Figure 1.
Classification of DIPPR molecules per chemical family (the numbers on the right of the bars correspond to the number of molecules within each family).
Figure 1.
Classification of DIPPR molecules per chemical family (the numbers on the right of the bars correspond to the number of molecules within each family).
Figure 2.
Classification of DIPPR molecules (a) per number of atoms and (b) per number of rings, within each molecule.
Figure 2.
Classification of DIPPR molecules (a) per number of atoms and (b) per number of rings, within each molecule.
Figure 3.
Distribution of (a) the enthalpy and (b) the entropy values of the DIPPR database. A total of 2147 and 2119 values are present in the database for the enthalpy and the entropy, respectively.
Figure 3.
Distribution of (a) the enthalpy and (b) the entropy values of the DIPPR database. A total of 2147 and 2119 values are present in the database for the enthalpy and the entropy, respectively.
Figure 4.
Procedure for converting the initial SMILES notation, of the DIPPR database, to molecular descriptor values.
Figure 4.
Procedure for converting the initial SMILES notation, of the DIPPR database, to molecular descriptor values.
Figure 5.
Heatmap of DescMVs (white = Desc-MVs; black = defined values; molecules are classified by their chemical family).
Figure 5.
Heatmap of DescMVs (white = Desc-MVs; black = defined values; molecules are classified by their chemical family).
Figure 6.
Graph theory-based method for the elimination of correlations between descriptors (nodes and edges correspond to descriptors and correlations (above a given threshold for the value of the correlation coefficient), respectively). Case 1: non correlated descriptors; Case 2: pairwise correlated descriptors; Case 3: multiple correlations between descriptors. Descriptors in green are selected while those in red are removed.
Figure 6.
Graph theory-based method for the elimination of correlations between descriptors (nodes and edges correspond to descriptors and correlations (above a given threshold for the value of the correlation coefficient), respectively). Case 1: non correlated descriptors; Case 2: pairwise correlated descriptors; Case 3: multiple correlations between descriptors. Descriptors in green are selected while those in red are removed.
Figure 7.
Overview of feature selection methods with their advantages and limits in green and red, respectively.
Figure 7.
Overview of feature selection methods with their advantages and limits in green and red, respectively.
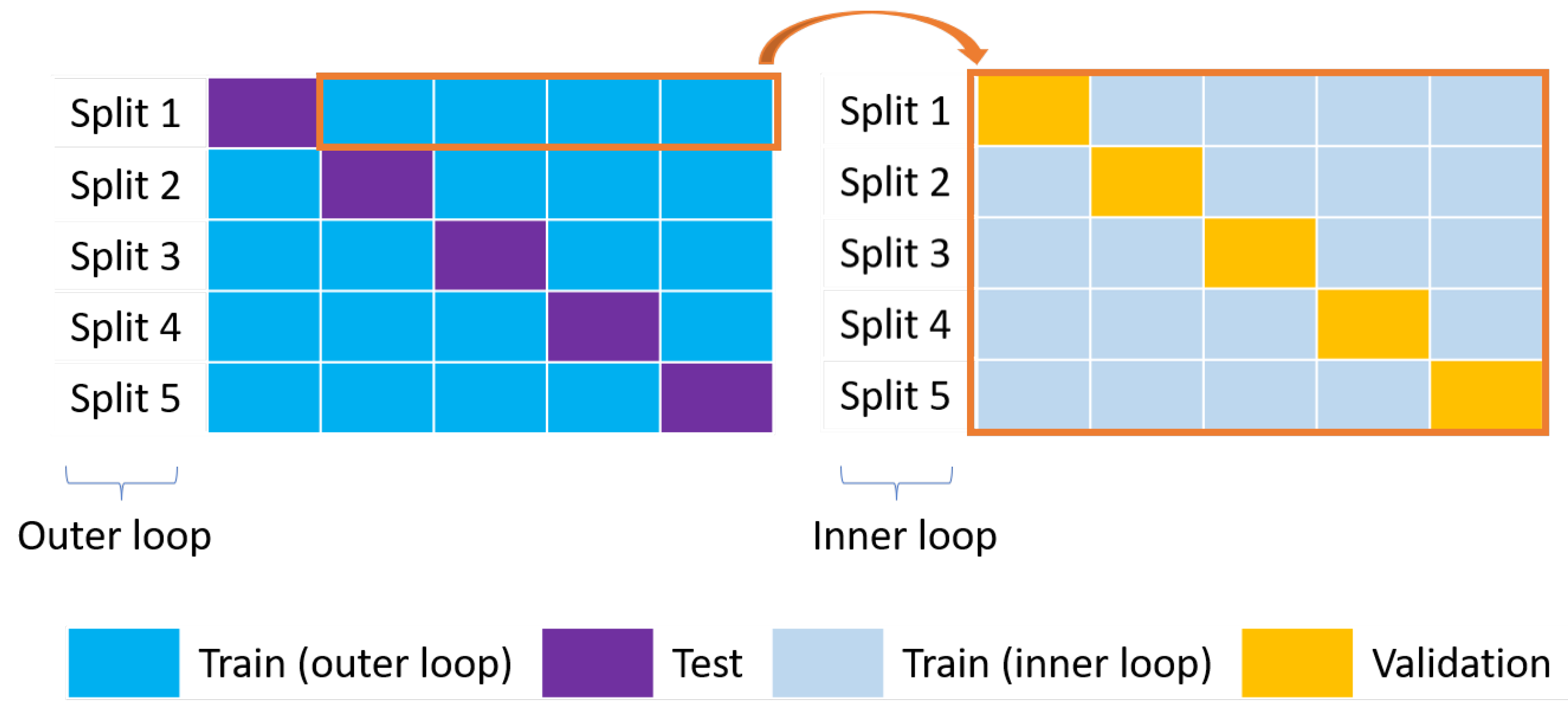
Figure 8.
Nested CV. The outer loop on the left (blue and purple boxes for the training and test sets, respectively) is used for model selection while the inner loop on the right (grey and yellow boxes for the training and validation sets, respectively) is used for the optimization of the HPs.
Figure 8.
Nested CV. The outer loop on the left (blue and purple boxes for the training and test sets, respectively) is used for model selection while the inner loop on the right (grey and yellow boxes for the training and validation sets, respectively) is used for the optimization of the HPs.
Figure 9.
Performance of the different ML models during the preliminary screening for the enthalpy: (a) ; (b) ; (c) (preprocessing: default, splitting: 5-fold external CV, scaling: standard, dimensionality reduction: none, HP optimization: none).
Figure 9.
Performance of the different ML models during the preliminary screening for the enthalpy: (a) ; (b) ; (c) (preprocessing: default, splitting: 5-fold external CV, scaling: standard, dimensionality reduction: none, HP optimization: none).
Figure 10.
Parity plots of the selected ML models during the preliminary screening, for different splits, for the enthalpy (preprocessing: default, splitting: 5-fold external CV, scaling: standard, dimensionality reduction: none, HP optimization: none).
Figure 10.
Parity plots of the selected ML models during the preliminary screening, for different splits, for the enthalpy (preprocessing: default, splitting: 5-fold external CV, scaling: standard, dimensionality reduction: none, HP optimization: none).
Figure 11.
Distribution of the coefficients in various linear regression models during the preliminary screening, for split 1, for the enthalpy (preprocessing: default, splitting: 5-fold external CV, scaling: standard, dimensionality reduction: none, HP optimization: none).
Figure 11.
Distribution of the coefficients in various linear regression models during the preliminary screening, for split 1, for the enthalpy (preprocessing: default, splitting: 5-fold external CV, scaling: standard, dimensionality reduction: none, HP optimization: none).
Figure 12.
Effect of the value of k’, for the external CV, on the (a) train (b), test , (c) training time, of the different ML models during the preliminary screening for the enthalpy (preprocessing: default, splitting: 5 and 10-fold external CV, scaling: standard, dimensionality reduction: none, HP optimization: none).
Figure 12.
Effect of the value of k’, for the external CV, on the (a) train (b), test , (c) training time, of the different ML models during the preliminary screening for the enthalpy (preprocessing: default, splitting: 5 and 10-fold external CV, scaling: standard, dimensionality reduction: none, HP optimization: none).
Figure 13.
Effect of the data scaling technique on the (a) train (b), test , of the different ML models during the preliminary screening for the enthalpy (preprocessing: default, splitting: 5-fold external CV, scaling: standard/min-max/robust, dimensionality reduction: none, HP optimization: none). N.B. Robust scaler did not work with the SVR lin method (cf. red crosses).
Figure 13.
Effect of the data scaling technique on the (a) train (b), test , of the different ML models during the preliminary screening for the enthalpy (preprocessing: default, splitting: 5-fold external CV, scaling: standard/min-max/robust, dimensionality reduction: none, HP optimization: none). N.B. Robust scaler did not work with the SVR lin method (cf. red crosses).
Figure 14.
Effect of the value of (a) the variance threshold (b) the correlation coefficient threshold, on the number of retained descriptors and Lasso model test for the enthalpy (preprocessing: default for (a) and default with low variance threshold = 0.0001 for (b), splitting: 5-fold external CV, scaling: standard, dimensionality reduction: none, HP optimization: none).
Figure 14.
Effect of the value of (a) the variance threshold (b) the correlation coefficient threshold, on the number of retained descriptors and Lasso model test for the enthalpy (preprocessing: default for (a) and default with low variance threshold = 0.0001 for (b), splitting: 5-fold external CV, scaling: standard, dimensionality reduction: none, HP optimization: none).
Figure 15.
Explained variance as a function of the principal components obtained with PCA for (a) the enthalpy and (b) the entropy. (preprocessing: final, splitting: 5-fold external CV, scaling: standard, dimensionality reduction: PCA, HP optimization: none).
Figure 15.
Explained variance as a function of the principal components obtained with PCA for (a) the enthalpy and (b) the entropy. (preprocessing: final, splitting: 5-fold external CV, scaling: standard, dimensionality reduction: PCA, HP optimization: none).
Figure 16.
Parity plots of the selected ML models after HP optimization, for different splits, for the enthalpy (preprocessing: final, splitting: 5-fold external CV, scaling: standard, dimensionality reduction: wrapper-GA Lasso, HP optimization: yes).
Figure 16.
Parity plots of the selected ML models after HP optimization, for different splits, for the enthalpy (preprocessing: final, splitting: 5-fold external CV, scaling: standard, dimensionality reduction: wrapper-GA Lasso, HP optimization: yes).
Figure 17.
Parity plots of the selected ML models after HP optimization, for different splits, for the entropy (preprocessing: final, splitting: 5-fold external CV, scaling: standard, dimensionality reduction: wrapper-GA Lasso, HP optimization: yes).
Figure 17.
Parity plots of the selected ML models after HP optimization, for different splits, for the entropy (preprocessing: final, splitting: 5-fold external CV, scaling: standard, dimensionality reduction: wrapper-GA Lasso, HP optimization: yes).
Table 1.
Classification of DIPPR data per uncertainty.
Table 1.
Classification of DIPPR data per uncertainty.
| Property | Uncertainty |
|---|
|
<0.2%
|
<1%
|
<3%
|
<5%
|
<10%
|
<25%
|
<50%
|
<100%
|
NaN
|
|---|
| Enthalpy | 50 | 401 | 1013 | 242 | 197 | 188 | 33 | 4 | 19 |
| Entropy | 66 | 184 | 1019 | 419 | 184 | 199 | 20 | 0 | 28 |
Table 2.
AlvaDesc descriptors per category.
Table 2.
AlvaDesc descriptors per category.
| Category | Category | Number of | Category | Category | Number of |
|---|
|
n°
|
Name
|
Descriptors
|
n°
|
Name
|
Descriptors
|
|---|
| 1 | Constitutional indices | 50 | 18 | WHIM descriptors | 114 |
| 2 | Ring descriptors | 35 | 19 | GETAWAY descriptors | 273 |
| 3 | Topological indices | 79 | 20 | Randic molecular profiles | 41 |
| 4 | Walk and path counts | 46 | 21 | Functional group counts | 154 |
| 5 | Connectivity indices | 37 | 22 | Atom-centred fragments | 115 |
| 6 | Information indices | 51 | 23 | Atom-type E-state indices | 346 |
| 7 | 2D matrix-based descriptors | 608 | 24 | Pharmacophore descriptors | 165 |
| 8 | 2D autocorrelations | 213 | 25 | 2D Atom Pairs | 1596 |
| 9 | Burden eigenvalues | 96 | 26 | 3D Atom Pairs | 36 |
| 10 | P_VSA-like descriptors | 69 | 27 | Charge descriptors | 15 |
| 11 | ETA indices | 40 | 28 | Molecular properties | 27 |
| 12 | Edge adjacency indices | 324 | 29 | Drug-like indices | 30 |
| 13 | Geometrical descriptors | 38 | 30 | CATS 3D descriptors | 300 |
| 14 | 3D matrix-based descriptors | 132 | 31 | WHALES descriptors | 33 |
| 15 | 3D autocorrelations | 80 | 32 | MDE descriptors | 19 |
| 16 | RDF descriptors | 210 | 33 | Chirality descriptors | 70 |
| 17 | 3D-MoRSE descriptors | 224 | | | |
Table 3.
Summary of the tested and default preprocessing options.
Table 3.
Summary of the tested and default preprocessing options.
| Preprocessing Step | Tested | Default |
|---|
| Elimination of Desc-MVs | - By row | |
| - By column | Alternating row or column |
| - Alternating row or column | |
| Elimination of descriptors | [0, 0.0001, 0.001, 0.01, | 0.001 |
| with low variance | 0.1, 1, 10, 100, 1000] | |
| Elimination of correlated | [0.6, 0.7, 0.8, 0.9, | 0.95 |
| descriptors | 0.92, 0.95, 0.98, 0.99] | |
Table 4.
Methods tested for dimensionality reduction.
Table 4.
Methods tested for dimensionality reduction.
| Feature Extraction | Feature Selection |
|---|
|
Filter
|
Wrapper
|
Embedded
|
|---|
| | Pearson coefficient | GA | Lasso |
| PCA | MI | SFS | SVR lin |
| | | | ET |
Table 5.
Configurations tested during ML model construction.
Table 5.
Configurations tested during ML model construction.
| Data Scaling | Data Splitting | ML Models | Performance Metrics |
|---|
| Standard | 5-fold internal CV | LR | Coefficient of determination |
| 5 and 10-fold external CV | Ridge | |
| | | Lasso | |
| Min-Max | | SVR lin | Root Mean Squared Error |
| | GP | |
| | | kNN | |
| Robust | | DT | Mean Absolute Error |
| | RF | |
| | | ET | |
| | | AB | |
| | | GB | |
| | | MLP | |
Table 6.
Configurations selected for the study of the effects of data preprocessing and dimensionality reduction, and for HP optimization.
Table 6.
Configurations selected for the study of the effects of data preprocessing and dimensionality reduction, and for HP optimization.
| Data Scaling | Data Splitting | ML Models | Performance Metrics |
|---|
| Standard | 5-fold external CV | Lasso | |
| SVR lin |
| ET |
| MLP |
Table 7.
Effect of the algorithms for the elimination of Desc-MVs on the data set size and Lasso model test for the enthalpy (preprocessing: default, splitting: 5-fold external CV, scaling: standard, dimensionality reduction: none, HP optimization: none).
Table 7.
Effect of the algorithms for the elimination of Desc-MVs on the data set size and Lasso model test for the enthalpy (preprocessing: default, splitting: 5-fold external CV, scaling: standard, dimensionality reduction: none, HP optimization: none).
| Elimination | Data Set | Data Set | Data Set after | MAE Train | MAE Test |
|---|
|
Procedure
|
with Desc-MVs
|
without Desc-MVs
|
Preprocessing
|
(kJ/mol)
|
(kJ/mol)
|
|---|
| Alg.1: by row | 1903 × 5666 | 236 × 5666 | 236 × 1378 | 7.6 | 20.4 |
| mol. desc. | 0 duplicates |
| Alg.2: by column | 1903 × 5666 | 1903 × 2855 | 1903 × 988 | 21.1 | 32.9 |
| 73 duplicates |
| Alg.3: alternating | 1903 × 5666 | 1785 × 5531 | 1785 × 1961 | 16.7 | 27.6 |
| row or column | 0 duplicates |
Table 8.
Summary of the final preprocessing options.
Table 8.
Summary of the final preprocessing options.
| Preprocessing Step | Final |
|---|
| Elimination of Desc-MVs | Alternating row or column |
| Elimination of descriptors with low variance | 0.0001 |
| Elimination of correlated descriptors | 0.98 |
Table 9.
Effect of the different dimensionality reduction methods on the test of the selected ML models for the enthalpy (preprocessing: final, splitting: 5-fold external CV, scaling: standard, dimensionality reduction: different methods, HP optimization: none).
Table 9.
Effect of the different dimensionality reduction methods on the test of the selected ML models for the enthalpy (preprocessing: final, splitting: 5-fold external CV, scaling: standard, dimensionality reduction: different methods, HP optimization: none).
| Dimensionality Reduction Method | Nb of Desc. | Time/Split (s) | MAE Test (kJ/mol) | Nb of | Nb of Desc. |
|---|
|
Lasso
|
SVR lin
|
ET
|
MLP
|
Pairwise Correlations ≥ 0.9
|
with Variance ≤ 0.01
|
|---|
| None (reference case) | 2506 | 0 | 27.0 | 28.6 | 42.6 | 37.9 | 4473 | 512 |
| Filter-Pearson | 100 | 19.4 | 61.6 | 75.0 | 56.7 | 116.0 | 124 | 2 |
| Filter-MI | 100 | 18.7 | 55.8 | 62.5 | 43.8 | 90.8 | 72 | 4 |
| Wrapper-SFS Lasso | 100 | 7795 | 31.1 | 34.9 | 42.9 | 84.6 | 3 | 21 |
| Wrapper-GA Lasso | 100 | 49573 | 24.2 | 31.0 | 43.8 | 76.1 | 5 | 26 |
| Embedded-Lasso | 100 | 1.5 | 29.0 | 29.0 | 38.8 | 88.7 | 14 | 24 |
| Embedded-SVR lin | 100 | 7.0 | 39.8 | 40.0 | 41.1 | 84.1 | 34 | 18 |
| Embedded-ET | 100 | 3.7 | 50.3 | 51.2 | 41.4 | 85.5 | 46 | 4 |
| PCA 95% (261–265 PC) | 2506 | 2.9 | 37.3 | 34.2 | 76.8 | 38.0 | - | - |
Table 10.
Top five descriptor categories identified by the different dimensionality reduction methods for the enthalpy (preprocessing: final, splitting: 5-fold external CV, scaling: standard, dimensionality reduction: different methods, HP optimization: none). The percentages correspond to the proportion of a descriptor category among the descriptors obtained with each method.
Table 10.
Top five descriptor categories identified by the different dimensionality reduction methods for the enthalpy (preprocessing: final, splitting: 5-fold external CV, scaling: standard, dimensionality reduction: different methods, HP optimization: none). The percentages correspond to the proportion of a descriptor category among the descriptors obtained with each method.
| Dimensionality Reduction Method | Top 5 Descriptor Categories |
|---|
| None (reference case) | 25 | 19 | 8 | 30 | 17 |
| Filter-Pearson | 8 | 3 | 16 | 19 | 7 |
| Filter-MI | 8 | 7 | 3 | 11 | 27 |
| Wrapper-SFS Lasso | 25 | 23 | 22 | 21 | 17 |
| Wrapper-GA Lasso | 25 | 23 | 22 | 21 | 10 |
| Embedded-Lasso | 25 | 22 | 7 | 10 | 23 |
| Embedded-SVR lin | 25 | 23 | 10 | 22 | 1 |
| Embedded-ET | 12 | 8 | 7 | 3 | 11 |
| PCA 95% (261–265 PC) | 25 | 19 | 8 | 30 | 17 |
Table 11.
Effect of the different dimensionality reduction methods on the test of the selected ML models for the entropy (preprocessing: final, splitting: 5-fold external CV, scaling: standard, dimensionality reduction: different methods, HP optimization: none).
Table 11.
Effect of the different dimensionality reduction methods on the test of the selected ML models for the entropy (preprocessing: final, splitting: 5-fold external CV, scaling: standard, dimensionality reduction: different methods, HP optimization: none).
| Dimensionality Reduction Method | Nb of Desc. | Time/Split (s) | MAE Test (J/mol/K) | Nb of | Nb of Desc. |
|---|
|
Lasso
|
SVR lin
|
ET
|
MLP
|
Pairwise Correlations ≥ 0.9
|
with Variance ≤ 0.01
|
|---|
| None (reference case) | 2479 | 0 | 18.7 | 24.3 | 19.6 | 27.1 | 4469 | 487 |
| Filter-Pearson | 100 | 18.1 | 20.9 | 18.5 | 20.3 | 46.9 | 909 | 2 |
| Filter-MI | 100 | 16.3 | 21.4 | 19.1 | 20.0 | 31.4 | 514 | 6 |
| Wrapper-SFS Lasso | 100 | 8294 | 19.2 | 18.9 | 19.6 | 55.8 | 15 | 19 |
| Wrapper-GA Lasso | 100 | 53,315 | 17.4 | 18.1 | 19.6 | 57.5 | 9 | 25 |
| Embedded-Lasso | 100 | 1.4 | 18.8 | 18.5 | 19.7 | 52.6 | 21 | 20 |
| Embedded-SVR lin | 100 | 5.6 | 24.2 | 23.5 | 21.3 | 53.0 | 8 | 17 |
| Embedded-ET | 100 | 3.7 | 21.6 | 19.7 | 20.8 | 31.0 | 338 | 8 |
| PCA 95% (254–260 PCs) | 2479 | 3.0 | 19.7 | 18.3 | 23.0 | 29.1 | - | - |
Table 12.
Top 5 descriptor categories identified by the different dimensionality reduction methods for the entropy (preprocessing: final, splitting: 5-fold external CV, scaling: standard, dimensionality reduction: different methods, HP optimization: none).
Table 12.
Top 5 descriptor categories identified by the different dimensionality reduction methods for the entropy (preprocessing: final, splitting: 5-fold external CV, scaling: standard, dimensionality reduction: different methods, HP optimization: none).
| Dimensionality Reduction Method | Top 5 Descriptor Categories |
|---|
| None (reference case) | 25 | 19 | 8 | 17 | 30 |
| Filter-Pearson | 7 | 16 | 19 | 3 | 14 |
| Filter-MI | 7 | 14 | 3 | 8 | 1 |
| Wrapper-SFS Lasso | 25 | 30 | 21 | 7 | 24 |
| Wrapper-GA Lasso | 25 | 21 | 30 | 24 | 7 |
| Embedded-Lasso | 25 | 21 | 16 | 30 | 23 |
| Embedded-SVR lin | 25 | 30 | 16 | 24 | 21 |
| Embedded-ET | 7 | 8 | 14 | 9 | 19 |
| PCA 95% (254–260 PCs) | 25 | 19 | 8 | 17 | 30 |
Table 13.
HP optimization settings and results for the selected ML models for the enthalpy (preprocessing: final, splitting: 5-fold external CV, scaling: standard, dimensionality reduction: wrapper-GA Lasso, HP optimization: yes).
Table 13.
HP optimization settings and results for the selected ML models for the enthalpy (preprocessing: final, splitting: 5-fold external CV, scaling: standard, dimensionality reduction: wrapper-GA Lasso, HP optimization: yes).
| ML | HPs | Screening Ranges | Optimal HP Settings per Split |
|---|
|
Model
| |
(Blue = Default Value)
|
Split 1
|
Split 2
|
Split 3
|
Split 4
|
Split 5
|
|---|
| Lasso | alpha | [0.001, 0.01, 0.1, 0.5, 1, 1.5, 2] | 0.1 | 0.001 | 0.1 | 0.5 | 0.1 |
| | kernel | [‘linear’] | linear | linear | linear | linear | linear |
| SVR lin | C | [0.1, 0.5, 1, 1.5, 2] | 2 | 2 | 2 | 2 | 2 |
| | epsilon | [0.01, 0.1, 1] | 0.1 | 0.1 | 1 | 1 | 1 |
| | n_estimators | [50, 100, 200] | 200 | 200 | 100 | 100 | 200 |
| | max_features | [‘sqrt’, ‘log2’, None] | None | None | None | None | None |
| | min_samples_split | [2, 5] | 2 | 2 | 2 | 2 | 2 |
| ET | min_samples_leaf | [1, 5] | 1 | 1 | 1 | 1 | 1 |
| | max_depth | [10, None] | None | None | None | None | None |
| | criterion | [‘absolute error’, | squared | squared | squared | squared | squared |
| | | ‘squared error’] | | | | | |
| | activation | [‘relu’] | relu | relu | relu | relu | relu |
| | hidden_layer_sizes | 1 hidden layer: [(i)], | (100) | (10,10) | (15,15) | (10,10) | (15,15) |
| | | i = 100, 200, 400; | | | | | |
| MLP | | 2 hidden layers: [(i, i)], | | | | | |
| | | i = 10, 15, 20 | | | | | |
| | solver | [‘adam’, ‘lbfgs’] | lbfgs | lbfgs | lbfgs | lbfgs | lbfgs |
| | learning_rate_init | [0.001, 0.01, 0.1, 0.5] | 0.001 | 0.001 | 0.001 | 0.001 | 0.001 |
| | max_iter | [200, 500] | 200 | 500 | 500 | 200 | 200 |
Table 14.
Performance of the selected ML models with and without HP optimization for the enthalpy (preprocessing: final, splitting: 5-fold external CV, scaling: standard, dimensionality reduction: wrapper-GA Lasso, HP optimization: none/yes).
Table 14.
Performance of the selected ML models with and without HP optimization for the enthalpy (preprocessing: final, splitting: 5-fold external CV, scaling: standard, dimensionality reduction: wrapper-GA Lasso, HP optimization: none/yes).
| Model | Data Set | | MAE (kJ/mol) | RMSE (kJ/mol) |
|---|
|
HP Not Opt.
|
HP Opt.
|
HP Not Opt.
|
HP Opt.
|
HP Not Opt.
|
HP Opt.
|
|---|
| Lasso | Train (internal) | 0.995 | 0.996 | 15.8 | 14.6 | 35.9 | 33.7 |
| Validation | 0.987 | 0.989 | 24.8 | 22.3 | 52.2 | 47.8 |
| Train (external) | 0.995 | 0.996 | 15.5 | 14.6 | 36.9 | 34.6 |
| Test | 0.978 | 0.976 | 24.2 | 25.1 | 70.8 | 74.2 |
| SVR lin | Train (internal) | 0.987 | 0.993 | 23.1 | 17.9 | 58.2 | 44.6 |
| Validation | 0.975 | 0.984 | 35.6 | 27.8 | 75.6 | 60.1 |
| Train (external) | 0.990 | 0.993 | 21.4 | 17.3 | 53.5 | 43.7 |
| Test | 0.968 | 0.971 | 31.0 | 27.8 | 85.8 | 82.4 |
| ET | Train (internal) | 1.000 | 1.000 | 0.0 | 0.0 | 0.0 | 0.0 |
| Validation | 0.933 | 0.933 | 61.6 | 61.3 | 114.5 | 114.4 |
| Train (external) | 1.000 | 1.000 | 0.0 | 0.0 | 0.0 | 0.0 |
| Test | 0.955 | 0.955 | 43.8 | 43.6 | 112.3 | 112.3 |
| MLP | Train (internal) | 0.955 | 0.998 | 79.7 | 11.8 | 112.2 | 20.1 |
| Validation | 0.764 | 0.964 | 125.9 | 42.5 | 197.0 | 81.3 |
| Train (external) | 0.968 | 0.999 | 65.2 | 10.3 | 95.3 | 17.7 |
| Test | 0.943 | 0.976 | 76.1 | 34.9 | 117.6 | 78.3 |
Table 15.
Performance of Lasso model with HP optimization for the entropy (preprocessing: final, splitting: 5-fold external CV, scaling: standard, dimensionality reduction: wrapper-GA Lasso, HP optimization: yes).
Table 15.
Performance of Lasso model with HP optimization for the entropy (preprocessing: final, splitting: 5-fold external CV, scaling: standard, dimensionality reduction: wrapper-GA Lasso, HP optimization: yes).
| Model | Data Set | | MAE (J/mol/K) | RMSE (J/mol/K) |
|---|
| Lasso | Train (internal) | 0.982 | 13.8 | 27.6 |
| Validation | 0.966 | 17.5 | 34.5 |
| Train (external) | 0.982 | 13.7 | 28.0 |
| Test | 0.968 | 17.9 | 36.2 |
Table 16.
Hydrocarbons, oxygenated, nitrogenated, chlorinated, fluorinated, brominated, iodinated, phosphorus containing, sulfonated, silicon containing, multifunctional. GroupGAT (group-contribution-based graph attention). Probabilistic vector learned from interatomic distances, bond angles, and dihedral angles histograms with GMM (Gaussian Mixture Model). N/A: not available.
Table 16.
Hydrocarbons, oxygenated, nitrogenated, chlorinated, fluorinated, brominated, iodinated, phosphorus containing, sulfonated, silicon containing, multifunctional. GroupGAT (group-contribution-based graph attention). Probabilistic vector learned from interatomic distances, bond angles, and dihedral angles histograms with GMM (Gaussian Mixture Model). N/A: not available.
| Property | Reference | Data Source | Type of Molecules | Nb of Molecules | Molecular Representation | ML Model | Test
| MAE Test | RMSE Test
| k |
|---|
| H (kJ/mol) | [109] | DIPPR exp. | Diverse | 741 | GNN | GNN | 0.99 | 18.6 | 30.5 | 10 |
| This work | 100 descriptors | Lasso | 0.99 | 12.4 | 21.6 |
| [51] | Literature exp., ab initio | Noncyclic hydrocarbons | 310 | 261 descriptors | SVR | 0.995 | 5.703 | N/A | 10 |
| This work | 100 descriptors | Lasso | 0.998 | 4.426 | 6.520 |
| [52] | Literature exp. | Cyclic hydrocarbons | 192 | 47 descriptors | SVR | 0.986 | 9.71 | N/A | 10 |
| [56] | GauL HDAD | ANN | N/A | 9.6 | 12.9 |
| This work | 100 descriptors | Lasso | 0.985 | 10.01 | 14.51 |
| [56] | Lignin QM ab initio | (Poly)cyclic hydrocarbons and oxygenates | 3926 | GauL HDAD | ANN | N/A | 9.34 | 15.89 | 10 |
| This work | 100 descriptors | Lasso | 0.98 | 21.48 | 30.81 |
| [110] | SPEED exp. | Diverse | 1059 | 240 groups | GP | 0.987 | N/A | 42.74 | 20 |
| This work | 100 descriptors | Lasso | 0.976 | 11.30 | 28.20 |
| S (J/mol/K) | [56] | Lignin QM ab initio | (Poly)cyclic hydrocarbons and oxygenates | 3926 | GauL HDAD | ANN | N/A | 3.86 | 5.32 | 10 |
| This work | 100 descriptors | Lasso | 0.99 | 5.57 | 7.43 |
| [53] | Literature exp., theo. | Hydrocarbons | 310 | 252 descriptors | SVR | 0.99 | 6.3 | 9 | 10 |
| This work | 100 descriptors | Lasso | 0.98 | 8.3 | 10.8 |
| [111] | DIPPR exp. | Organic | 511 | GNN | GNN | 0.99 | 5.3 | N/A | 10 |
| This work | 100 descriptors | Lasso | 0.99 | 6.1 | 9.4 |






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}