
Digital Twin for HIV-Gag VLP Production in HEK293 Cells

 ,
,  ,
,  , , and
, , and 
Abstract
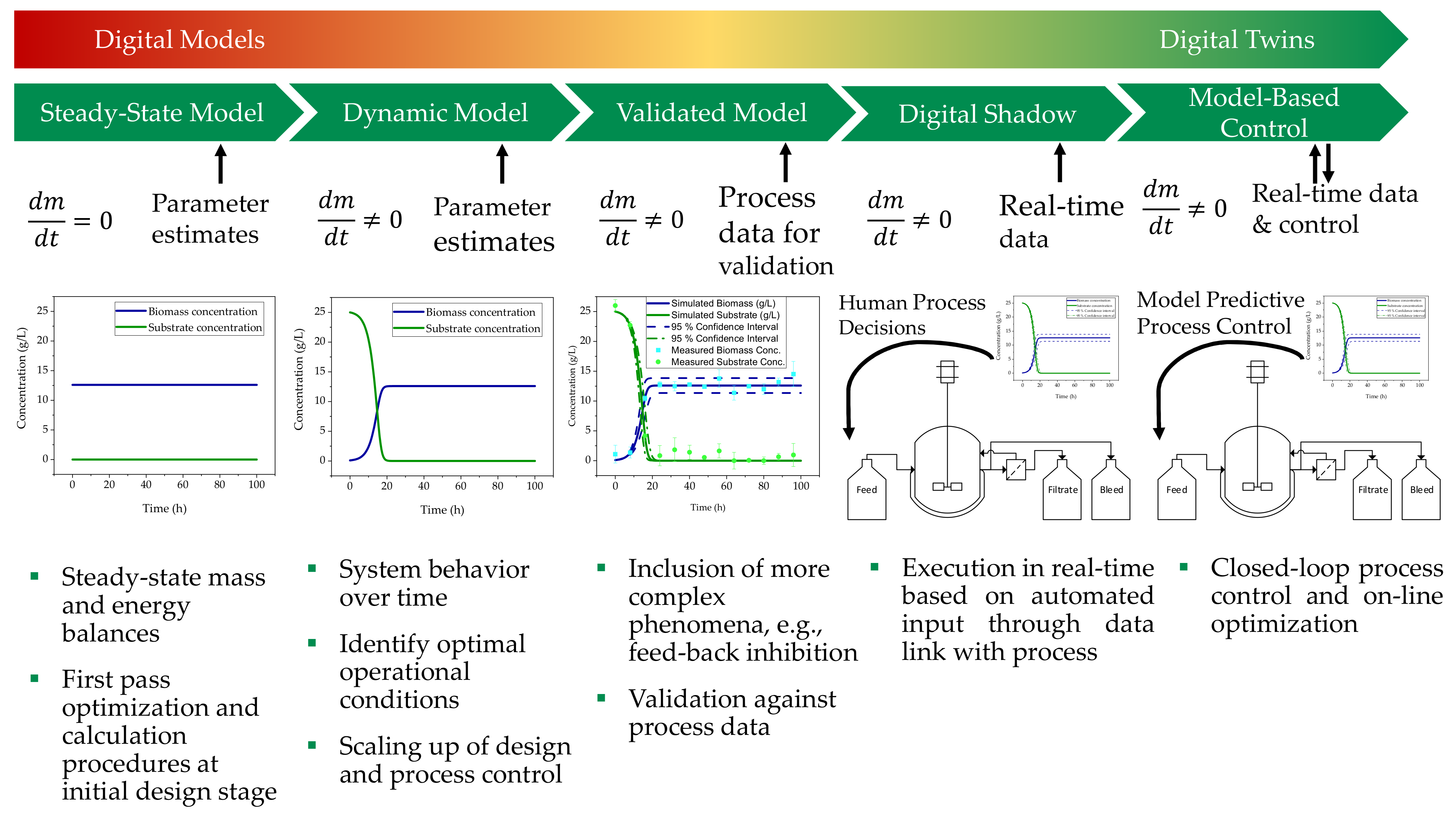
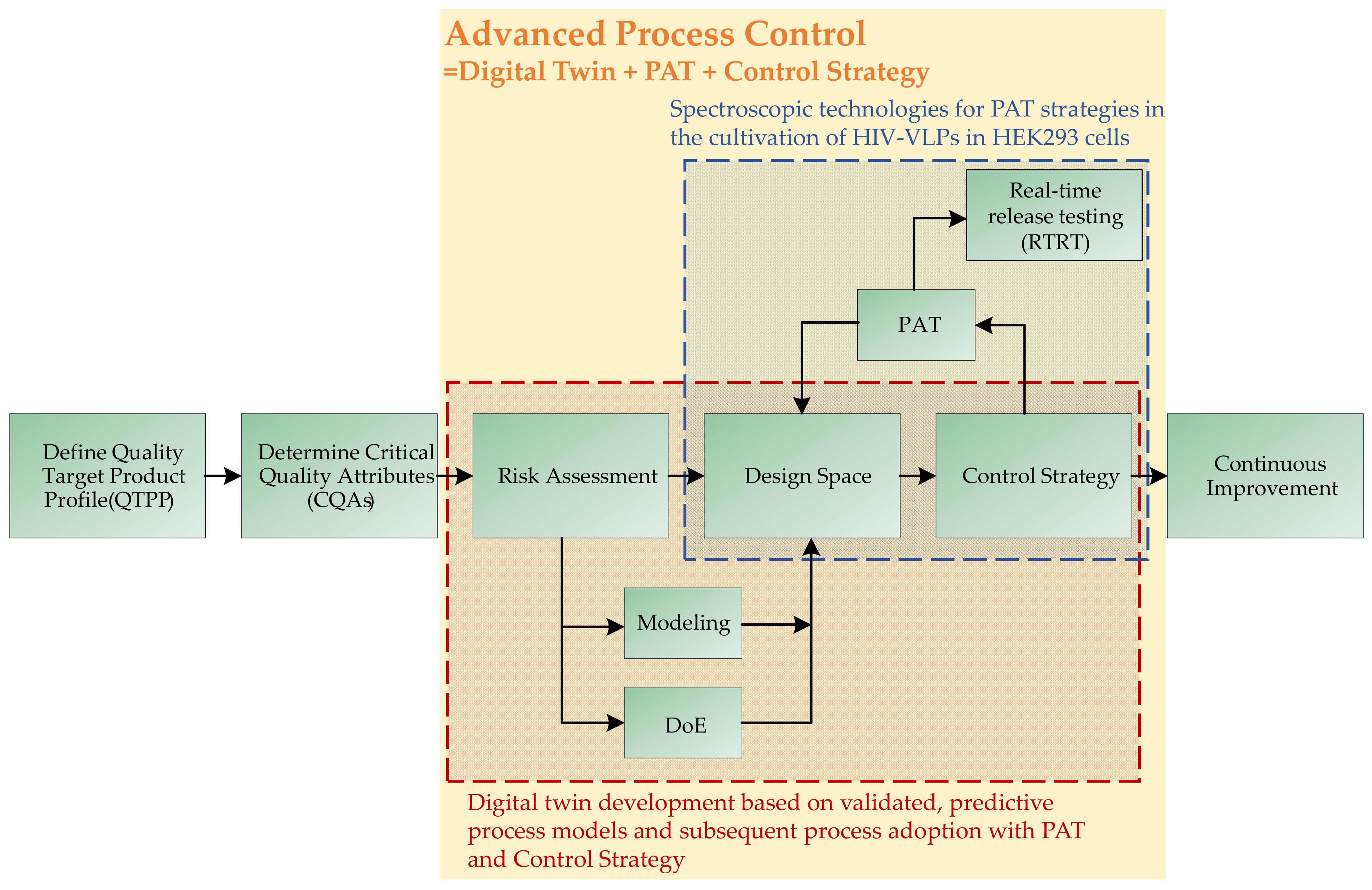
:1. Introduction
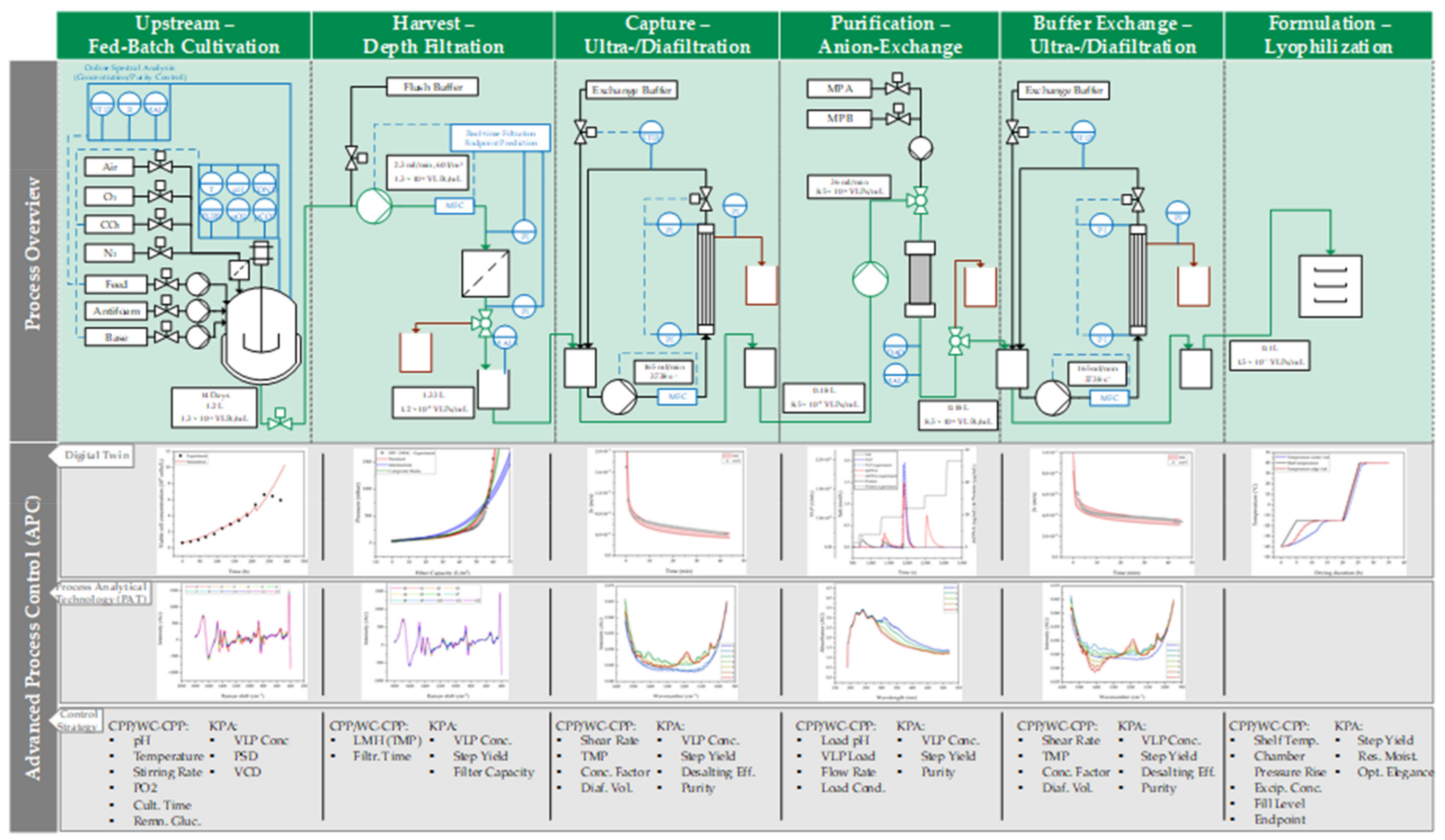
2. Materials and Methods
2.1. Fed-Batch Cultivation
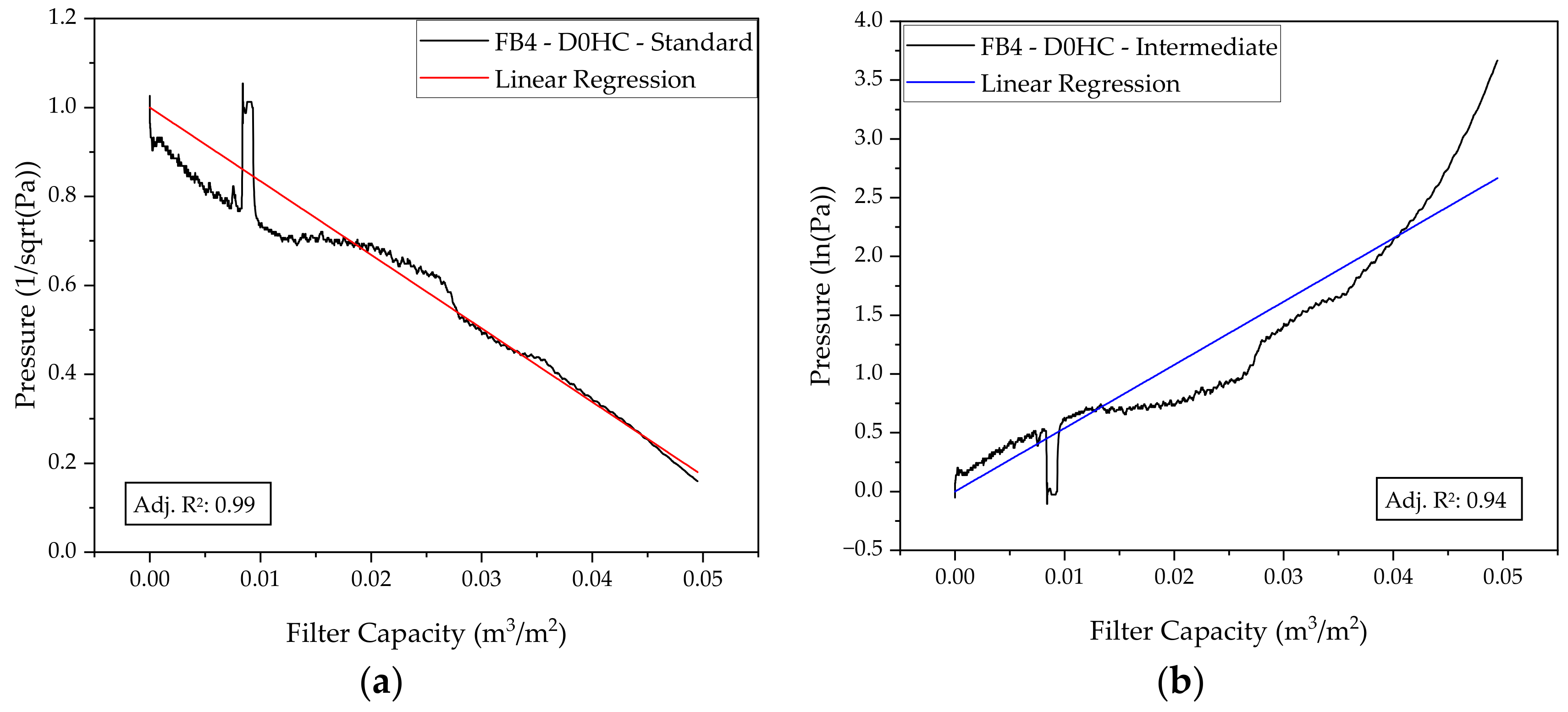
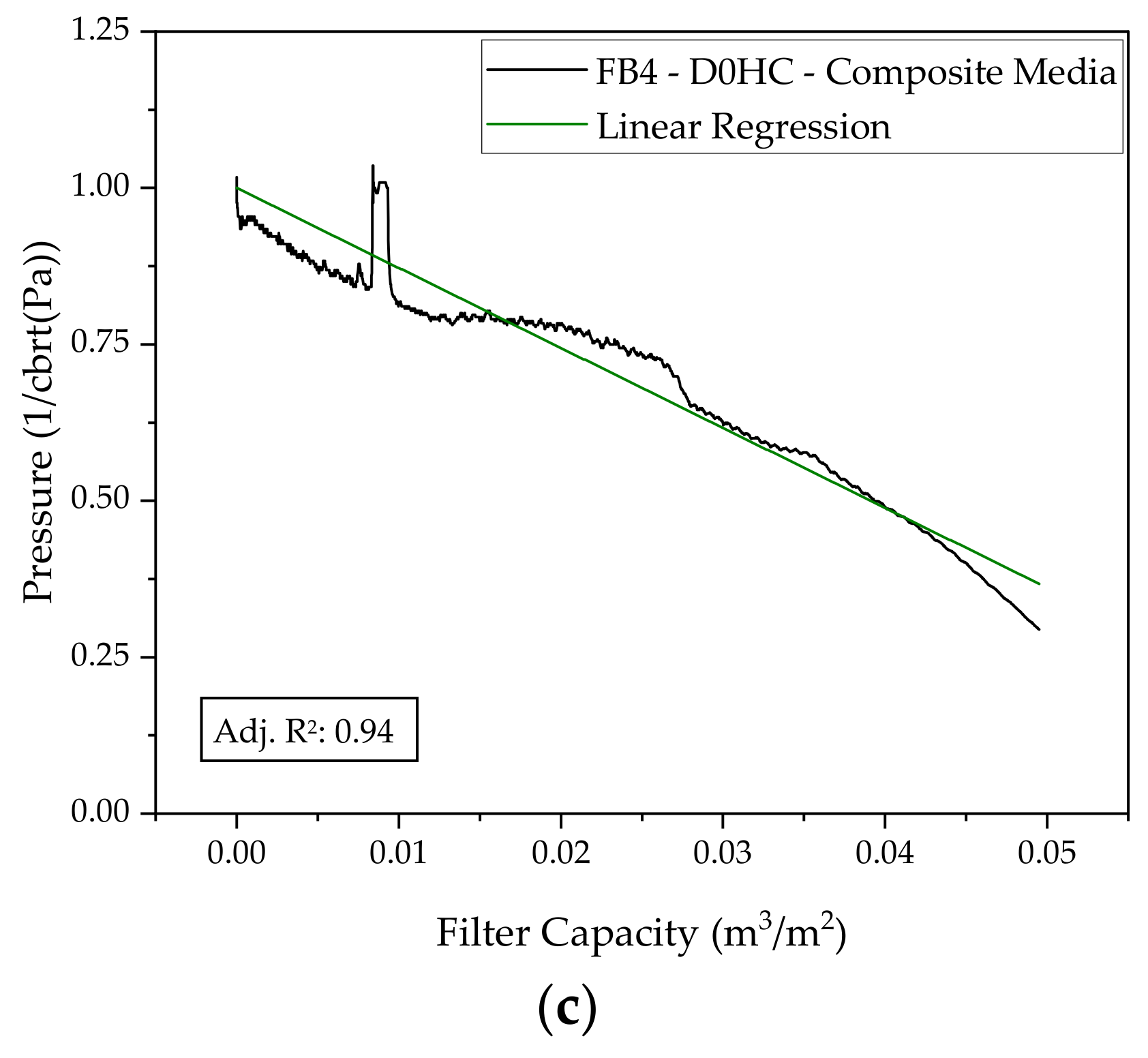
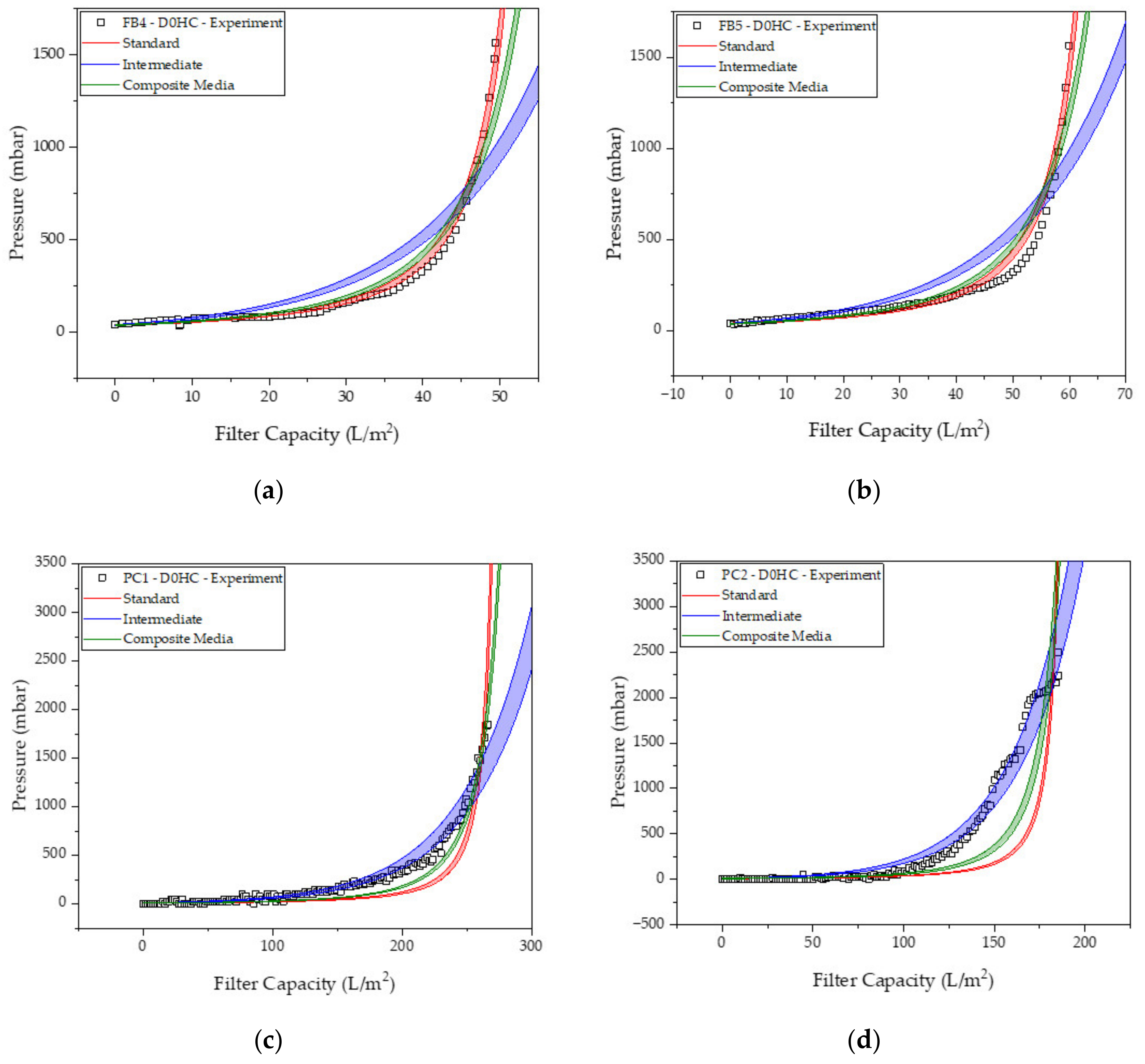
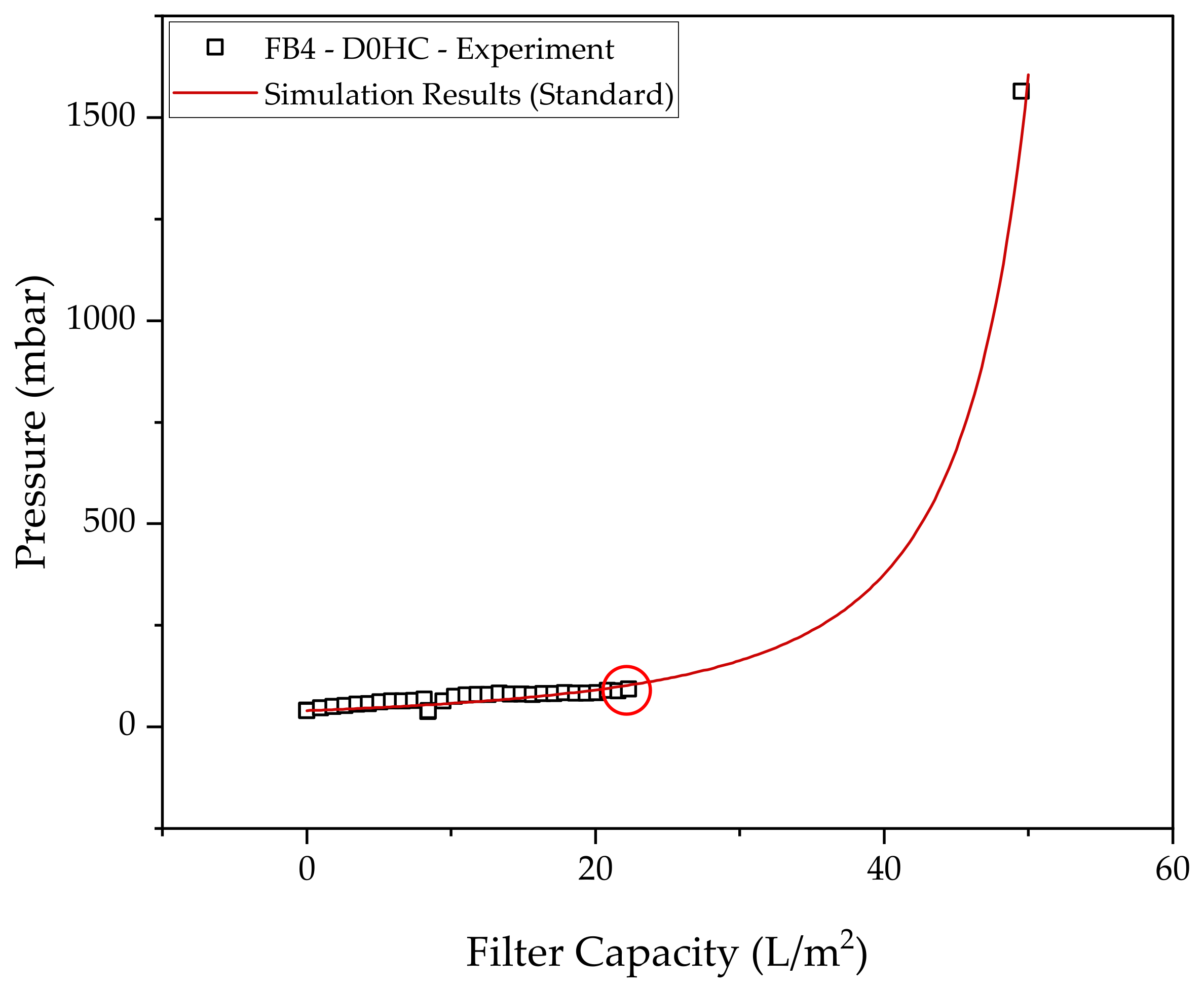
2.2. Depth Filtration
2.3. Ultra- and Diafiltration
2.4. Anion-Exchange Chromatography
2.5. Lyophilization
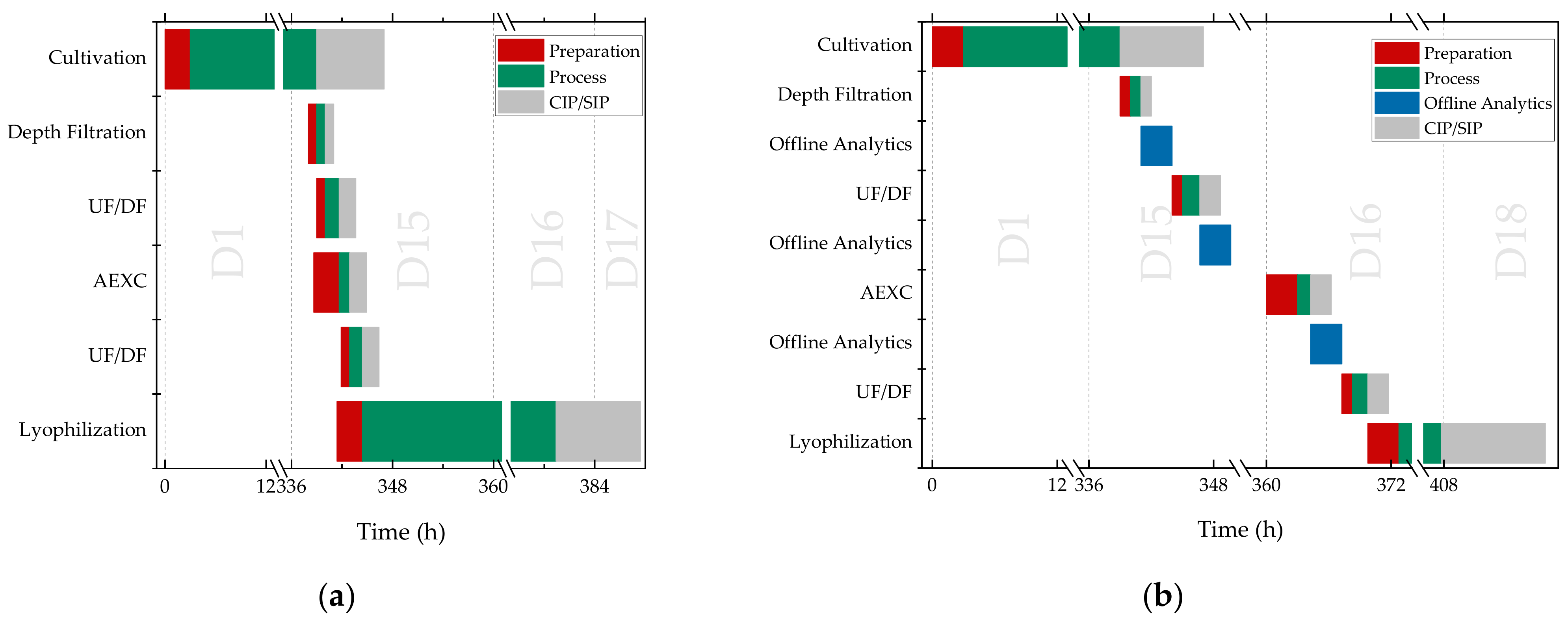
3. Results and Discussion
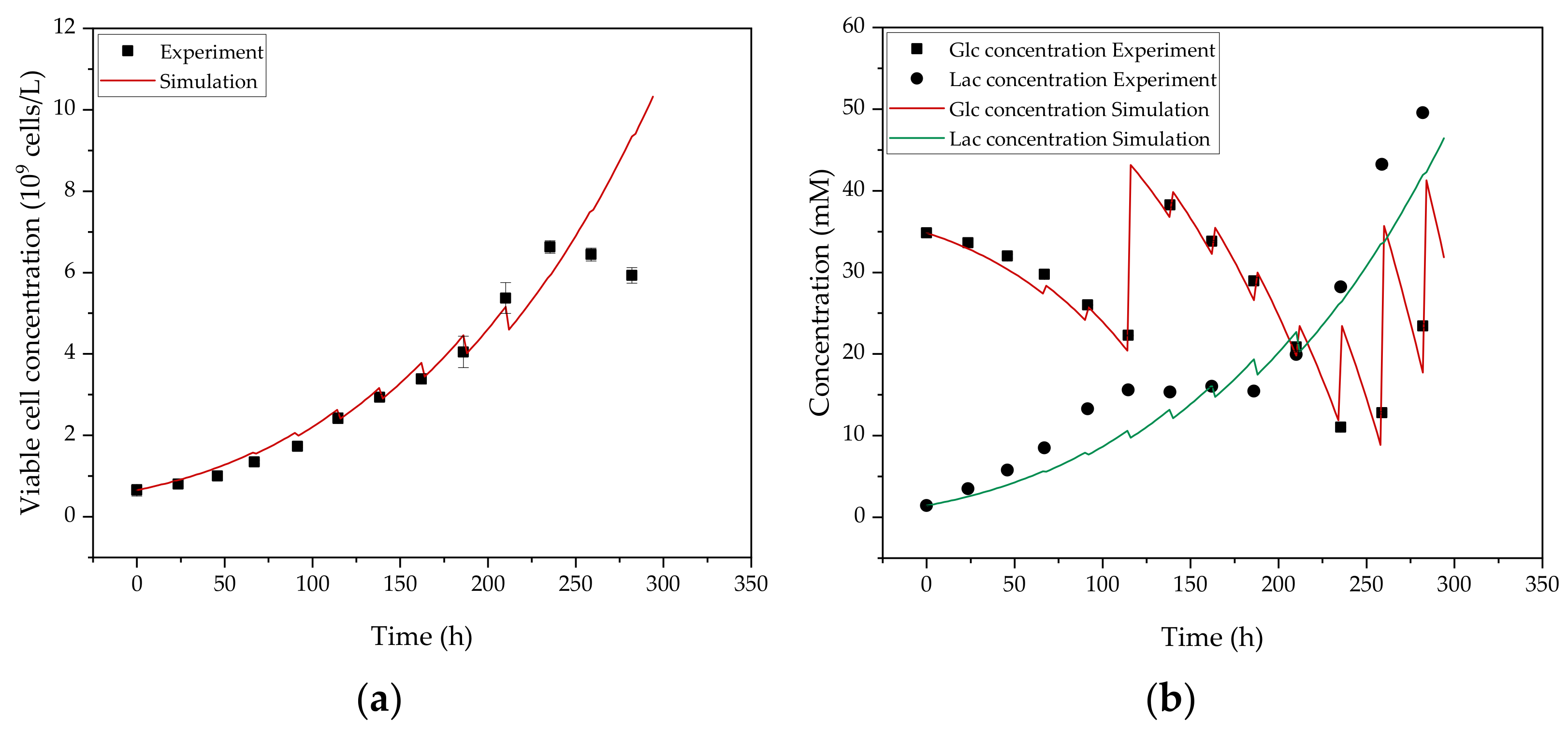
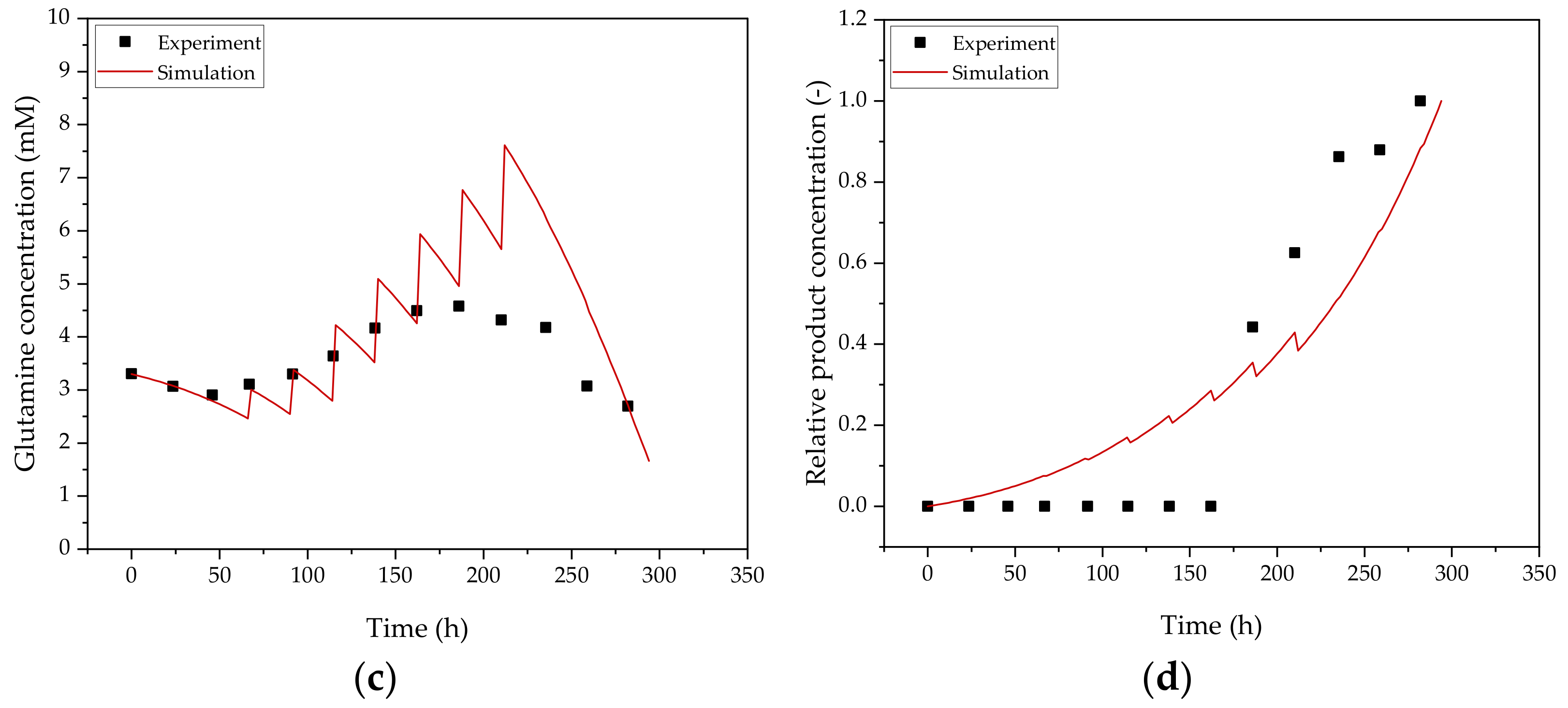
3.1. Cultivation of HIV-Gag Producing HEK293 Cells
3.2. Harvest via Depth Filtration
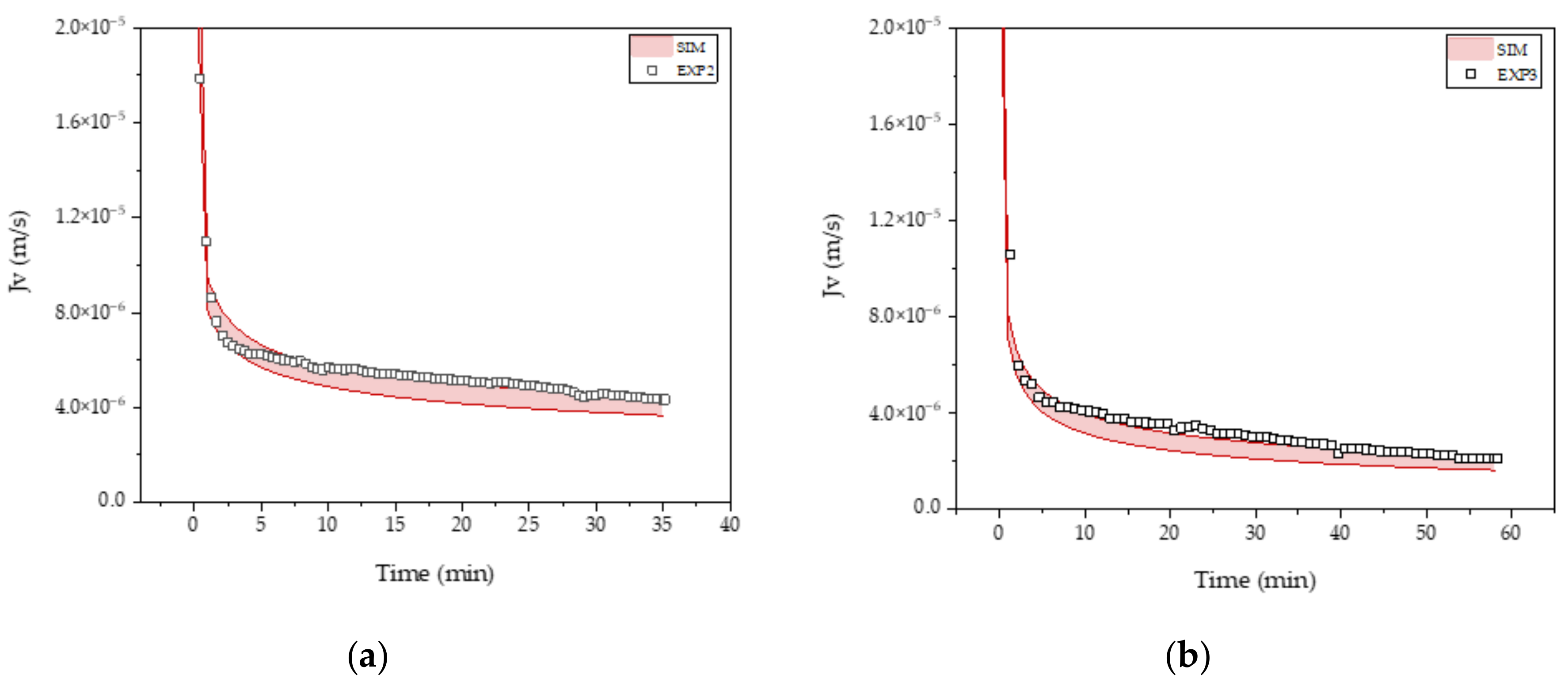
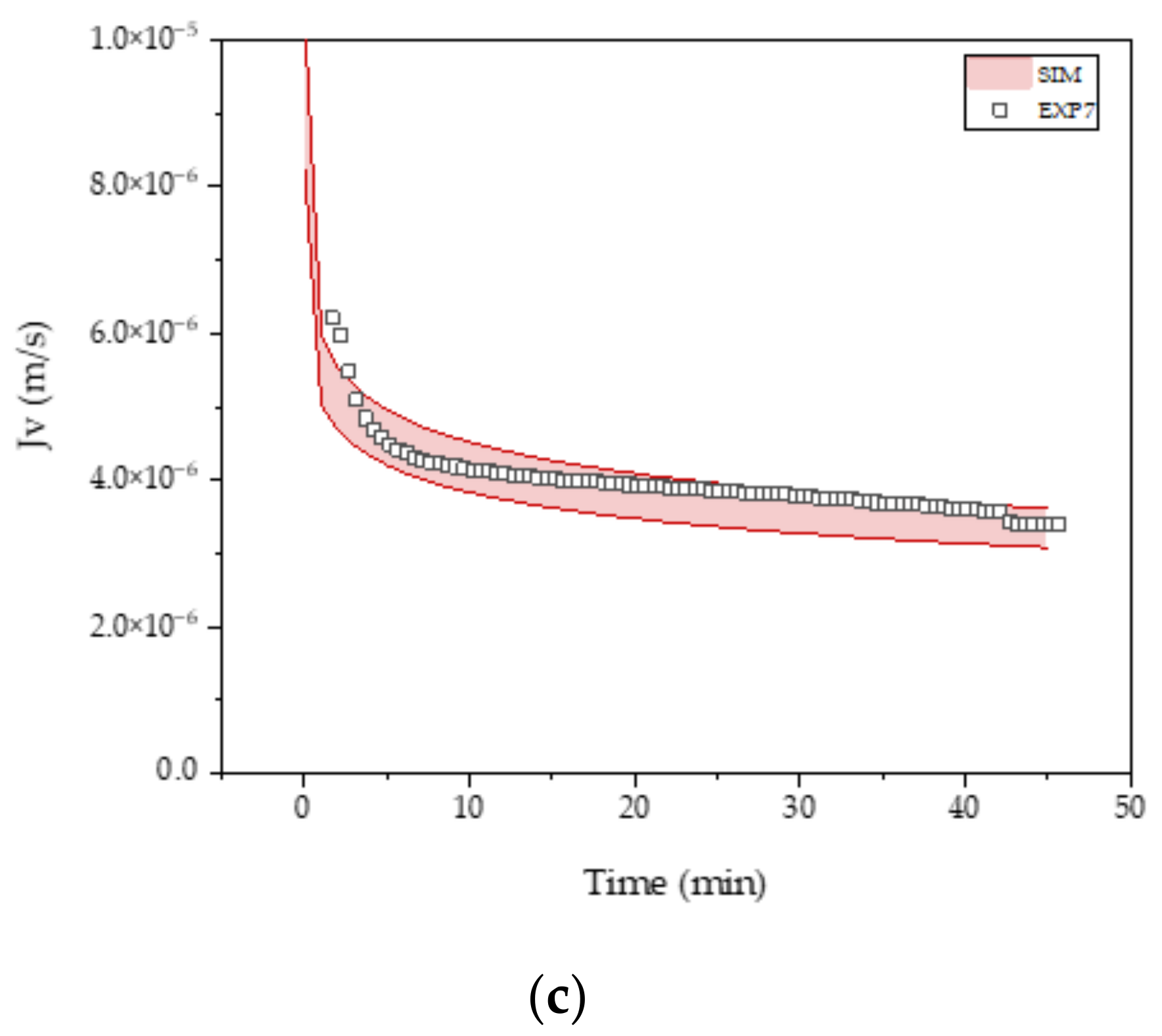
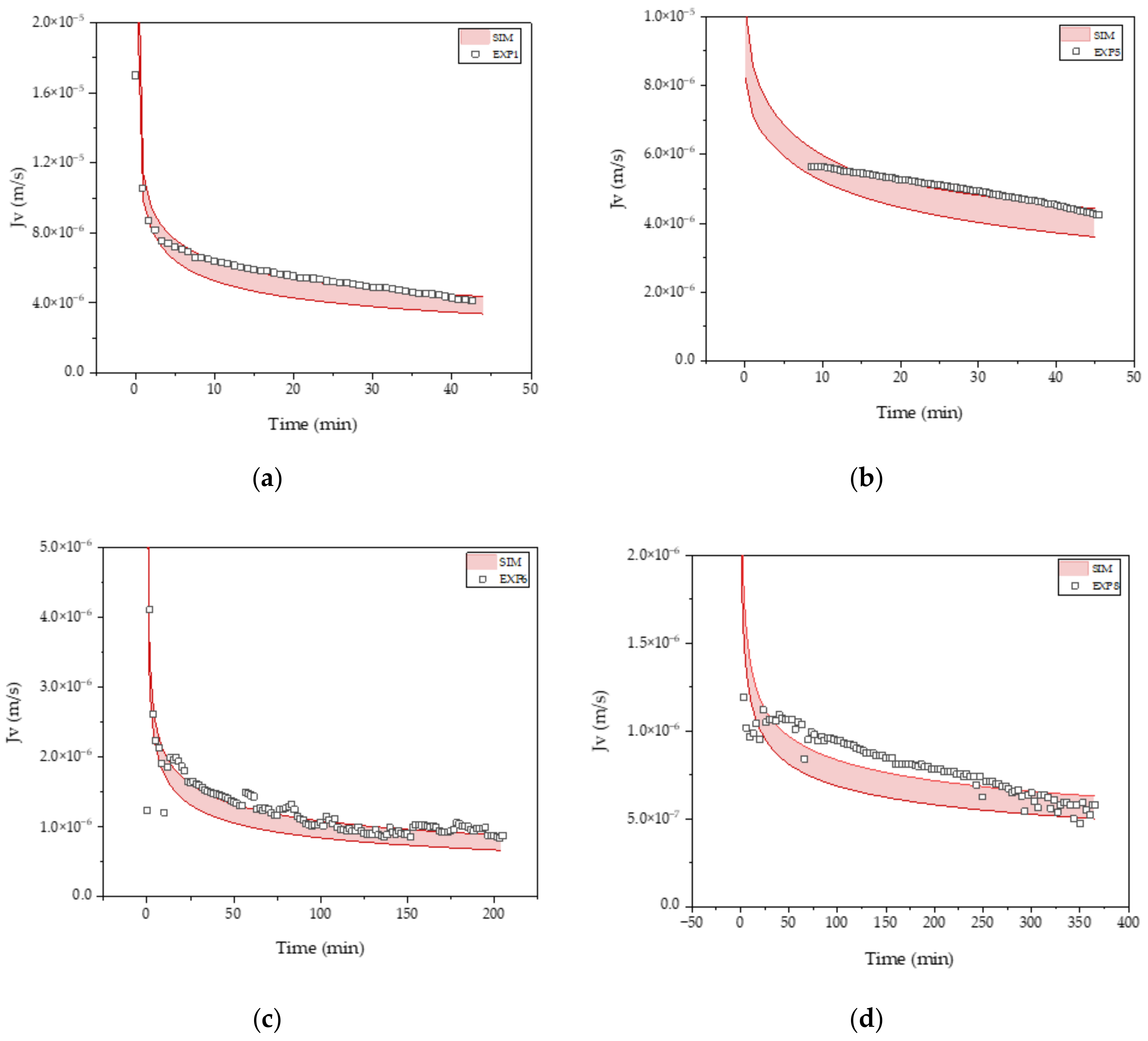
3.3. Intermediate Purification and Concentration via Ultra-/Diafiltration
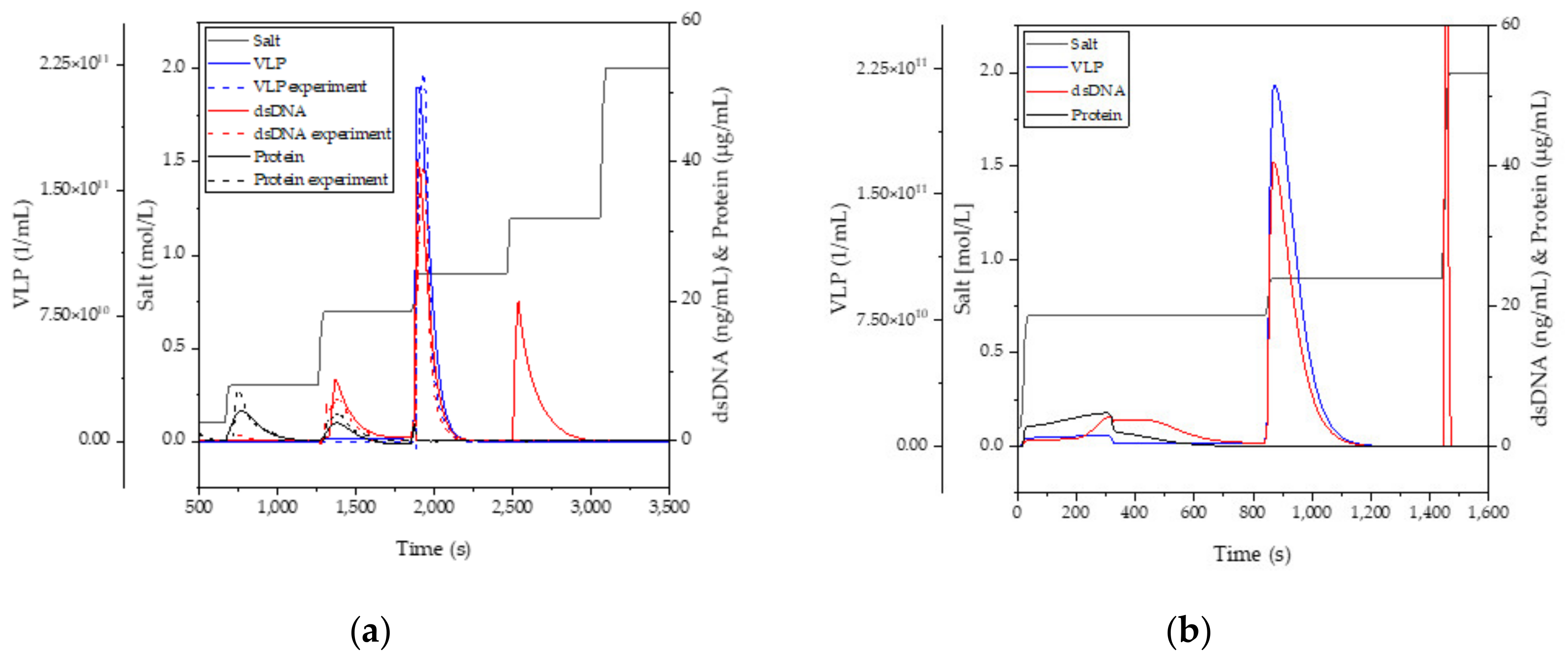
3.4. Anion-Exchange Chromatography
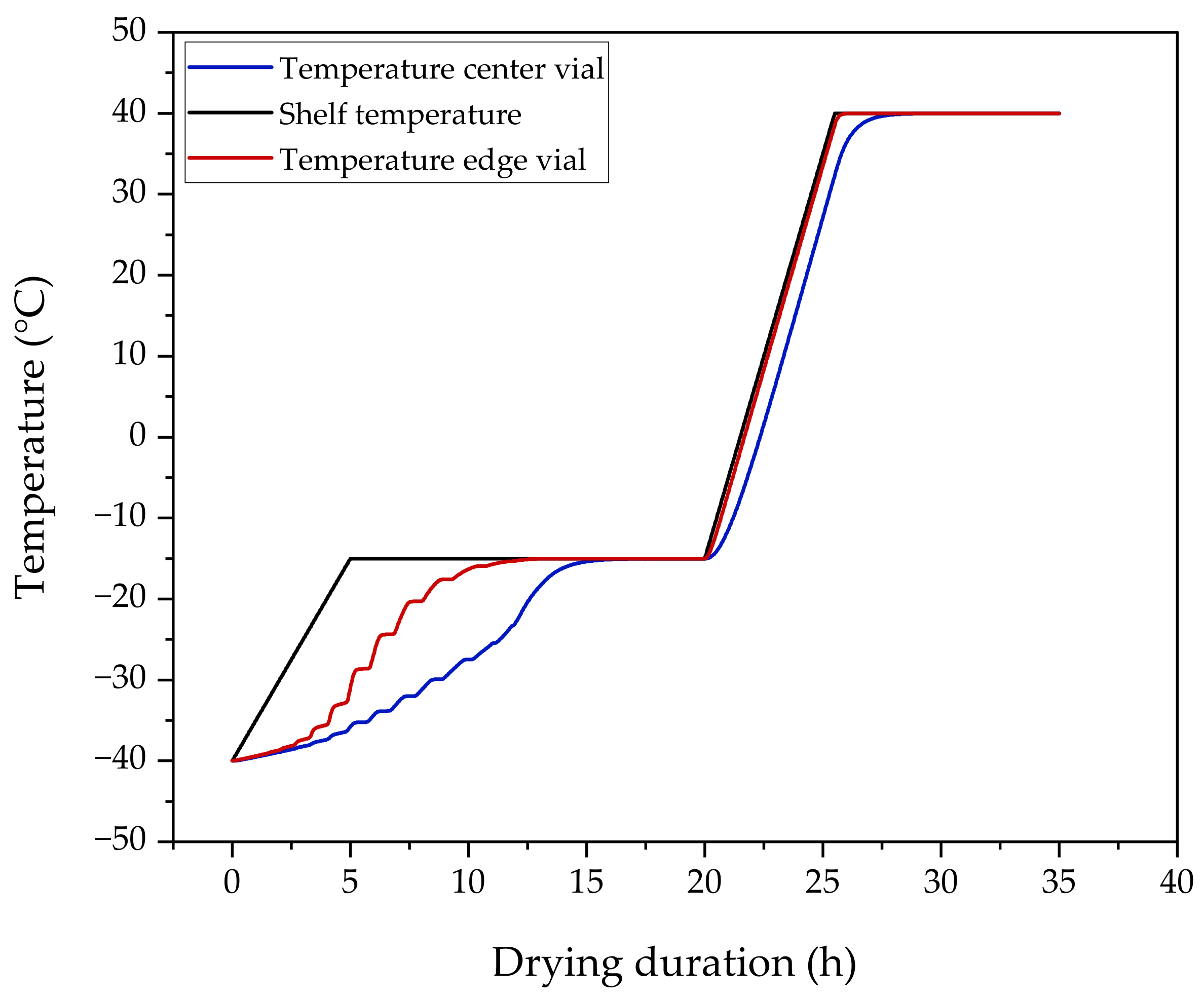
3.5. Formulation via Lyophilization
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Iqbal, N.; Karaca, Y. Complex Fractional-Order HIV Diffusion Model Based on Amplitude Equations with Turing Patterns and Turing Instability. Fractals 2021, 29, 2140013. [Google Scholar] [CrossRef]
- Yasmin, H. Effect of vaccination on non-integer dynamics of pneumococcal pneumonia infection. Chaos Solitons Fractals 2022, 158, 112049. [Google Scholar] [CrossRef]
- Flynn, N.M.; Forthal, D.N.; Harro, C.D.; Judson, F.N.; Mayer, K.H.; Para, M.F. Placebo-controlled phase 3 trial of a recombinant glycoprotein 120 vaccine to prevent HIV-1 infection. J. Infect. Dis. 2005, 191, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Hammonds, J.; Chen, X.; Zhang, X.; Lee, F.; Spearman, P. Advances in methods for the production, purification, and characterization of HIV-1 Gag-Env pseudovirion vaccines. Vaccine 2007, 25, 8036–8048. [Google Scholar] [CrossRef] [PubMed]
- Deml, L.; Speth, C.; Dierich, M.P.; Wolf, H.; Wagner, R. Recombinant HIV-1 Pr55gag virus-like particles: Potent stimulators of innate and acquired immune responses. Mol. Immunol. 2005, 42, 259–277. [Google Scholar] [CrossRef]
- Mohsen, M.O.; Zha, L.; Cabral-Miranda, G.; Bachmann, M.F. Major findings and recent advances in virus-like particle (VLP)-based vaccines. Semin. Immunol. 2017, 34, 123–132. [Google Scholar] [CrossRef]
- Roldão, A.; Mellado, M.C.M.; Castilho, L.R.; Carrondo, M.J.T.; Alves, P.M. Virus-like particles in vaccine development. Expert Rev. Vaccines 2010, 9, 1149–1176. [Google Scholar] [CrossRef]
- Cervera, L.; Gòdia, F.; Tarrés-Freixas, F.; Aguilar-Gurrieri, C.; Carrillo, J.; Blanco, J.; Gutiérrez-Granados, S. Production of HIV-1-based virus-like particles for vaccination: Achievements and limits. Appl. Microbiol. Biotechnol. 2019, 103, 7367–7384. [Google Scholar] [CrossRef]
- Rosengarten, J.F.; Schatz, S.; Wolf, T.; Barbe, S.; Stitz, J. Components of a HIV-1 vaccine mediate virus-like particle (VLP)-formation and display of envelope proteins exposing broadly neutralizing epitopes. Virology 2022, 568, 41–48. [Google Scholar] [CrossRef]
- Ludwig, C.; Wagner, R. Virus-like particles-universal molecular toolboxes. Curr. Opin. Biotechnol. 2007, 18, 537–545. [Google Scholar] [CrossRef]
- Boix-Besora, A.; Lorenzo, E.; Lavado-García, J.; Gòdia, F.; Cervera, L. Optimization, Production, Purification and Characterization of HIV-1 GAG-Based Virus-like Particles Functionalized with SARS-CoV-2. Vaccines 2022, 10, 250. [Google Scholar] [CrossRef] [PubMed]
- Doan, L.X.; Li, M.; Chen, C.; Yao, Q. Virus-like particles as HIV-1 vaccines. Rev. Med. Virol. 2005, 15, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Cruz, P.E.; Cunha, A.; Peixoto, C.C.; Clemente, J.; Moreira, J.L.; Carrondo, M.J. Optimization of the production of virus-like particles in insect cells. Biotechnol. Bioeng. 1998, 60, 408–418. [Google Scholar] [CrossRef]
- Pillay, S.; Meyers, A.; Williamson, A.-L.; Rybicki, E.P. Optimization of chimeric HIV-1 virus-like particle production in a baculovirus-insect cell expression system. Biotechnol. Prog. 2009, 25, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Puente-Massaguer, E.; Grau-Garcia, P.; Strobl, F.; Grabherr, R.; Striedner, G.; Lecina, M.; Gòdia, F. Accelerating HIV-1 VLP production using stable High Five insect cell pools. Biotechnol. J. 2021, 16, e2000391. [Google Scholar] [CrossRef]
- Visciano, M.L.; Diomede, L.; Tagliamonte, M.; Tornesello, M.L.; Asti, V.; Bomsel, M.; Buonaguro, F.M.; Lopalco, L.; Buonaguro, L. Generation of HIV-1 Virus-Like Particles expressing different HIV-1 glycoproteins. Vaccine 2011, 29, 4903–4912. [Google Scholar] [CrossRef]
- Zhu, J. Mammalian cell protein expression for biopharmaceutical production. Biotechnol. Adv. 2012, 30, 1158–1170. [Google Scholar] [CrossRef]
- Cervera, L.; Gutiérrez-Granados, S.; Martínez, M.; Blanco, J.; Gòdia, F.; Segura, M.M. Generation of HIV-1 Gag VLPs by transient transfection of HEK 293 suspension cell cultures using an optimized animal-derived component free medium. J. Biotechnol. 2013, 166, 152–165. [Google Scholar] [CrossRef]
- Durocher, Y.; Pham, P.L.; St-Laurent, G.; Jacob, D.; Cass, B.; Chahal, P.; Lau, C.J.; Nalbantoglu, J.; Kamen, A. Scalable serum-free production of recombinant adeno-associated virus type 2 by transfection of 293 suspension cells. J. Virol. Methods 2007, 144, 32–40. [Google Scholar] [CrossRef]
- Kamen, A.; Henry, O. Development and optimization of an adenovirus production process. J. Gene Med. 2004, 6 (Suppl. 1), S184–S192. [Google Scholar] [CrossRef]
- Le Ru, A.; Jacob, D.; Transfiguracion, J.; Ansorge, S.; Henry, O.; Kamen, A.A. Scalable production of influenza virus in HEK-293 cells for efficient vaccine manufacturing. Vaccine 2010, 28, 3661–3671. [Google Scholar] [CrossRef] [PubMed]
- Segura, M.M.; Garnier, A.; Durocher, Y.; Coelho, H.; Kamen, A. Production of lentiviral vectors by large-scale transient transfection of suspension cultures and affinity chromatography purification. Biotechnol. Bioeng. 2007, 98, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Helgers, H.; Hengelbrock, A.; Schmidt, A.; Rosengarten, J.; Stitz, J.; Strube, J. Process Design and Optimization towards Digital Twins for HIV-Gag VLP Production in HEK293 Cells, including Purification. Processes 2022, 10, 419. [Google Scholar] [CrossRef]
- Beg, S.; Hasnain, M.S.; Rahman, M.; Swain, S. Introduction to Quality by Design (QbD): Fundamentals, Principles, and Applications. In Pharmaceutical Quality by Design; Elsevier: Amsterdam, The Netherlands, 2019; pp. 1–17. ISBN 9780128157992. [Google Scholar]
- Rathore, A.S.; Winkle, H. Quality by design for biopharmaceuticals. Nat. Biotechnol. 2009, 27, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.X. Pharmaceutical quality by design: Product and process development, understanding, and control. Pharm. Res. 2008, 25, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yang, O.; Sampat, C.; Bhalode, P.; Ramachandran, R.; Ierapetritou, M. Digital Twins in Pharmaceutical and Biopharmaceutical Manufacturing: A Literature Review. Processes 2020, 8, 1088. [Google Scholar] [CrossRef]
- Schmidt, A.; Helgers, H.; Vetter, F.L.; Juckers, A.; Strube, J. Digital Twin of mRNA-Based SARS-COVID-19 Vaccine Manufacturing towards Autonomous Operation for Improvements in Speed, Scale, Robustness, Flexibility and Real-Time Release Testing. Processes 2021, 9, 748. [Google Scholar] [CrossRef]
- Lopez, P.C.; Udugama, I.A.; Thomsen, S.T.; Roslander, C.; Junicke, H.; Mauricio-Iglesias, M.; Gernaey, K.V. Towards a digital twin: A hybrid data-driven and mechanistic digital shadow to forecast the evolution of lignocellulosic fermentation. Biofuels Bioprod. Bioref. 2020, 14, 1046–1060. [Google Scholar] [CrossRef]
- Matsunami, K.; Ryckaert, A.; Peeters, M.; Badr, S.; Sugiyama, H.; Nopens, I.; de Beer, T. Analysis of the Effects of Process Parameters on Start-Up Operation in Continuous Wet Granulation. Processes 2021, 9, 1502. [Google Scholar] [CrossRef]
- Meitz, A.; Sagmeister, P.; Langemann, T.; Herwig, C. An Integrated Downstream Process Development Strategy along QbD Principles. Bioengineering 2014, 1, 213–230. [Google Scholar] [CrossRef] [Green Version]
- Udugama, I.A.; Lopez, P.C.; Gargalo, C.L.; Li, X.; Bayer, C.; Gernaey, K.V. Digital Twin in biomanufacturing: Challenges and opportunities towards its implementation. Syst. Microbiol. Biomanuf. 2021, 1, 257–274. [Google Scholar] [CrossRef]
- Brunet, R.; Guillén-Gosálbez, G.; Pérez-Correa, J.R.; Caballero, J.A.; Jiménez, L. Hybrid simulation-optimization based approach for the optimal design of single-product biotechnological processes. Comput. Chem. Eng. 2012, 37, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Brunef, R.; Kumar, K.S.; Guillen-Gosalbez, G.; Jimenez, L. Integrating process simulation, multi-objective optimization and LCA for the development of sustainable processes. In 21st European Symposium on Computer Aided Process Engineering; Elsevier: Amsterdam, The Netherlands, 2011; pp. 1271–1275. ISBN 9780444538956. [Google Scholar]
- Del Castillo-Romo, A.Á.; Morales-Rodriguez, R.; Román-Martínez, A. Multi-objective optimization for the biotechnological conversion of lingocellulosic biomass to value-added products. In 26th European Symposium on Computer Aided Process Engineering; Elsevier: Amsterdam, The Netherlands, 2016; pp. 1515–1520. ISBN 9780444634283. [Google Scholar]
- Pérez, A.D.; van der Bruggen, B.; Fontalvo, J. Modeling of a liquid membrane in Taylor flow integrated with lactic acid fermentation. Chem. Eng. Process. Process Intensif. 2019, 144, 107643. [Google Scholar] [CrossRef]
- Mat Isham, N.K.; Mokhtar, N.; Fazry, S.; Lim, S.J. The development of an alternative fermentation model system for vinegar production. LWT 2019, 100, 322–327. [Google Scholar] [CrossRef]
- Da Costa Basto, R.M.; Casals, M.P.; Mudde, R.F.; van der Wielen, L.A.; Cuellar, M.C. A mechanistic model for oil recovery in a region of high oil droplet concentration from multiphasic fermentations. Chem. Eng. Sci. X 2019, 3, 100033. [Google Scholar] [CrossRef]
- Udugama, A.; Öner, I.; Lopez, M.; Beenfeldt, P.C.; Bayer, C.; Huusom, C.; Gernaey, J.K.; Sin, K.V. Towards Digitalization in Bio-Manufacturing Operations: A Survey on Application of Big Data and Digital Twin Concepts in Denmark. Front. Chem. Eng. 2021, 3, 727152. [Google Scholar] [CrossRef]
- Zurdo, J.; Arnell, A.; Obrezanova, O.; Smith, N.; La Gómez de Cuesta, R.; Gallagher, T.R.A.; Michael, R.; Stallwood, Y.; Ekblad, C.; Abrahmsén, L.; et al. Early implementation of QbD in biopharmaceutical development: A practical example. BioMed Res. Int. 2015, 2015, 605427. [Google Scholar] [CrossRef] [Green Version]
- ICH Expert Working Group. Pharmaceuticel Development Q8(R2): ICH Harmonised Tripartite Guideline; ICH Expert Working Group: Geneva, Switzerland, 2009. [Google Scholar]
- Keith Pugh. Prior Knowledge in Product Development/Design. Available online: https://www.google.com/url?sa=t&rct=j&q=&esrc=s&source=web&cd=&ved=2ahUKEwjE5a-Y9vbzAhU0g_0HHSUvCk4QFnoECAoQAQ&url=https%3A%2F%2Fwww.ema.europa.eu%2Fdocuments%2Fpresentation%2Fpresentation-regulators-perspective-session-2-keith-pugh_en.pdf&usg=AOvVaw0NxD1cs8-EMstl-sdm9IxA (accessed on 13 March 2022).
- Yu, L.X.; Amidon, G.; Khan, M.A.; Hoag, S.W.; Polli, J.; Raju, G.K.; Woodcock, J. Understanding pharmaceutical quality by design. AAPS J. 2014, 16, 771–783. [Google Scholar] [CrossRef] [Green Version]
- Alt, N.; Zhang, T.Y.; Motchnik, P.; Taticek, R.; Quarmby, V.; Schlothauer, T.; Beck, H.; Emrich, T.; Harris, R.J. Determination of critical quality attributes for monoclonal antibodies using quality by design principles. Biologicals 2016, 44, 291–305. [Google Scholar] [CrossRef]
- Schmidt, A.; Strube, J. Distinct and Quantitative Validation Method for Predictive Process Modeling with Examples of Liquid-Liquid Extraction Processes of Complex Feed Mixtures. Processes 2019, 7, 298. [Google Scholar] [CrossRef] [Green Version]
- Zobel-Roos, S.; Schmidt, A.; Mestmäcker, F.; Mouellef, M.; Huter, M.; Uhlenbrock, L.; Kornecki, M.; Lohmann, L.; Ditz, R.; Strube, J. Accelerating Biologics Manufacturing by Modeling or: Is Approval under the QbD and PAT Approaches Demanded by Authorities Acceptable Without a Digital-Twin? Processes 2019, 7, 94. [Google Scholar] [CrossRef] [Green Version]
- Kis, Z.; Kontoravdi, C.; Shattock, R.; Shah, N. Resources, Production Scales and Time Required for Producing RNA Vaccines for the Global Pandemic Demand. Vaccines 2020, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- Kornecki, M.; Schmidt, A.; Strube, J. PAT as key-enabling technology for QbD in pharmaceutical manufacturing A conceptual review on upstream and downstream processing. Chim. Oggi-Chem. Today 2018, 36, 44–48. [Google Scholar]
- Woodcock, J. Modernizing Pharmaceutical Manufacturing—Continuous Manufacturing as a Key Enabler: MIT-CMAC International Symposium on Continuous Manufacturing of Pharmaceuticals. Available online: https://iscmp.mit.edu/sites/default/files/documents/ISCMP%202014%20-%20Keynote_Slides.pdf (accessed on 6 December 2021).
- Helgers, H.; Hengelbrock, A.; Schmidt, A.; Strube, J. Digital Twins for Continuous mRNA Production. Processes 2021, 9, 1967. [Google Scholar] [CrossRef]
- Schmidt, A.; Helgers, H.; Vetter, F.L.; Juckers, A.; Strube, J. Fast and Flexible mRNA Vaccine Manufacturing as a Solution to Pandemic Situations by Adopting Chemical Engineering Good Practice—Continuous Autonomous Operation in Stainless Steel Equipment Concepts. Processes 2021, 9, 1874. [Google Scholar] [CrossRef]
- Giglia, S.; Straeffer, G. Combined mechanism fouling model and method for optimization of series microfiltration performance. J. Membr. Sci. 2012, 417, 144–153. [Google Scholar] [CrossRef]
- Iritani, E.; Katagiri, N. Developments of Blocking Filtration Model in Membrane Filtration. KONA 2016, 33, 179–202. [Google Scholar] [CrossRef] [Green Version]
- Zydney, A.L. Development of a new blocking model for membrane fouling based on a composite media model. J. Membr. Sci. Lett. 2022, 2, 100018. [Google Scholar] [CrossRef]
- Grote, F.; Fröhlich, H.; Strube, J. Integration of Ultrafiltration Unit Operations in Biotechnology Process Design. Chem. Eng. Technol. 2011, 34, 673–687. [Google Scholar] [CrossRef]
- Weisbach, J.L. Lehrbuch der Ingenieur-und Maschinen-Mechanik. In Theoretische Mechanik; Druck und Verlag von Friedrich Vieweg und Sohn: Berlin, Germany, 1845. [Google Scholar]
- Darcy, H. Les Fontaines Publiques de la Ville de Dijon in Exposition et Application; Victor Dalmont: Paris, France, 1856. [Google Scholar]
- Brown, G.O. The History of the Darcy-Weisbach Equation for Pipe Flow Resistance. In Environmental and Water Resources History, Proceedings of the Environmental and Water Resources History Sessions at ASCE Civil Engineering Conference and Exposition, Washington, DC, USA, 3–7 November 2002; Rogers, J.R., Fredrich, A.J., Eds.; American Society of Civil Engineers: Reston, VA, USA, 2002; pp. 34–43. ISBN 9780784406502. [Google Scholar]
- Van den Berg, G.B.; Smolders, C.A. Flux decline in ultrafiltration processes. Desalination 1990, 77, 101–133. [Google Scholar] [CrossRef] [Green Version]
- Wijmans, J.G.; Nakao, S.; van den Berg, J.; Troelstra, F.R.; Smolders, C.A. Hydrodynamic resistance of concentration polarization boundary layers in ultrafiltration. J. Membr. Sci. 1985, 22, 117–135. [Google Scholar] [CrossRef] [Green Version]
- Huter, M.J.; Strube, J. Model-Based Design and Process Optimization of Continuous Single Pass Tangential Flow Filtration Focusing on Continuous Bioprocessing. Processes 2019, 7, 317. [Google Scholar] [CrossRef] [Green Version]
- Effio, C.L.; Hubbuch, J. Next generation vaccines and vectors: Designing downstream processes for recombinant protein-based virus-like particles. Biotechnol. J. 2015, 10, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Ladd Effio, C.; Hahn, T.; Seiler, J.; Oelmeier, S.A.; Asen, I.; Silberer, C.; Villain, L.; Hubbuch, J. Modeling and simulation of anion-exchange membrane chromatography for purification of Sf9 insect cell-derived virus-like particles. J. Chromatogr. A 2016, 1429, 142–154. [Google Scholar] [CrossRef]
- McNally, D.J.; Darling, D.; Farzaneh, F.; Levison, P.R.; Slater, N.K.H. Optimised concentration and purification of retroviruses using membrane chromatography. J. Chromatogr. A 2014, 1340, 24–32. [Google Scholar] [CrossRef] [Green Version]
- Pereira Aguilar, P.; Reiter, K.; Wetter, V.; Steppert, P.; Maresch, D.; Ling, W.L.; Satzer, P.; Jungbauer, A. Capture and purification of Human Immunodeficiency Virus-1 virus-like particles: Convective media vs porous beads. J. Chromatogr. A 2020, 1627, 461378. [Google Scholar] [CrossRef]
- Pereira Aguilar, P.; Schneider, T.A.; Wetter, V.; Maresch, D.; Ling, W.L.; Tover, A.; Steppert, P.; Jungbauer, A. Polymer-grafted chromatography media for the purification of enveloped virus-like particles, exemplified with HIV-1 gag VLP. Vaccine 2019, 37, 7070–7080. [Google Scholar] [CrossRef]
- Steppert, P.; Burgstaller, D.; Klausberger, M.; Berger, E.; Aguilar, P.P.; Schneider, T.A.; Kramberger, P.; Tover, A.; Nöbauer, K.; Razzazi-Fazeli, E.; et al. Purification of HIV-1 gag virus-like particles and separation of other extracellular particles. J. Chromatogr. A 2016, 1455, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Vicente, T.; Sousa, M.F.; Peixoto, C.; Mota, J.P.; Alves, P.M.; Carrondo, M.J. Anion-exchange membrane chromatography for purification of rotavirus-like particles. J. Membr. Sci. 2008, 311, 270–283. [Google Scholar] [CrossRef]
- Guiochon, G.; Felinger, A.; Shirazi, D.G.; Katti, A.M. Fundamentals of Preparative and Nonlinear Chromatography, 2nd ed.; Elsevier Academic Press: Cambridge, MA, USA, 2006; ISBN 9780123705372. [Google Scholar]
- Zobel-Roos, S.; Mouellef, M.; Ditz, R.; Strube, J. Distinct and Quantitative Validation Method for Predictive Process Modelling in Preparative Chromatography of Synthetic and Bio-Based Feed Mixtures Following a Quality-by-Design (QbD) Approach. Processes 2019, 7, 580. [Google Scholar] [CrossRef] [Green Version]
- Seidel-Morgenstern, A. Experimental determination of single solute and competitive adsorption isotherms. J. Chromatogr. A 2004, 1037, 255–272. [Google Scholar] [CrossRef] [PubMed]
- Carta, G.; Rodrigues, A.E. Diffusion and convection in chromatographic processes using permeable supports with a bidisperse pore structure. Chem. Eng. Sci. 1993, 48, 3927–3935. [Google Scholar] [CrossRef]
- Wilson, E.J.; Geankoplis, C.J. Liquid Mass Transfer at Very Low Reynolds Numbers in Packed Beds. Ind. Eng. Chem. Fund. 1966, 5, 9–14. [Google Scholar] [CrossRef]
- Leśko, M.; Åsberg, D.; Enmark, M.; Samuelsson, J.; Fornstedt, T.; Kaczmarski, K. Choice of Model for Estimation of Adsorption Isotherm Parameters in Gradient Elution Preparative Liquid Chromatography. Chromatographia 2015, 78, 1293–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klepzig, L.S.; Juckers, A.; Knerr, P.; Harms, F.; Strube, J. Digital Twin for Lyophilization by Process Modeling in Manufacturing of Biologics. Processes 2020, 8, 1325. [Google Scholar] [CrossRef]
- Juckers, A.; Knerr, P.; Harms, F.; Strube, J. Advanced Process Analytical Technology in Combination with Process Modeling for Endpoint and Model Parameter Determination in Lyophilization Process Design and Optimization. Processes 2021, 9, 1600. [Google Scholar] [CrossRef]
- Martin Christ Gefriertrocknungsanlagen Gmbh. Pilot-Gefriertrocknungsanlagen Innovative Technologie. Available online: https://www.martinchrist.de/fileadmin/user_upload/christ/PDF/Broschueren/Pilot/Christ_Pilot_GT_dt_2021-08.pdf (accessed on 22 February 2022).
- Rambhatla, S.; Pikal, M.J. Heat and mass transfer scale-up issues during freeze-drying, I: Atypical radiation and the edge vial effect. AAPS PharmSciTech 2003, 4, E14. [Google Scholar] [CrossRef] [Green Version]
- Lang, R.; Winter, G.; Vogt, L.; Zurcher, A.; Dorigo, B.; Schimmele, B. Rational design of a stable, freeze-dried virus-like particle-based vaccine formulation. Drug Dev. Ind. Pharm. 2009, 35, 83–97. [Google Scholar] [CrossRef]
- Helgers, H.; Schmidt, A.; Lohmann, L.J.; Vetter, F.L.; Juckers, A.; Jensch, C.; Mouellef, M.; Zobel-Roos, S.; Strube, J. Towards Autonomous Operation by Advanced Process Control—Process Analytical Technology for Continuous Biologics Antibody Manufacturing. Processes 2021, 9, 172. [Google Scholar] [CrossRef]
- Helgers, H.; Schmidt, A.; Strube, J. Towards Autonomous Process Control—Digital Twin for CHO Cell-Based Antibody Manufacturing Using a Dynamic Metabolic Model. Processes 2022, 10, 316. [Google Scholar] [CrossRef]
- Matthews, T.E.; Berry, B.N.; Smelko, J.; Moretto, J.; Moore, B.; Wiltberger, K. Closed loop control of lactate concentration in mammalian cell culture by Raman spectroscopy leads to improved cell density, viability, and biopharmaceutical protein production. Biotechnol. Bioeng. 2016, 113, 2416–2424. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.M.; Kessler, J.-M.; Salou, P.; Menezes, J.C.; Peinado, A. Monitoring mAb cultivations with in-situ raman spectroscopy: The influence of spectral selectivity on calibration models and industrial use as reliable PAT tool. Biotechnol. Prog. 2018, 34, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Van Reis, R.; Zydney, A. Bioprocess membrane technology. J. Membr. Sci. 2007, 297, 16–50. [Google Scholar] [CrossRef]
- Taylor, N.; Ma, W.; Kristopeit, A.; Wang, S.-C.; Zydney, A.L. Enhancing the performance of sterile filtration for viral vaccines and model nanoparticles using an appropriate prefilter. J. Membr. Sci. 2022, 647, 120264. [Google Scholar] [CrossRef]
- Dryden, W.A.; Larsen, L.M.; Britt, D.W.; Smith, M.T. Technical and economic considerations of cell culture harvest and clarification technologies. Biochem. Eng. J. 2021, 167, 107892. [Google Scholar] [CrossRef]
- Negrete, A.; Pai, A.; Shiloach, J. Use of hollow fiber tangential flow filtration for the recovery and concentration of HIV virus-like particles produced in insect cells. J. Virol. Methods 2014, 195, 240–246. [Google Scholar] [CrossRef]
- Loewe, D.; Häussler, J.; Grein, T.A.; Dieken, H.; Weidner, T.; Salzig, D.; Czermak, P. Forced Degradation Studies to Identify Critical Process Parameters for the Purification of Infectious Measles Virus. Viruses 2019, 11, 725. [Google Scholar] [CrossRef] [Green Version]
- Bayer, B.; Dalmau Diaz, R.; Melcher, M.; Striedner, G.; Duerkop, M. Digital Twin Application for Model-Based DoE to Rapidly Identify Ideal Process Conditions for Space-Time Yield Optimization. Processes 2021, 9, 1109. [Google Scholar] [CrossRef]
- Chopda, V.; Gyorgypal, A.; Yang, O.; Singh, R.; Ramachandran, R.; Zhang, H.; Tsilomelekis, G.; Chundawat, S.P.S.; Ierapetritou, M.G. Recent advances in integrated process analytical techniques, modeling, and control strategies to enable continuous biomanufacturing of monoclonal antibodies. J. Chem. Technol. Biotechnol. 2021. [Google Scholar] [CrossRef]
- Walther, J.; Godawat, R.; Hwang, C.; Abe, Y.; Sinclair, A.; Konstantinov, K. The business impact of an integrated continuous biomanufacturing platform for recombinant protein production. J. Biotechnol. 2015, 213, 3–12. [Google Scholar] [CrossRef]
- Aristoteleio Panepistēmio Thessalonikēs; Associazione Italiana di Ingegneria Chimica. In Proceedings of the PRES’13: 16th Conference on Process Integration, Modelling and Optimisation for Energy Saving and Pollution Reduction, Rhodes, Greece, 29 September–2 October 2013; AIDIC: Milano, Italy, 2013. ISBN 9788895608266.
- Andersen, T.M.; Toftegaard, O. CIP Technology. In Challenges and Trends; MORK Process Inc.: Copenhagen, Denmark, 2010. [Google Scholar]
- Roy, K.; Undey, C.; Mistretta, T.; Naugle, G.; Sodhi, M. Multivariate statistical monitoring as applied to clean-in-place (CIP) and steam-in-place (SIP) operations in biopharmaceutical manufacturing. Biotechnol. Prog. 2014, 30, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Uhlenbrock, L.; Strube, J. Technical Potential for Energy and GWP Reduction in Chemical–Pharmaceutical Industry in Germany and EU—Focused on Biologics and Botanicals Manufacturing. Processes 2020, 8, 818. [Google Scholar] [CrossRef]
- Dakin, J. Supply Chain Challenges Creating Hurdles to COVID-19 Vaccine Production No. 4. 2021. Available online: https://www.pharmtech.com/view/supply-chain-challenges-creating-hurdles-to-covid-19-vaccine-production (accessed on 7 March 2022).
- Badman, C.; Cooney, C.L.; Florence, A.; Konstantinov, K.; Krumme, M.; Mascia, S.; Nasr, M.; Trout, B.L. Why We Need Continuous Pharmaceutical Manufacturing and How to Make It Happen. J. Pharm. Sci. 2019, 108, 3521–3523. [Google Scholar] [CrossRef] [PubMed]
- Deng, F. Advances and challenges in enveloped virus-like particle (VLP)-based vaccines. J. Immunol. Sci. 2018, 2, 36–41. [Google Scholar] [CrossRef] [Green Version]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| R0 (m−1) | KS (m−1) | KI (m−1) | KCM (m−1) | |
|---|---|---|---|---|
| FB4 | 2.4 × 1011 | 33.7 ± 0.01 | 64.2 ± 0.13 | 27.5 ± 0.02 |
| FB5 | 2.3 × 1011 | 27.9 ± 0.01 | 53.5 ± 0.12 | 22.9 ± 0.02 |
| PC1 | 6.0 × 1010 | 7.0 ± 0.001 | 18.8 ± 0.01 | 6.3 ± 0.001 |
| PC2 | 6.0 × 1010 | 10.2 ± 0.003 | 30.2 ± 0.01 | 9.3 ± 0.004 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hengelbrock, A.; Helgers, H.; Schmidt, A.; Vetter, F.L.; Juckers, A.; Rosengarten, J.F.; Stitz, J.; Strube, J. Digital Twin for HIV-Gag VLP Production in HEK293 Cells. Processes 2022, 10, 866. https://doi.org/10.3390/pr10050866
Hengelbrock A, Helgers H, Schmidt A, Vetter FL, Juckers A, Rosengarten JF, Stitz J, Strube J. Digital Twin for HIV-Gag VLP Production in HEK293 Cells. Processes. 2022; 10(5):866. https://doi.org/10.3390/pr10050866
Chicago/Turabian StyleHengelbrock, Alina, Heribert Helgers, Axel Schmidt, Florian Lukas Vetter, Alex Juckers, Jamila Franca Rosengarten, Jörn Stitz, and Jochen Strube. 2022. "Digital Twin for HIV-Gag VLP Production in HEK293 Cells" Processes 10, no. 5: 866. https://doi.org/10.3390/pr10050866
APA StyleHengelbrock, A., Helgers, H., Schmidt, A., Vetter, F. L., Juckers, A., Rosengarten, J. F., Stitz, J., & Strube, J. (2022). Digital Twin for HIV-Gag VLP Production in HEK293 Cells. Processes, 10(5), 866. https://doi.org/10.3390/pr10050866