



Multicomponent Synthesis of Unsymmetrical Derivatives of 4-Methyl-Substituted 5-Nitropyridines

 ,
, 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
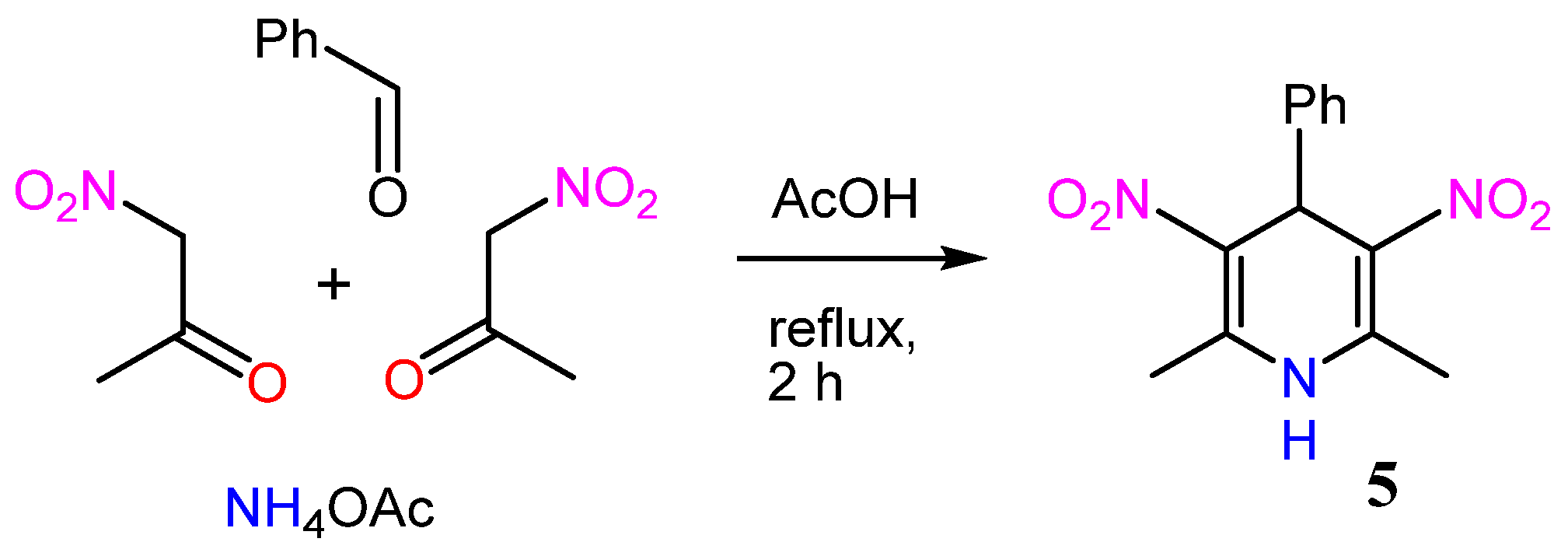
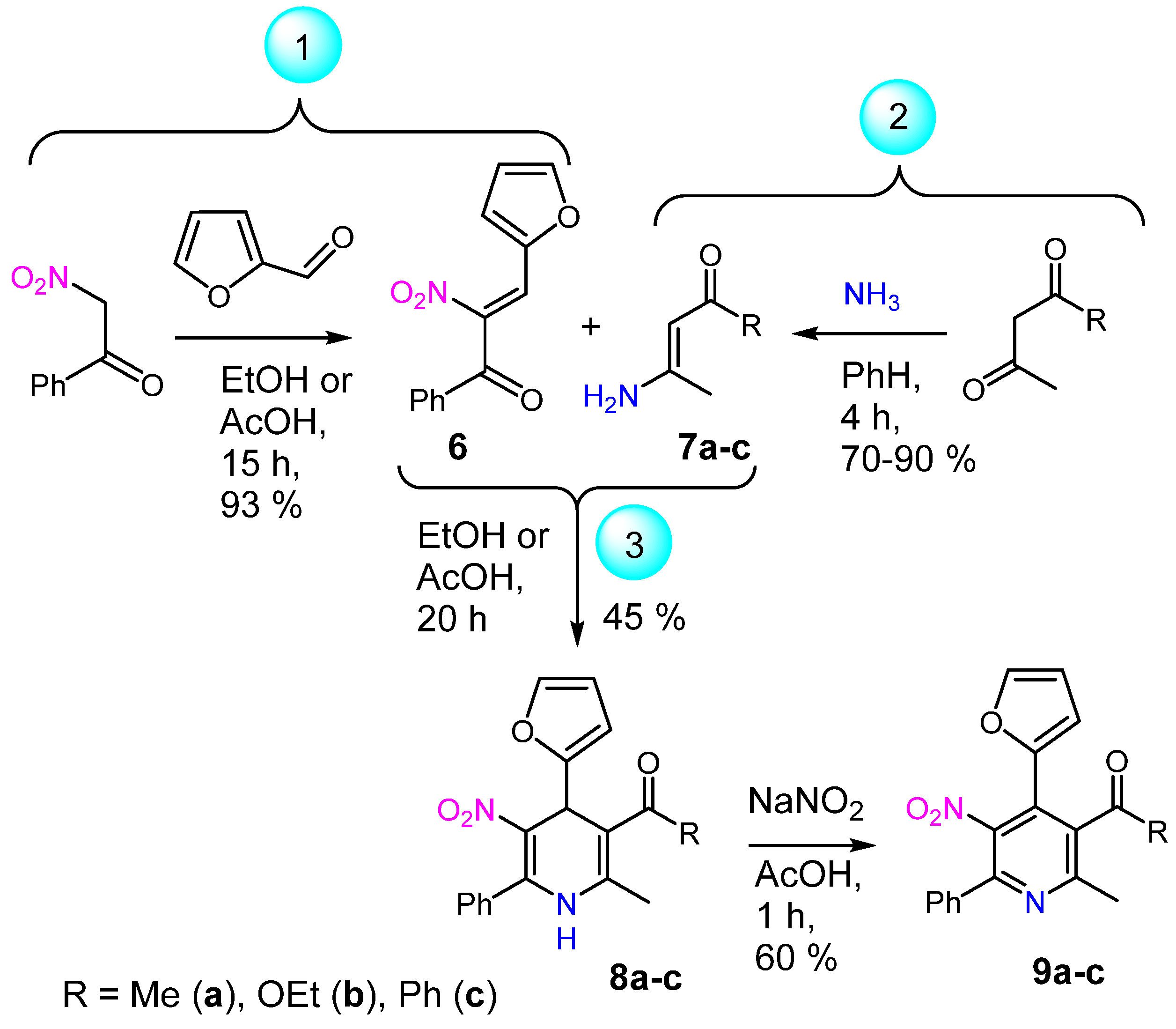
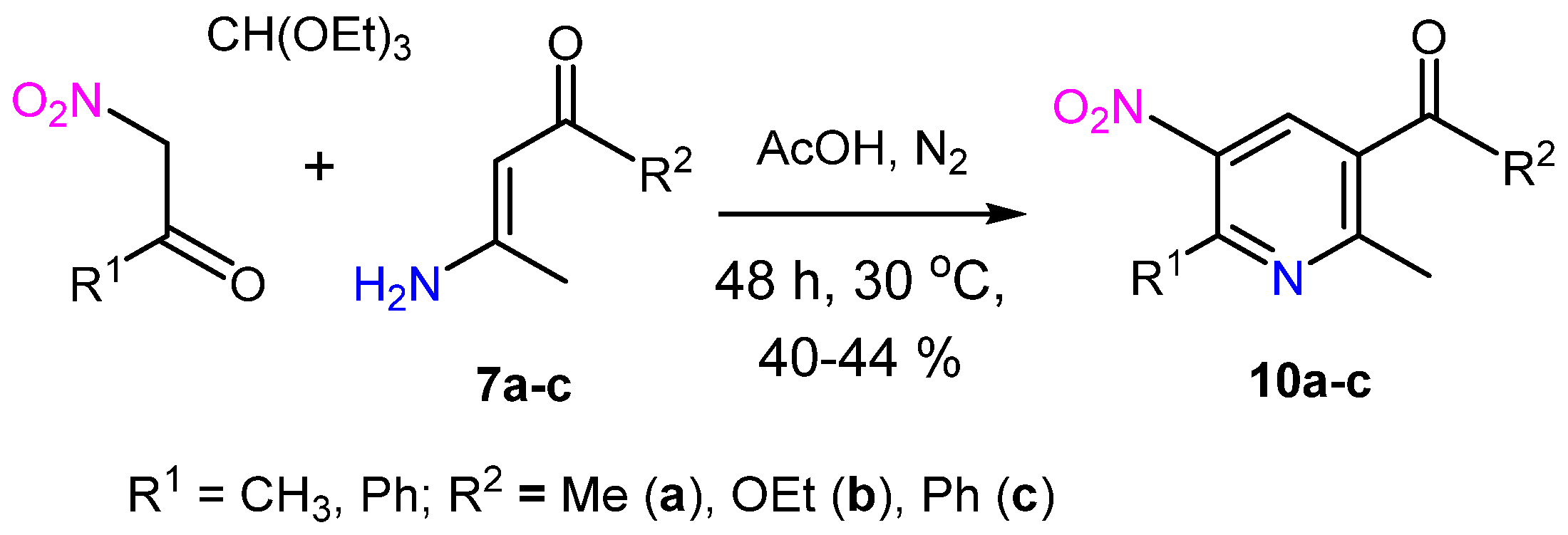
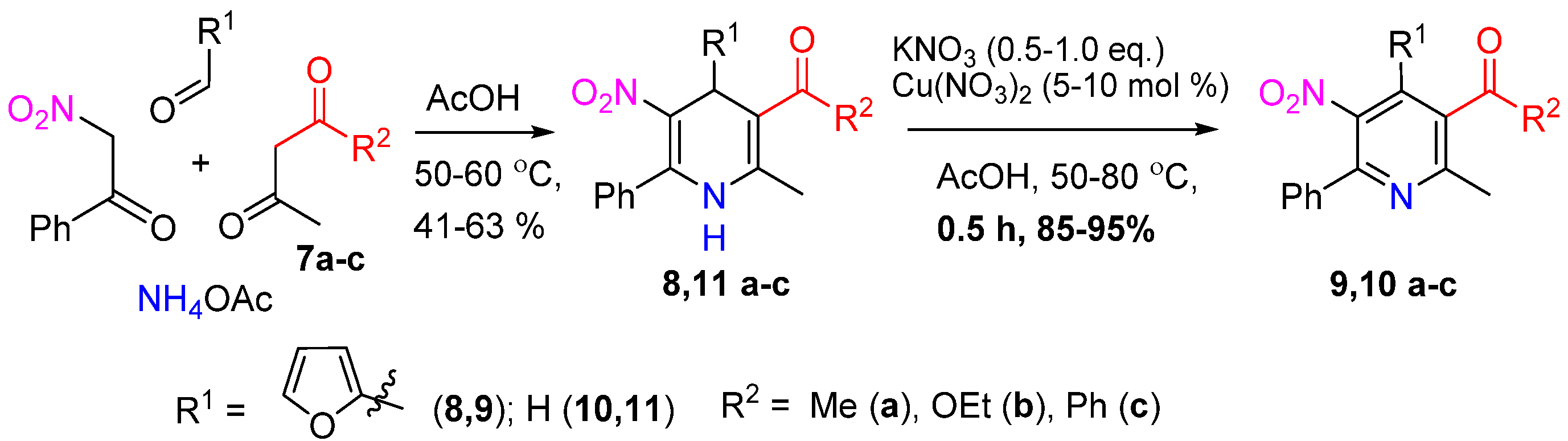
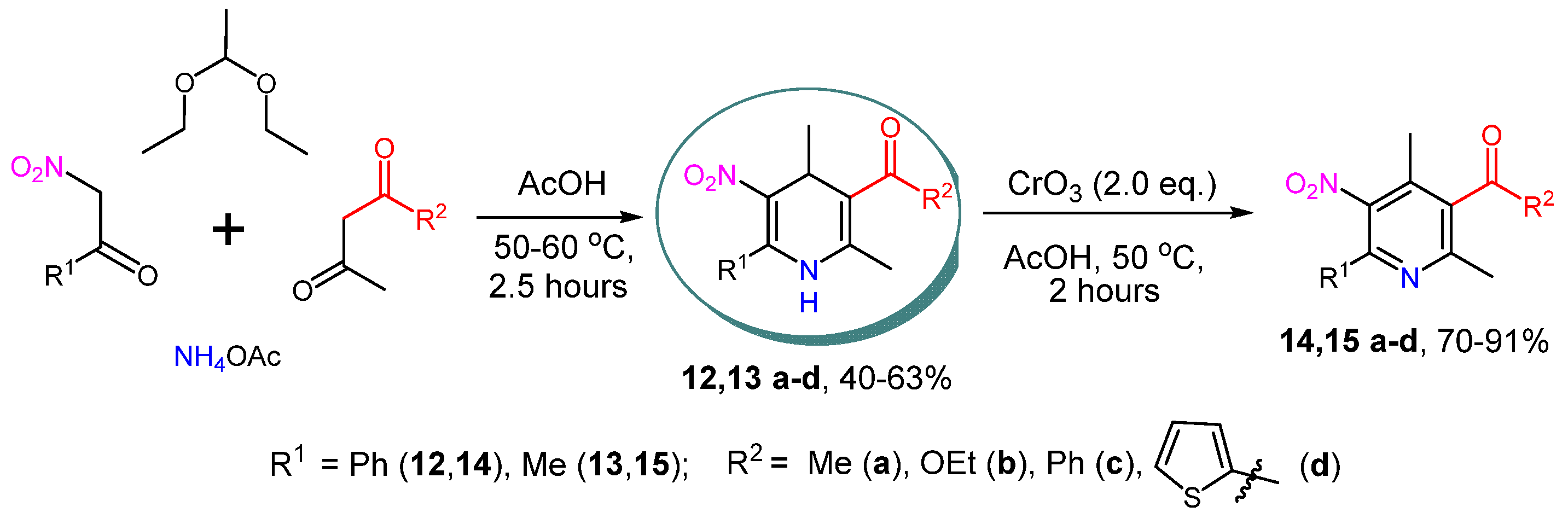
2. Results and Discussion
3. Conclusions
4. Materials and Methods
Synthesis of Compounds
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mishra, M.; Sharma, M.; Dubey, R.; Kumari, P.; Ranjan, V.; Pandey, J. Green synthesis interventions of pharmaceutical industries for sustainable development. Curr. Res. Green Sustain. Chem. 2021, 4, 100174. [Google Scholar] [CrossRef]
- Meyer, M.F.; Powers, S.M.; Hampton, S.E. An Evidence Synthesis of Pharmaceuticals and Personal Care Products (PPCPs) in the Environment: Imbalances among Compounds, Sewage Treatment Techniques, and Ecosystem Types. Environ. Sci. Technol. 2019, 53, 12961–12973. [Google Scholar] [CrossRef] [PubMed]
- Caban, M.; Stepnowski, P. How to decrease pharmaceuticals in the environment? A review. Environ. Chem. Lett. 2021, 19, 3115–3138. [Google Scholar] [CrossRef]
- Kar, S.; Sanderson, H.; Roy, K.; Benfenati, E.; Leszczynski, J. Green Chemistry in the Synthesis of Pharmaceuticals. Chem. Rev. 2022, 122, 3637–3710. [Google Scholar] [CrossRef]
- Hu, C.; Testa, C.J.; Born, S.C.; Wu, W.; Shvedova, K.; Sayin, R.; Halkude, B.S.; Casati, F.; Ramnath, A.; Hermant, P. E-factor analysis of a pilot plant for end-to-end integrated continuous manufacturing (ICM) of pharmaceuticals. Green Chem. 2020, 22, 4350–4356. [Google Scholar] [CrossRef]
- Bttps:, A.C.; Longstreet, A.R.; Britton, J.; Wang, Y.; Moriguchi, H.; Hicklin, R.W.; Green, W.H.; Jamison, T.F. Minimizing E-factor in the continuous-flow synthesis of diazepam and atropine. Bioorgan. Med. Chem. 2016, 25, 6233–6241. [Google Scholar]
- Ameta, K.L.; Dandia, A. Multicomponent Reactions: Synthesis of Bioactive Heterocycles, 1st ed.; Taylor & Francis: Boca Raton, FL, USA, 2017; pp. 33–54. [Google Scholar]
- Dömling, A.; Wang, W.; Wang, K. Chemistry and biology of multicomponent reactions. Chem. Rev. 2012, 112, 3083–3135. [Google Scholar] [CrossRef] [PubMed]
- Cimarelli, C. Multicomponent Reactions. Molecules 2019, 24, 2372. [Google Scholar] [CrossRef] [PubMed]
- Stephy, E.J.; Shivani, G.; Nagula, S. Recent advances in multi-component reactions and their mechanistic insights: A triennium review. Org. Chem. Front. 2021, 8, 4237–4287. [Google Scholar]
- Graebin, C.S.; Ribeiro, F.V.; Rogério, K.R.; Kümmerle, A.E. Multicomponent Reactions for the Synthesis of Bioactive Compounds: A Review. Curr. Org. Synth. 2019, 16, 855–899. [Google Scholar] [CrossRef] [PubMed]
- Younus, H.A.; Al-Rashida, M.; Hameed, A.; Uroos, M.; Salar, U.; Rana, S.; Khan, K.M. Multicomponent reactions (MCR) in medicinal chemistry: A patent review (2010-2020). Expert Opin. Ther. Pat. 2021, 31, 267–289. [Google Scholar] [CrossRef] [PubMed]
- Becerra, D.; Abonia, R.; Castillo, J.C. Recent Applications of the Multicomponent Synthesis for Bioactive Pyrazole Derivatives. Molecules 2022, 27, 4723. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Domling, A. Multicomponent Reactions in Medicinal Chemistry. In Multicomponent Reactions towards Heterocycles; van der Eycken, E., Sharma, U.K., Eds.; Wiley-VCH: Weinhiem, Germany, 2022; pp. 91–137. [Google Scholar]
- Cioc, R.C.; Ruijter, E.; Orru, R.V. Multicomponent Reactions: Advanced Tools for Sustainable Organic Synthesis. Green Chem. 2014, 16, 2959. [Google Scholar] [CrossRef]
- Xu, J.; Fan, W.; Popowycz, F.; Queneau, Y.; Gu, Y. Multicomponent Reactions: A New Strategy for Enriching the Routes of Value-Added Conversions of Bio-platform Molecules. Chin. J. Org. Chem. 2019, 39, 2131–2138. [Google Scholar] [CrossRef]
- Bosica, G.; Baldacchino, K.; Abdilla, R.; De Nittis, R. Green Organic Synthesis via Multicomponent Reactions. Sci. J. Malta Chamb. Sci. 2021, 9, 173–179. [Google Scholar]
- Lee, M.; Vosburg, N.J.; Shimizu, E.A.; Rentería-Gómez, M.A.; Gámez-Montaño, R.; Vosburg, D.A. Multicomponent Synthesis of Lidocaine at Room Temperature. J. Chem. Educ. 2022, 99, 2399–2402. [Google Scholar] [CrossRef]
- Cores, Á.; Clerigué, J.; Orocio-Rodríguez, E.; Menéndez, J.C. Multicomponent Reactions for the Synthesis of Active Pharmaceutical Ingredients. Pharmaceuticals 2022, 15, 1009. [Google Scholar] [CrossRef]
- Ruijter, E.; Orru, R. Synthetic and BioOrganic Chemistry Group. Multicomponent reactions in drug discovery and medicinal chemistry. Drug Discov. Today Technol. 2018, 29, 1–2. [Google Scholar] [CrossRef]
- Slobbe, P.; Ruijter, E.; Orru, R.V.A. Recent applications of multicomponent reactions in medicinal chemistry. Med. Chem. Commun. 2012, 3, 1189–1218. [Google Scholar] [CrossRef]
- Pedrola, M.; Jorba, M.; Jardas, E.; Jardi, F.; Ghashghaei, O.; Viñas, M.; Lavilla, R. Multicomponent Reactions Upon the Known Drug Trimethoprim as a Source of Novel Antimicrobial Agents. Front. Chem. 2019, 7, 475. [Google Scholar] [CrossRef]
- Bosica, G.; Cachia, F.; De Nittis, R.; Mariotti, N. Efficient One-Pot Synthesis of 3,4-Dihydropyrimidin-2(1H)-ones via a Three-Component Biginelli Reaction. Molecules 2021, 26, 3753. [Google Scholar] [CrossRef] [PubMed]
- Qu, C.; Hao, J.; Ding, H.; Lv, Y.; Zhao, X.-E.; Zhao, X.; Wei, W. Visible-Light-Initiated Multicomponent Reactions of α-Diazoesters to Access Organophosphorus Compounds. J. Org. Chem. 2022, 87, 12921–12931. [Google Scholar] [CrossRef] [PubMed]
- Hantzsch, A. Condensationprodukte aus Aldehydammoniak und Ketonartigen Verbindungen. Chem. Ber. 1881, 14, 1637–1638. [Google Scholar] [CrossRef] [Green Version]
- Katritzky, A.R.; Ostercamp, D.L.; Yousaf, T.I. The Mechanism of the Hantzsch Pyridine Synthesis: A study by 15N and 13C NMR spectroscopy. Tetrahedron 1986, 42, 5729–5738. [Google Scholar] [CrossRef]
- Santos, V.G.; Godoi, M.N.; Regiani, T.; Gama, F.H.S.; Coelho, M.B.; de Souza, R.; Eberlin, M.N.; Garden, S.J. The Multicomponent Hantzsch Reaction: Comprehensive Mass Spectrometry Monitoring Using Charge-Tagged Reagants. Chemistry 2014, 20, 12808–12816. [Google Scholar] [CrossRef] [PubMed]
- Alvim, H.G.; da Silva Júnior, E.N.; Neto, B.A. What do we know about multicomponent reactions? Mechanisms and trends for the Biginelli, Hantzsch, Mannich, Passerini and Ugi MCRs. RSC Adv. 2014, 4, 54282–54299. [Google Scholar] [CrossRef]
- Vanden Eynde, J.J.; Mayence, A. Synthesis and Aromatization of Hantzsch 1,4-Dihydropyridines under Microwave Irradiation. An Overview. Molecules 2003, 8, 381–391. [Google Scholar] [CrossRef]
- Dolzhenko, A.V. Microwave-assisted multicomponent reactions. In Green Sustainable Process for Chemical and Environmental Engineering and Science; Elsevier: Amsterdam, The Netherlands, 2021. [Google Scholar]
- Kerru, N.; Maddila, S.; Jonnalagadda, S.B. A Facile and Catalyst-Free Microwave-Promoted Multicomponent Reaction for the Synthesis of Functionalised 1,4-Dihydropyridines with Superb Selectivity and Yields. Front. Chem. 2021, 9, 638832. [Google Scholar] [CrossRef]
- Penteado, F.; Monti, B.; Sancineto, L.; Perin, G.; Jacob, R.G.; Santi, C.; Lenardão, E.J. Ultrasound-assisted Multicomponent Reactions and Organochalcogen Chemistry. Asian J. Org. Chem. 2018, 7, 2368. [Google Scholar] [CrossRef]
- Wang, S.X.; Li, Z.Y.; Zhang, J.C.; Li, J.T. The Solvent-free Synthesis of 1,4-dihydropyridines under Ultrasound Irradiation without Catalyst. Ultrason. Sonochem. 2008, 15, 677–680. [Google Scholar] [CrossRef]
- Moradi, L.; Zare, M. Ultrasound-promoted Green Synthesis of 1,4-dihydropyridines Using Fuctionalized MWCNTs as a Highly Efficient Heterogeneous Catalyst. Green Chem. Lett. Rev. 2018, 11, 197–208. [Google Scholar] [CrossRef]
- Gu, Y. Multicomponent Reactions in Unconventional Solvents: State of the Art. Green Chem. 2012, 14, 2091–2128. [Google Scholar] [CrossRef]
- Reddy, G.M.; Garcia, R.J.; Reddy, N.B.; Zyryanov, V.G.; Yuvaraja, G. An Efficient One-Pot, Multicomponent, and Green Solvent Protocol for the Synthesis of Dihydropyridine Derivatives. J. Heterocycl. Chem. 2019, 56, 845–849. [Google Scholar] [CrossRef]
- Shirini, F.; Rad-Moghadam, K.; Akbari-Dadamahaleh, S. Application of Ionic Liquids in Multicomponent Reactions. In Green Solvents II; Springer: Amsterdam, The Netherlands, 2012; pp. 289–334. [Google Scholar]
- Ren, Y.-M.; Cai, C.; Yang, R.-C. Molecular Iodine-catalyzed Multicomponent Reactions: An Efficient Catalyst for Organic Synthesis. RSC Adv. 2013, 3, 7182–7204. [Google Scholar] [CrossRef]
- Prajapati, N.; Vekariya, R.; Patel, H. Ceric Ammonium Nitrate (CAN)–Catalyzed Multicomponent Reactions: An Efficient Catalyst for Green Organic Synthesis. Synth. Commun. 2015, 45, 2399–2425. [Google Scholar] [CrossRef]
- Koolivand, M.; Nikoorazm, M.; Ghorbani-Choghamarani, A. Ni–citric acid coordination polymer as a practical catalyst for multicomponent reactions. Sci. Rep. 2021, 11, 24475. [Google Scholar] [CrossRef] [PubMed]
- Bosica, G.; Abdilla, R. Recent Advances in Multicomponent Reactions Catalysed under Operationally Heterogeneous Conditions. Catalysts 2022, 12, 725. [Google Scholar] [CrossRef]
- Neto, B.A.D.; Rocha, R.O.; Rodrigues, M.O. Catalytic Approaches to Multicomponent Reactions: A Critical Review and Perspectives on the Roles of Catalysis. Molecules 2022, 27, 132. [Google Scholar] [CrossRef] [PubMed]
- Jumbam, N.D.; Masamba, W. Bio-Catalysis in Multicomponent Reactions. Molecules 2020, 25, 5935. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.M.; Kunze, D.L.; Yatani, A. The agonist effect of dihydropyridines on Ca channels. Nature 1984, 311, 570–572. [Google Scholar] [CrossRef]
- DrugBank Online. Available online: https://go.drugbank.com/drugs/DB00381 (accessed on 10 December 2022).
- DrugBank Online. Available online: https://go.drugbank.com/drugs/DB01054 (accessed on 10 December 2022).
- DrugBank Online. Available online: https://go.drugbank.com/drugs/DB00270 (accessed on 10 December 2022).
- Keating, G.M. Clevidipine: A review of its use for managing blood pressure in perioperative and intensive care settings. Drugs 2014, 74, 1947–1960. [Google Scholar] [CrossRef] [PubMed]
- Toal, C.B.; Meredith, P.A.; Elliott, H.L. Long-acting dihydropyridine calcium-channel blockers and sympathetic nervous system activity in hypertension: A literature review comparing amlodipine and nifedipine GITS. Blood Press 2012, 21, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Bucci, C.; Mamdani, M.M.; Juurlink, D.N.; Tu, J.V. Dihydropyridine calcium channel blockers and cardiovascular outcomes in elderly patients: A population-based study. Can. J. Cardiol. 2008, 24, 629–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Straits Research. Available online: https://straitsresearch.com/report/nifedipine-market (accessed on 10 December 2022).
- Takahashi, D.; Oyunzul, L.; Onoue, S.; Ito, Y.; Uchida, S.; Simsek, R.; Gunduz, M.G.; Safak, C.; Yamada, S. Structure-activity relationships of receptor binding of 1,4-dihydropyridine derivatives. Biol. Pharm. Bull. 2008, 31, 473. [Google Scholar] [CrossRef] [PubMed]
- Garaliene, V.; Barsys, V.; Mačys, A.; Vigante, B.; Krauze, A. Effect of 4-aryl-2-methyl-5-nitro-1,4-dihydropyridine-3-carboxylates on the guinea pig papillary muscle and isolated human vena saphena magna that is used for coronary artery bypass grafting. Eur. J. Med. Chem. 2011, 46, 4441–4447. [Google Scholar] [CrossRef]
- Shan, R.; Velazquez, C.; Knaus, E.E. Syntheses, Calcium Channel Agonist-Antagonist Modulation Activities, and Nitric Oxide Release Studies of Nitrooxyalkyl 1,4-Dihydro-2,6-dimethyl-3-nitro-4-(2,1,3-benzoxadiazol-4-yl)pyridine-5-carboxylate Racemates, Enantiomers, and Diastereomers. J. Med. Chem. 2004, 47, 254. [Google Scholar] [CrossRef]
- Visentin, S.; Rolando, B.; Di Stilo, A.; Frutterro, R.; Novara, M.; Carbone, E.; Roussel, C.; Vanthuyne, N.; Gasco, A. New 1,4-dihydropyridines endowed with NO-donor and calcium channel agonist properties. J. Med. Chem. 2004, 47, 2688. [Google Scholar] [CrossRef] [PubMed]
- Kluљa, V. Cerebrocrast. Neuroprotectant cognition enhancer. Drugs Future 1995, 20, 135–138. [Google Scholar]
- Duburs, G.; Vigante, B.; Plotniece, A.; Krauze, A.; Sobolevs, A.; Briede, J.; Klufă, V.; Velena, A. Dihydropyridine derivatives as bioprotectors. Chim. Oggi 2008, 26, 68–70. [Google Scholar]
- Sharma, G.V.; Reddy, K.L.; Lakshmi, P.S.; Krishna, P.R. “In situ” Generated “HCl”—An Efficient Catalyst for Solvent-Free Hantzsch Reaction at Room Temperature: Synthesis of New Dihydropyridine Glycoconjugates. Synthesis 2006, 1, 55–58. [Google Scholar] [CrossRef]
- Debache, A.; Boulcina, R.; Belfaitah, A.; Rhouati, S.; Carboni, B. One-pot synthesis of 1, 4-dihydropyridines via a phenylboronic acid catalyzed Hantzsch three-component reaction. Synlett 2008, 4, 509–512. [Google Scholar] [CrossRef]
- Kumar, A.; Maurya, R.A. Efficient Synthesis of Hantzsch Esters and Polyhydroquinoline Derivatives in Aqueous Micelles. Synlett 2008, 6, 883–885. [Google Scholar] [CrossRef]
- Vigante, B.A.; Terekhova, M.I.; Ozols, Y.Y.; Petrov, E.S.; Dubur, G.Y. Equilibrium NH-acidity of 1,4-dihydropyridines and 4,5-dihydroindeno[1,2-b]pyridines. Chem. Heterocycl. Comp. 1989, 25, 1028. [Google Scholar] [CrossRef]
- Herrera, R.P.; Marquez-Lopez, E. Multicomponent Reactions: Concepts and Applications for Design and Synthesis; Wiley-VCH: Weinhiem, Germany, 2015; p. 477. [Google Scholar]
- Zhou, Y.; Kijima, T.; Kuwahara, S.; Watanabe, M.; Izumi, T. Synthesis of ethyl 5-cyano-6-hydroxy-2-methyl-4-(1-naphthyl)-nicotinate. Tetrahedron Lett. 2008, 49, 3757–3761. [Google Scholar] [CrossRef]
- Sagitullina, G.P.; Glizdinskaya, L.V.; Sagitullin, R.S. Nitropyridines: IV. Synthesis of 3-(2-furyl)biphenyls by recyclization of nitropyridinium salts. Russ. J. Org. Chem. 2007, 43, 602. [Google Scholar] [CrossRef]
- Baraldi, P.G.; Simoni, D.; Manfredini, S. An Improved Preparation of Enaminones from 1,3-Diketones and Ammonium Acetate or Amine Acetates. Synthesis 1983, 11, 902–903. [Google Scholar] [CrossRef]
- Sagitullina, G.P.; Garkushenko, A.K.; Sagitullin, R.S. Synthesis of substituted 5-nitro-6-phenylpyridines by the cyclo-condensation of nitroacetophenone, ethyl orthoformate, and enamines. Chem. Heterocycl. Compd. 2009, 45, 1147. [Google Scholar] [CrossRef]
- Zobenko, Y.A.; Pozhidaeva, S.A.; Kuratova, A.K.; Glyzdinskaya, L.V.; Vorontsova, M.A.; Sagitullina, G.P. New method for the synthesis of nitrobiaryls. Chem. Heterocycl. Compd. 2017, 53, 1014–1025. [Google Scholar] [CrossRef]
- Sagitullina, G.P.; Garkushenko, A.K.; Atavin, E.G.; Sagitullin, R.S. ChemInform Abstract: One-Pot Synthesis of 5-Nitropyridines by the Cyclocondensation of Nitroacetone, Triethyl Orthoformate and Enamines. Mendeleev Commun. 2009, 19, 155–156. [Google Scholar] [CrossRef]
- Shuvalov, V.Y.; Rupp, A.S.; Fisyuk, A.S.; Kuratova, A.K.; Nefedov, A.A.; Sagitullina, G.P. Synthesis and Optical Properties of Alkaloid Quindoline, Its Structural Analogues and Substituted δ-Carbolines. ChemistrySelect 2019, 4, 1696–1699. [Google Scholar] [CrossRef]
- Koveza, V.A.; Kulakov, I.V.; Shulgau, Z.T.; Seilkhanov, T.M. Multicomponent synthesis of unsymmetrical 5-nitropyridines. Chem. Heterocycl. Compd. 2018, 54, 1127–1130. [Google Scholar] [CrossRef]
- Kulakov, I.V.; Oleshchuk, A.L.; Koveza, V.A.; Palamarchuk, I.V. Multicomponent synthesis of 4-unsubstituted 5-nitropyridine derivatives. Synth. Commun. 2020, 50, 2432–2439. [Google Scholar] [CrossRef]
- Ryzhkova, Y.E.; Elinson, M.N. Multicomponent synthesis of 1,4-dihydropyridine-3,5-dicarbonitriles. Arkivoc 2022, v, 126–134. [Google Scholar] [CrossRef]
- Memarian, H.R.; Bagheri, M.; Döpp, D. Synthesis and photochemistry of novel 3, 5-diacetyl-1, 4-dihydropyridines. Monatsh. Chem. 2004, 135, 833–838. [Google Scholar] [CrossRef]
- D’alelio, G.F.; Kurosaki, T.; Schoenig, R.K. Polymeric Schiff Bases. IV. The Prototype Syntheses of Schiff Bases by Reactions of Aromatic Acetals with Aromatic Amines or Their Acyl Derivatives. J. Macromol. Sci. Part A—Chem. 1967, 1, 1279–1298. [Google Scholar] [CrossRef]
- Xiao, P.; Dumur, F.; Tehfe, M.-A.; Graff, B.; Gigmes, D.; Fouassier, J.P.; Lalevйe, J. Acridinediones: Effect of Substituents on Their Photoinitiating Abilities in Radical and Cationic Photopolymerization. Macromol. Chem. Phys. 2013, 214, 2276–2282. [Google Scholar] [CrossRef]
- Hurd, C.D.; Nilson, M.E. Aliphatic Nitro Ketones. J. Org. Chem. 1955, 20, 927. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turgunalieva, D.M.; Stalinskaya, A.L.; Kulakov, I.I.; Sagitullina, G.P.; Atuchin, V.V.; Elyshev, A.V.; Kulakov, I.V. Multicomponent Synthesis of Unsymmetrical Derivatives of 4-Methyl-Substituted 5-Nitropyridines. Processes 2023, 11, 576. https://doi.org/10.3390/pr11020576
Turgunalieva DM, Stalinskaya AL, Kulakov II, Sagitullina GP, Atuchin VV, Elyshev AV, Kulakov IV. Multicomponent Synthesis of Unsymmetrical Derivatives of 4-Methyl-Substituted 5-Nitropyridines. Processes. 2023; 11(2):576. https://doi.org/10.3390/pr11020576
Chicago/Turabian StyleTurgunalieva, Daria M., Alena L. Stalinskaya, Ilya I. Kulakov, Galina P. Sagitullina, Victor V. Atuchin, Andrey V. Elyshev, and Ivan V. Kulakov. 2023. "Multicomponent Synthesis of Unsymmetrical Derivatives of 4-Methyl-Substituted 5-Nitropyridines" Processes 11, no. 2: 576. https://doi.org/10.3390/pr11020576