefficients at 298.15 K. Processes 2020, 8, 623. [Google Scholar] [CrossRef]
- Roese, S.N.; Margulis, G.V.; Schmidt, A.J.; Uzat, C.B.; Heintz, J.D.; Paluch, A.S. A Simple Method to Predict and Interpret the Formation of Azeotropes in Binary Systems Using Conventional Solvation Free Energy Calculations. Ind. Eng. Chem. Res. 2019, 58, 22626–22632. [Google Scholar] [CrossRef]
- Gebhardt, J.; Kiesel, M.; Riniker, S.; Hansen, N. Combining Molecular Dynamics and Machine Learning to Predict Self-Solvation Free Energies and Limiting Activity Coefficients. J. Chem. Inf. Model 2020, 60, 5319–5330. [Google Scholar] [CrossRef]
- Fuerst, G.B.; Ley, R.T.; Paluch, A.S. Calculating the Fugacity of Pure, Low Volatile Liquids via Molecular Simulation with Application to Acetanilide, Acetaminophen, and Phenacetin. Ind. Eng. Chem. Res. 2015, 54, 9027–9037. [Google Scholar] [CrossRef]
- Winget, P.; Hawkins, G.D.; Cramer, C.J.; Truhlar, D.G. Predicting the Vapor Pressures from Self-Solvation Free Energies Calculated by the SM5 Series of Universal Solvation Models. J. Phys. Chem. B 2000, 104, 4726. [Google Scholar] [CrossRef]
- Horn, H.W.; Swope, W.C.; Pitera, J.W. Characterization of the TIP4P-Ew water model: Vapor pressure and boiling point. J. Chem. Phys. 2005, 123, 194504. [Google Scholar] [CrossRef] [PubMed]
- Hukkerikar, A.S.; Sarup, B.; Kate, A.T.; Abildskov, J.; Sin, G.; Gani, R. Group-contribution+ (GC+) based estimation of properties of pure components: Improved property estimation and uncertainty analysis. Fluid Phase Equilib. 2012, 321, 25–43. [Google Scholar] [CrossRef]
- Leach, A.R. Molecular Modelling: Principles and Applications, 2nd ed.; Pearson Education Limited: Harlow, UK, 2001. [Google Scholar]
- Frenkel, D.; Smit, B. Understanding Molecular Simulation: From Algorithms to Applications, 2nd ed.; Academic Press: San Diego, CA, USA, 2002. [Google Scholar]
- Rai, N.; Siepmann, J.I. Transferable Potentials for Phase Equilibria. 9. Explicit Hydrogen Description of Benzene and Five-Membered and Six-Membered Heterocyclic Aromatic Compounds. J. Phys. Chem. B 2007, 111, 10790–10799. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.G.; Siepmann, J.I. Transferable Potentials for Phase Equilibria. 1. United-Atom Description of n-Alkanes. J. Phys. Chem. B 1998, 102, 2569–2577. [Google Scholar] [CrossRef]
- Martin, M.G.; Siepmann, J.I. Novel Configurational-Bias Monte Carlo Method for Branched Molecules. Transferable Potentials for Phase Equilibria. 2. United-Atom Description of Branched Alkanes. J. Phys. Chem. B 1999, 103, 4508–4517. [Google Scholar] [CrossRef]
- Wick, C.D.; Martin, M.G.; Siepmann, J.I. Transferable Potentials for Phase Equilibria. 4. United-Atom Description of Linear and Branched Alkenes and Alkylbenzenes. J. Phys. Chem. B 2000, 104, 8008–8016. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Potoff, J.J.; Siepmann, J.I. Monte Carlo Calculations for Alcohols and Their Mixtures with Alkanes. Transferable Potentials for Phase Equilibria. 5. United-Atom Description of Primary, Secondary, and Tertiary Alcohols. J. Phys. Chem. B 2001, 105, 3093–3104. [Google Scholar] [CrossRef]
- Stubbs, J.M.; Potoff, J.J.; Siepmann, J.I. Transferable Potentials for Phase Equilibria. 6. United-Atom Description for Ethers, Glycols, Ketones, and Aldehydes. J. Phys. Chem. B 2004, 108, 17596–17605. [Google Scholar] [CrossRef]
- Wick, C.D.; Stubbs, J.M.; Rai, N.; Siepmann, J.I. Transferable Potentials for Phase Equilibria. 7. Primary, Secondary, and Tertiary Amines, Nitroalkanes and Nitrobenzene, Nitriles, Amides, Pyridine, and Pyrimidine. J. Phys. Chem. B 2005, 109, 18974–18982. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations. J. Mol. Graph. Modell. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindal, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- GROMACS: Fast, Flexible, Free. Available online: https://www.gromacs.org/ (accessed on 1 December 2020).
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926. [Google Scholar] [CrossRef]
- Chen, B.; Siepmann, J.I. Microscopic structure and solvation in dry and wet octanol. J. Phys. Chem. B 2006, 110, 3555–3563. [Google Scholar] [CrossRef]
- Rafferty, J.L.; Sun, L.; Siepmann, J.I.; Schure, M.R. Investigation of the driving forces for retention in reversed-phase liquid chromatography: Monte Carlo simulations of solute partitioning between n-hexadecane and various aqueous-organic mixtures. Fluid Phase Equilib. 2010, 290, 25–35. [Google Scholar] [CrossRef]
- Rai, N.; Bhatt, D.; Siepmann, J.I.; Fried, L.E. Monte Carlo simulations of 1,3,5-triamino-2,4,6-trinitrobenzene (TATB): Pressure and temperature effects for the solid phase and vapor-liquid phase equilibria. J. Chem. Phys. 2008, 129, 194510. [Google Scholar] [CrossRef]
- Rai, N.; Siepmann, J.I. Transferable Potentials for Phase Equilibria. 10. Explicit-Hydrogen Description of Substituted Benzenes and Polycyclic Aromatic Compounds. J. Phys. Chem. B 2013, 117, 273–288. [Google Scholar] [CrossRef]
- Caudle, M.A.; Cox, C.E.; Ley, R.T.; Paluch, A.S. A molecular study of the wastewater contaminants atenolol and atrazine in 1-n-butyl-3-methylimidazolium based ionic liquids for potential treatment applications. Mol. Phys. 2017, 115, 1264–1273. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 theory of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Account 2008, 120, 215–241. [Google Scholar]
- Cramer, C.J. Essentials of Computational Chemistry, 2nd ed.; John Wiley & Sons Ltd.: Chichester, UK, 2004. [Google Scholar]
- Marenich, A.V.; Olson, R.M.; Kelly, C.P.; Cramer, C.J.; Truhlar, D.G. Self-Consistent Reaction Field Model for Aqueous and Nonaqueous Solutions Based on Accurate Polarized Partial Charges. J. Chem. Theory. Comput. 2007, 3, 2011–2033. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.P.; Cramer, C.J.; Truhlar, D.G. SM6: A Density Functional Theory Continuum Solvation Model for Calculating Aqueous Solvation Free Energies of Neutrals, Ions, and Solute-Water Clusters. J. Chem. Theory. Comput. 2005, 1, 1133–1152. [Google Scholar] [CrossRef] [PubMed]
- Olson, R.M.; Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Charge Model 4 and Intramolecular Charge Polarization. J. Chem. Theory. Comput. 2007, 3, 2046–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, Y.; Gan, Z.; Epifanovsky, E.; Gilbert, A.T.B.; Wormit, M.; Kussmann, J.; Lange, A.W.; Behn, A.; Deng, J.; Feng, X.; et al. Advances in molecular quantum chemistry contained in the Q-Chem 4 program package. Mol. Phys. 2015, 113, 184–215. [Google Scholar] [CrossRef] [Green Version]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Bayly, C.I.; Cieplak, P.; Cornell, W.D.; Kollman, P.A. A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges: The RESP Model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Cieplak, P.; Cornell, W.D.; Bayly, C.; Kollman, P.A. Application of the multimolecule and multiconformational RESP methodology to biopolymers: Charge derivation for DNA, RNA, and proteins. J. Comput. Chem. 1995, 16, 1357–1377. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Darden, T.A.; Cheatham, T.E., III; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Walker, R.C.; Zhang, W.; Merz, K.M.; et al. AMBER 12; University of California: San Francisco, CA, USA, 2012. [Google Scholar]
- Comparison of Solvation Packages. Available online: https://comp.chem.umn.edu/solvation/comparison.htm (accessed on 28 February 2022).
- Mobley, D.L.; Dumont, E.; Chodera, J.D.; Dill, K.A. Comparison of Charge Models for Fixed-Charge Force Fields: Small-Molecule Hydration Free Energies in Explicit Solvents. J. Phys. Chem. B 2007, 111, 2242–2254. [Google Scholar] [CrossRef]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 367. Available online: https://bmcresnotes.biomedcentral.com/articles/10.1186/1756-0500-5-367 (accessed on 1 December 2020). [CrossRef] [Green Version]
- Sousa da Silva, A.W.; Vranken, W.F. acpype: AnteChamber PYthon Parser interfacE. Available online: https://pypi.org/project/acpype/ (accessed on 1 December 2020).
- Shirts, M.R.; Chodera, J.D. Statistically optimal analysis of samples from multiple equilibrium states. J. Chem. Phys. 2008, 129, 124105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beutler, T.C.; Mark, A.E.; van Schaik, R.C.; Gerber, P.R.; van Gunsteren, W.F. Avoiding singularities and numerical instabilities in free energy calculations based on molecular simulations. Chem. Phys. Lett. 1994, 222, 529–539. [Google Scholar] [CrossRef]
- Steinbrecher, T.; Mobley, D.L.; Case, D.A. Nonlinear scaling schemes for Lennard-Jones interactions in free energy calculations. J. Chem. Phys. 2007, 127, 214108. [Google Scholar] [CrossRef] [PubMed]
- PyMBAR: Python Implementation of the Multistate Bennett Acceptance Ratio (MBAR). Available online: https://github.com/choderalab/pymbar (accessed on 1 December 2020).
- Chodera, J.D.; Swope, W.C.; Pitera, J.W.; Seok, C.; Dill, K.A. Use of the Weighted Histogram Analysis Method for the Analysis of Simulated and Parallel Tempering Simulations. J. Chem. Theory Comput. 2007, 3, 26–41. [Google Scholar] [CrossRef] [Green Version]
- Klimovich, P.V.; Shirts, M.R.; Mobley, D.L. Guidelines for analysis of free energy calculations. J. Comput.-Aided Mol. Des. 2015, 29, 397–411. [Google Scholar] [CrossRef] [Green Version]
- Storn, R.; Price, K. Differential Evolution—A Simple and Efficient Heuristic for Global Optimization over Continuous Spaces. J. Global. Optim. 1997, 11, 341–359. [Google Scholar] [CrossRef]
- Eaton, J.W.; Bateman, D.; Hauberg, S. GNU Octave Version 3.0.1 Manual: A High-Level Interactive Language for Numerical Computations; CreateSpace Independent Publishing Platform: Charleston, SC, USA, 2009; ISBN 1441413006. [Google Scholar]
- Marrero, J.; Abildskov, J. (Eds.) Solubility and Related Properties of Large Complex Chemicals Part 1: Organic Solutes Ranging from C4 to C40; DECHEMA: Frankfurt am Main, Germany, 2003. [Google Scholar]
- Abildskov, J. (Ed.) Solubility and Related Properties of Large Complex Chemicals Part 2: Organic Solutes Ranging from C2 to C41; DECHEMA: Frankfurt am Main, Germany, 2005. [Google Scholar]
- Jia, D.; Li, Y.; Li, C. Measurement and Correlation of Solubility 2-Chloro-4-ethylamino-6-isopropylamino-1,3,5-triazine in Different Organic Solvents. J. Chem. Eng. Data 2013, 58, 3183–3189. [Google Scholar] [CrossRef]
- Donnelly, J.R.; Drewes, L.A.; Johnson, R.L.; Munslow, W.D.; Knapp, K.K.; Sovocool, G.W. Purity and Heat of Fusion Data for Environmental Standards as Determined by Differential Scanning Calorimetry. Thermochim. Acta 1990, 167, 155–187. [Google Scholar] [CrossRef]
- Anwer, M.K. Dissolution Thermodynamics and Solubility of Atenolol in Seven Different Solvents Useful in Dosage Form Design. Lat. Am. J. Pharm. 2015, 34, 1571–1575. [Google Scholar]
- Perlovich, G.L.; Volkova, T.V.; Bauer-Brandl, A. Thermodynamic Study of Sublimation, Solubility, Solvation, and Distribution Processes of Atenolol and Pindolol. Mol. Pharm. 2007, 4, 929–935. [Google Scholar] [CrossRef]
- Domańska, U.; Pobudkowska, A.; Pelczarska, A.; Winiarska-Tusznio, M. Solubility and pKa of select pharmaceuticals in water, ethanol, and 1-octanol. J. Chem. Thermodyn. 2010, 42, 1465–1472. [Google Scholar] [CrossRef]
- Ohio Supercomputer Center. Available online: http://osc.edu/ark:/19495/f5s1ph73 (accessed on 1 February 2022).
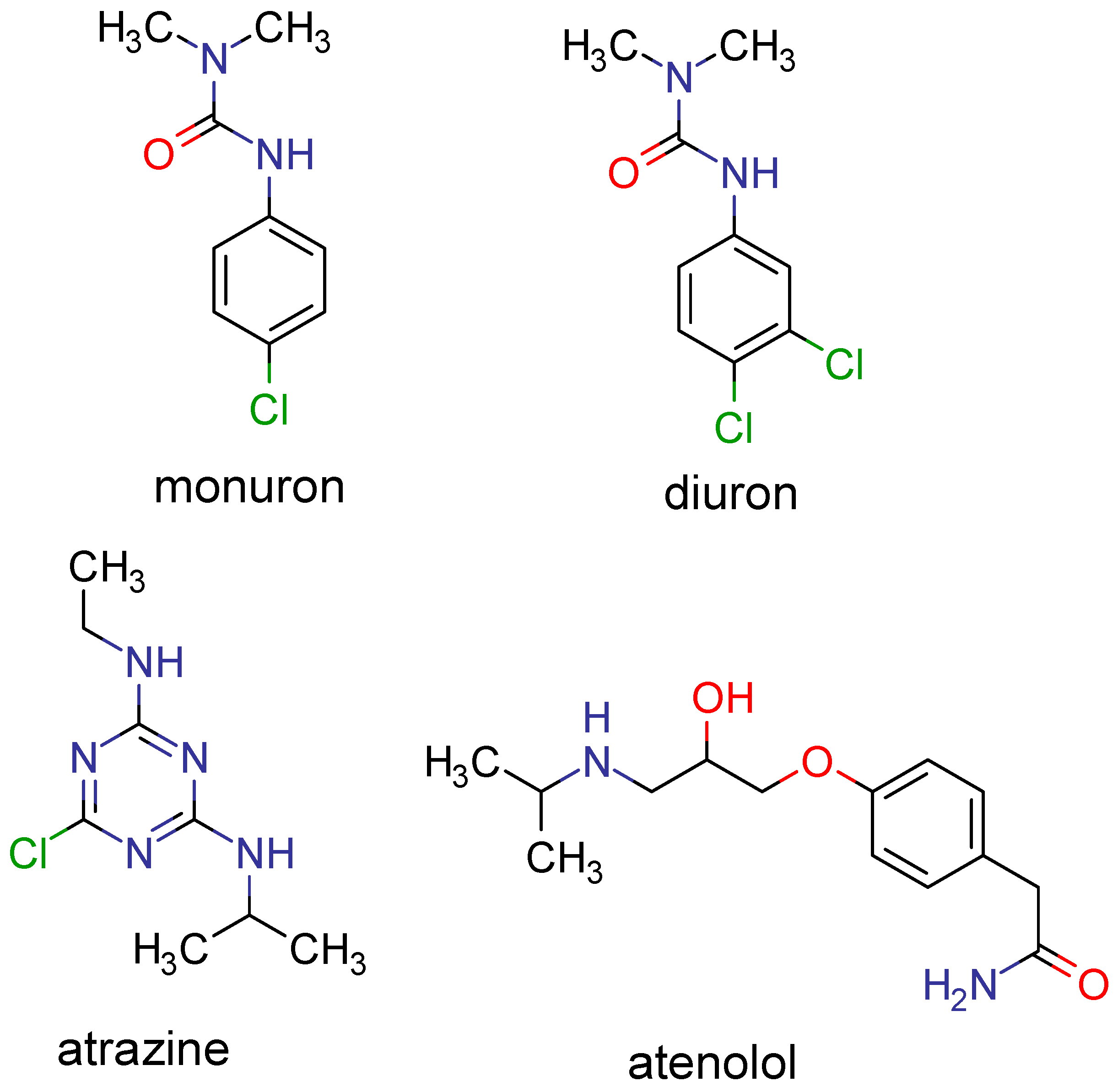
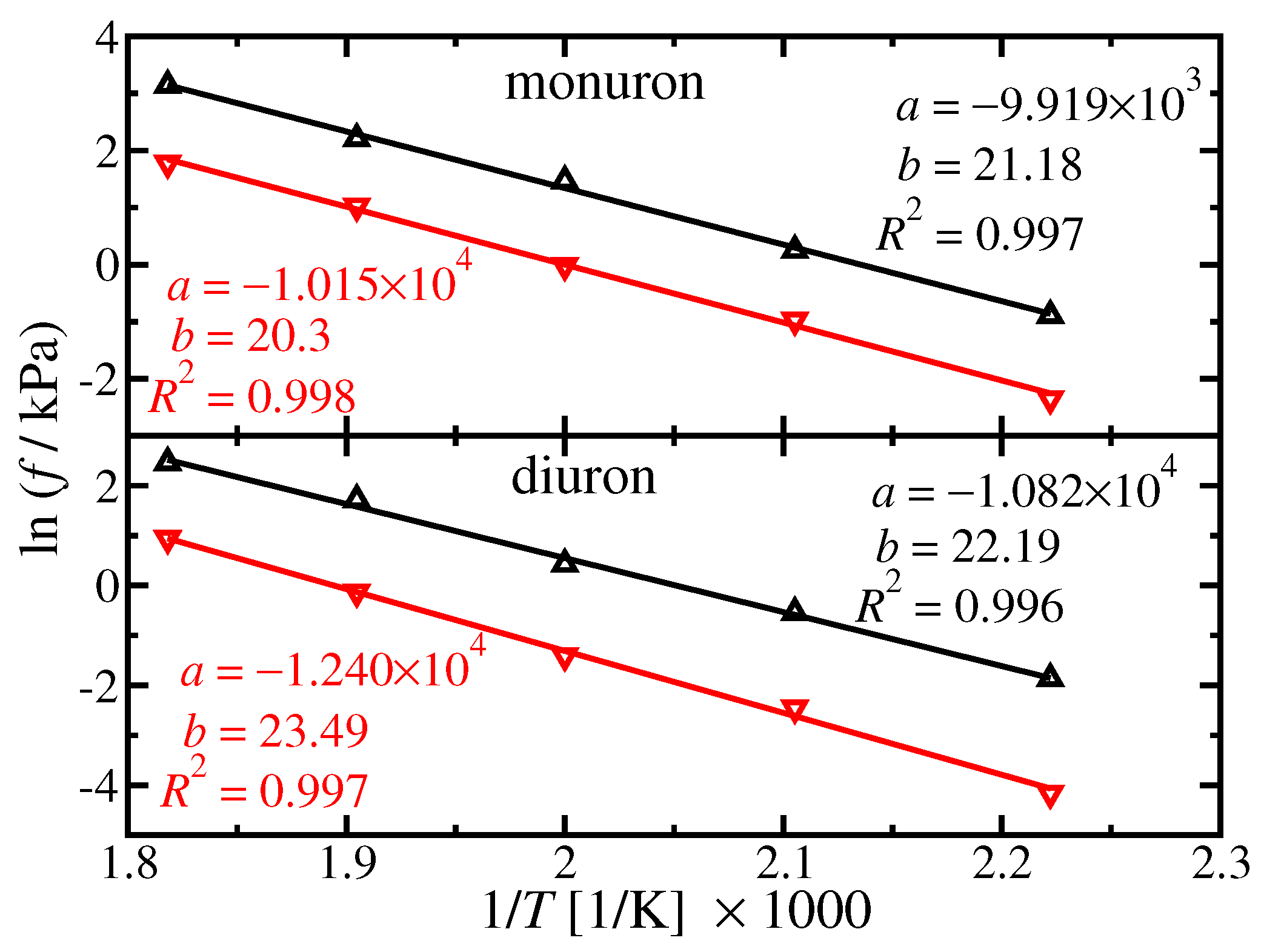
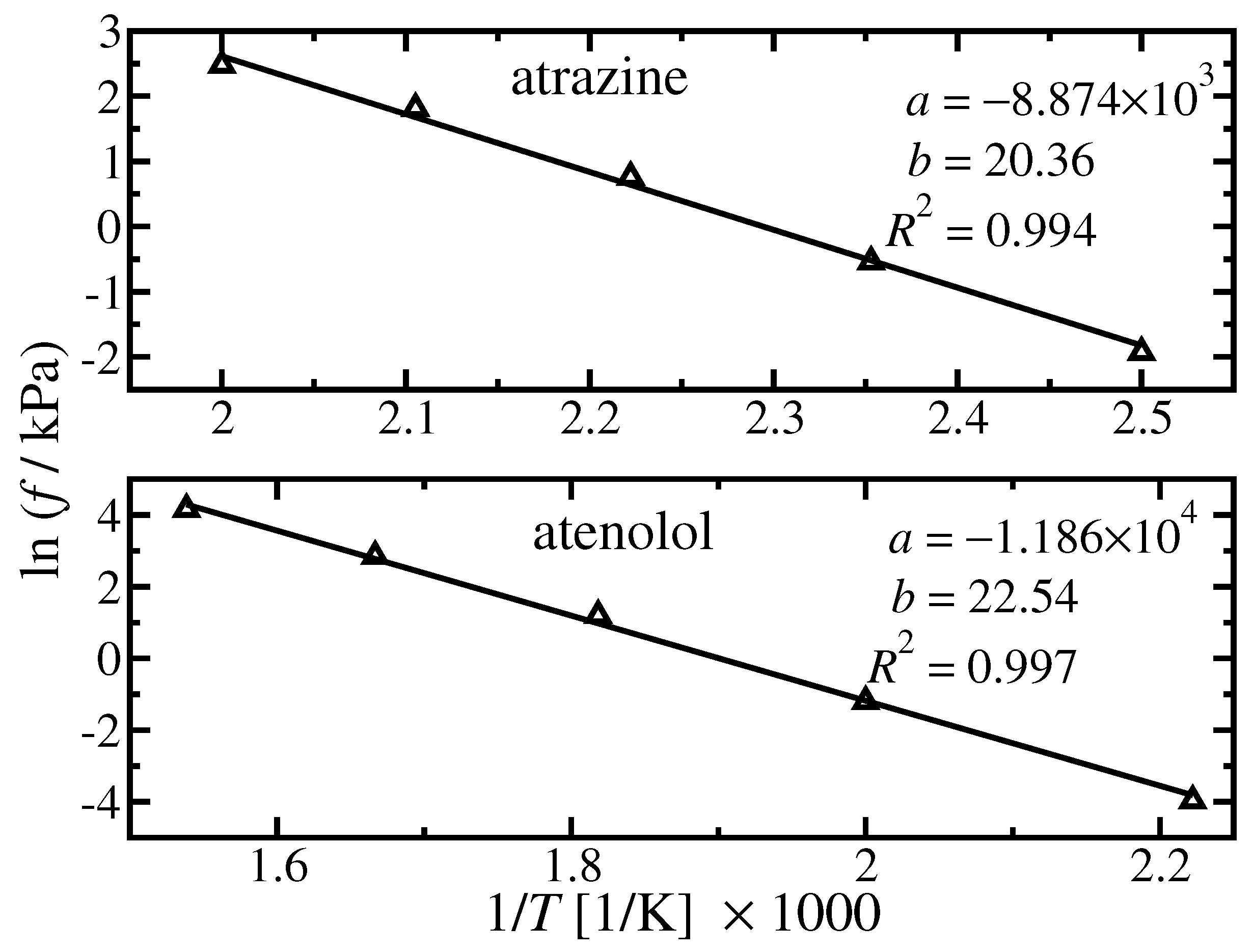

{kind=link}
{kind=link}
{kind=link}
| Solute | Method | N | RMSE | AAPD | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| monuron | ref | 152.80 | 16.44 | 5.48 | 7.16 | 9.65 | 32 | 22.0 | ||
| SM8 | 164.75 | 15.65 | 1.38 | 13.11 | 7.42 | 13 | 0.985 | 0.430 | 34.5 | |
| SMD | 162.98 | 12.49 | 0.00 | 18.63 | 6.87 | 13 | 0.931 | 1.233 | 156.7 | |
| diuron | ref | 164.80 | 16.99 | 4.12 | 7.88 | 9.88 | 37 | 36.3 | ||
| SM8 | 176.94 | 17.07 | 3.00 | 12.37 | 8.26 | 13 | 0.900 | 1.282 | 98.0 | |
| SMD | 176.32 | 17.14 | 2.72 | 14.91 | 9.83 | 13 | 0.984 | 0.607 | 56.6 | |
| atrazine | SM8 | 183.35 | 15.32 | 3.04 | 9.89 | 4.26 | 13 | 0.984 | 0.438 | 38.1 |
| atenolol | SM8 | 253.43 | 14.20 | 1.53 | 12.18 | 4.03 | 13 | 0.953 | 1.116 | 191.5 |
| Monuron | Diuron | Atrazine | Atenolol | |||
|---|---|---|---|---|---|---|
| Solvent | SM8 | SMD | SM8 | SMD | SM8 | SM8 |
| n-hexane | ||||||
| 2,5-dimethylhexane | ||||||
| 1-hexene | ||||||
| 1-octene | ||||||
| benzene | ||||||
| methanol | ||||||
| ethanol | ||||||
| 1-propanol | ||||||
| 2-propanol | ||||||
| acetone | ||||||
| butanone | ||||||
| diethylether | ||||||
| water | ||||||
| Solute | N Systems | N Solvents | Method | AAPD | RMSE | RMSE | Slope | ||
|---|---|---|---|---|---|---|---|---|---|
| monuron | 32 | 31 | ref | 0 | 21.97 | 0.26 | 0.53 | 0.94 | 0.93 |
| 1 | 110.15 | 0.91 | 0.81 | 0.94 | 0.93 | ||||
| opt | 325.15 | 2.68 | 1.44 | 0.95 | 0.94 | ||||
| SM8 | 0 | 95.83 | 1.29 | 1.30 | 0.82 | 1.17 | |||
| 1 | 190.63 | 3.01 | 1.29 | 0.82 | 1.16 | ||||
| opt | 401.98 | 5.86 | 1.59 | 0.83 | 1.14 | ||||
| SMD | 0 | 125.15 | 1.56 | 2.96 | 0.73 | 1.59 | |||
| 1 | 227.99 | 3.38 | 2.60 | 0.73 | 1.58 | ||||
| opt | 404.31 | 6.30 | 2.39 | 0.74 | 1.56 | ||||
| diuron | 36 | 36 | ref | 0 | 35.94 | 0.35 | 0.59 | 0.95 | 0.95 |
| 1 | 98.80 | 0.70 | 0.73 | 0.95 | 0.95 | ||||
| opt | 281.92 | 1.97 | 1.32 | 0.95 | 0.95 | ||||
| SM8 | 0 | 74.02 | 0.59 | 1.50 | 0.91 | 1.27 | |||
| 1 | 120.38 | 1.54 | 1.15 | 0.92 | 1.27 | ||||
| opt | 218.97 | 3.24 | 1.19 | 0.92 | 1.26 | ||||
| SMD | 0 | 76.75 | 0.40 | 3.48 | 0.88 | 1.70 | |||
| 1 | 114.17 | 0.93 | 2.95 | 0.88 | 1.70 | ||||
| opt | 196.85 | 2.36 | 2.55 | 0.88 | 1.70 | ||||
| atrazine | 63 | 6 | SM8 | 0 | 64.79 | 1.02 | 0.53 | 0.93 | 1.06 |
| 1 | 277.12 | 2.65 | 1.32 | 0.91 | 0.79 | ||||
| opt | 720.13 | 5.05 | 2.04 | 0.75 | 0.47 | ||||
| atenolol | 54 | 6 | SM8 | 0 | 6506.42 | 8.92 | 3.02 | 0.69 | 0.58 |
| 1 | 11,513.77 | 11.51 | 3.39 | 0.73 | 0.55 | ||||
| opt | 19,679.49 | 14.49 | 3.73 | 0.76 | 0.52 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ollier, R.C.; Nguyen, T.; Agarwal, H.; Phifer, J.R.; Ferreira da Silva, L.; Gonçalves Nogueira, G.; Pereira Barbosa, A.K.; Ley, R.T.; O’Loughlin, E.J.; Rygelski, B.T.; et al. Predicting the Solubility of Nonelectrolyte Solids Using a Combination of Molecular Simulation with the Solubility Parameter Method MOSCED: Application to the Wastewater Contaminants Monuron, Diuron, Atrazine and Atenolol. Processes 2022, 10, 538. https://doi.org/10.3390/pr10030538
Ollier RC, Nguyen T, Agarwal H, Phifer JR, Ferreira da Silva L, Gonçalves Nogueira G, Pereira Barbosa AK, Ley RT, O’Loughlin EJ, Rygelski BT, et al. Predicting the Solubility of Nonelectrolyte Solids Using a Combination of Molecular Simulation with the Solubility Parameter Method MOSCED: Application to the Wastewater Contaminants Monuron, Diuron, Atrazine and Atenolol. Processes. 2022; 10(3):538. https://doi.org/10.3390/pr10030538
Chicago/Turabian StyleOllier, Rachel C., Thomas Nguyen, Hrithik Agarwal, Jeremy R. Phifer, Larissa Ferreira da Silva, Gabriel Gonçalves Nogueira, Ana Karolyne Pereira Barbosa, Ryan T. Ley, Elizabeth J. O’Loughlin, Brett T. Rygelski, and et al. 2022. "Predicting the Solubility of Nonelectrolyte Solids Using a Combination of Molecular Simulation with the Solubility Parameter Method MOSCED: Application to the Wastewater Contaminants Monuron, Diuron, Atrazine and Atenolol" Processes 10, no. 3: 538. https://doi.org/10.3390/pr10030538