


NiO-MgO Prepared by the Complex-Decomposition Method as a Catalyst for Carbon Dioxide Reforming of Methane



Abstract
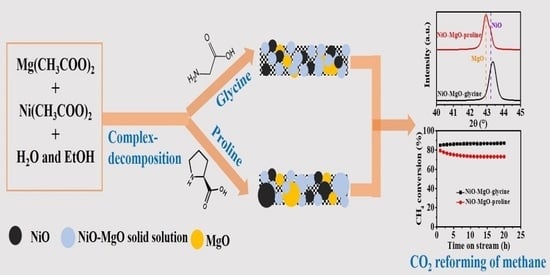
:
1. Introduction
2. Experimental Section
2.1. Catalysts Preparation
2.2. Catalysts Characterization
2.3. CRM Reaction
3. Results and Discussion
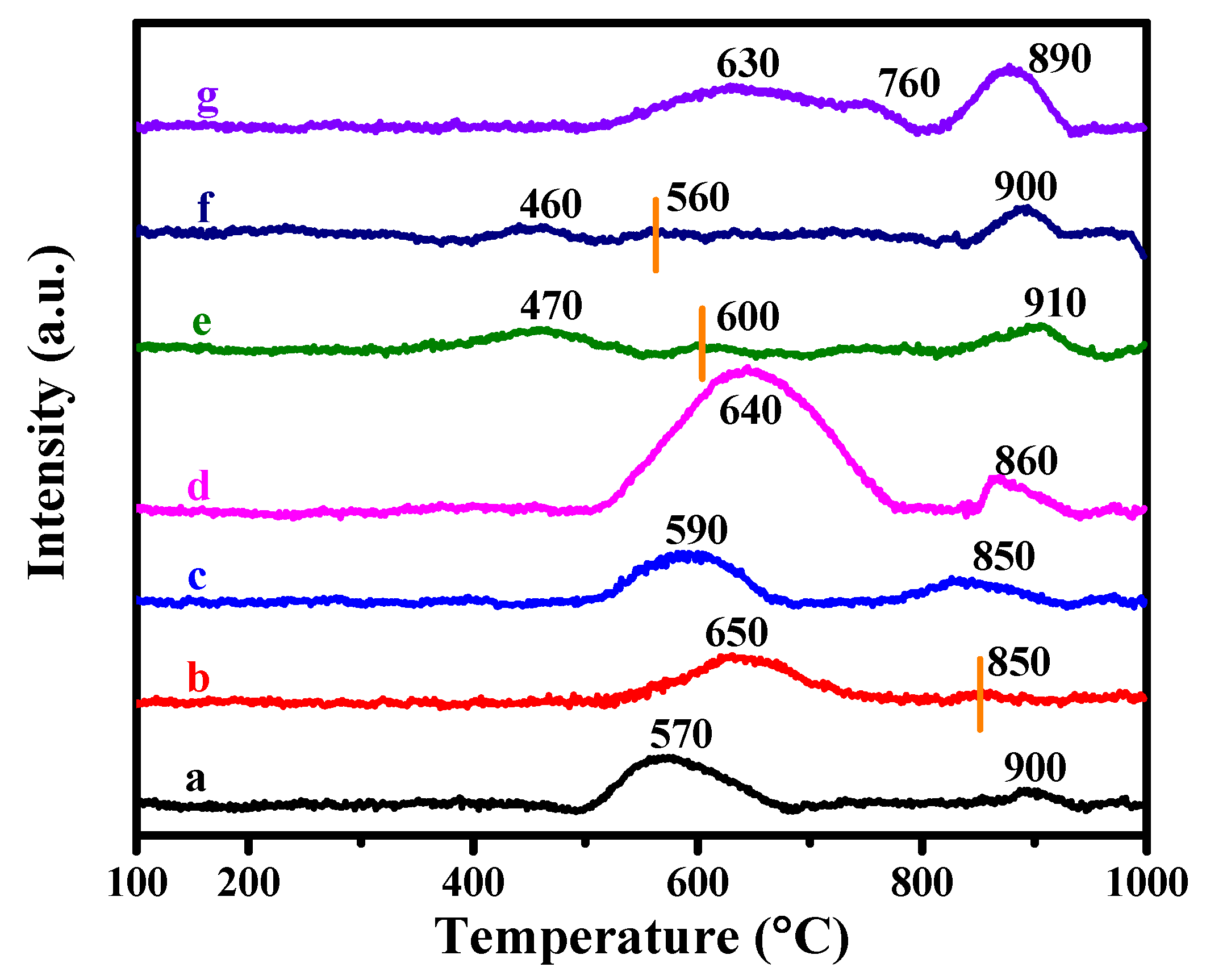
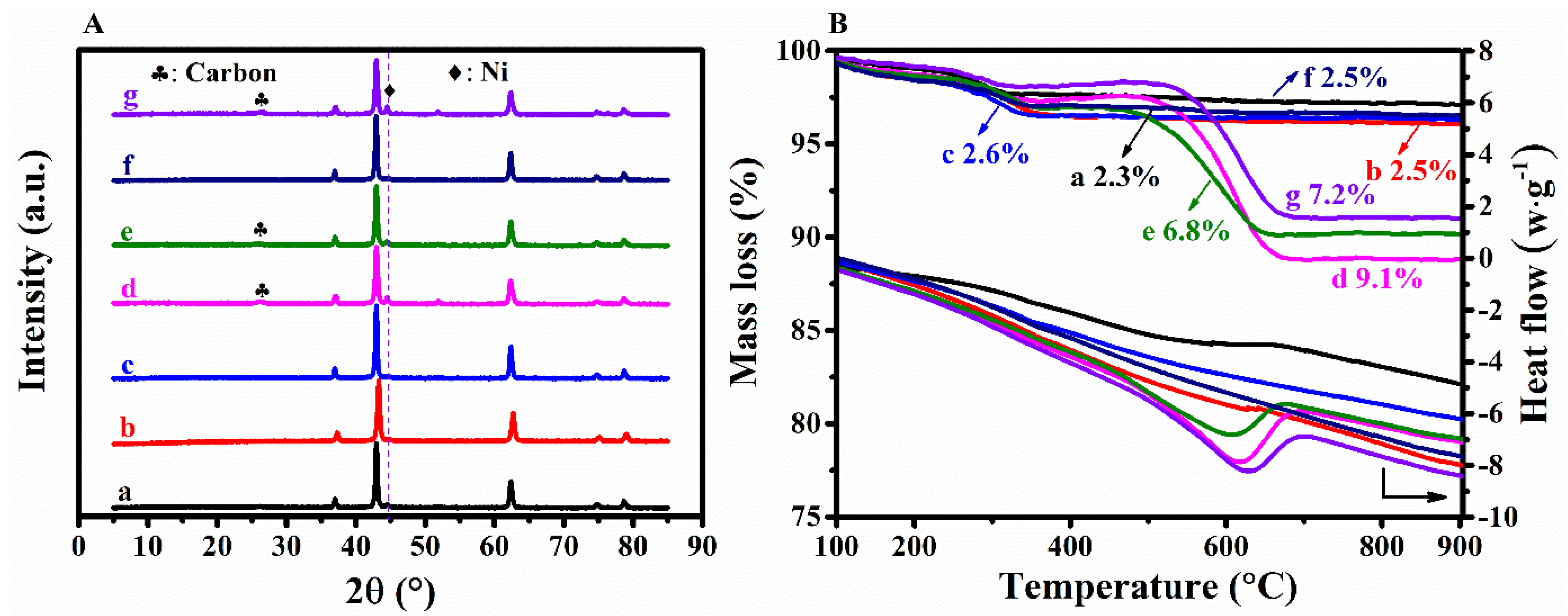
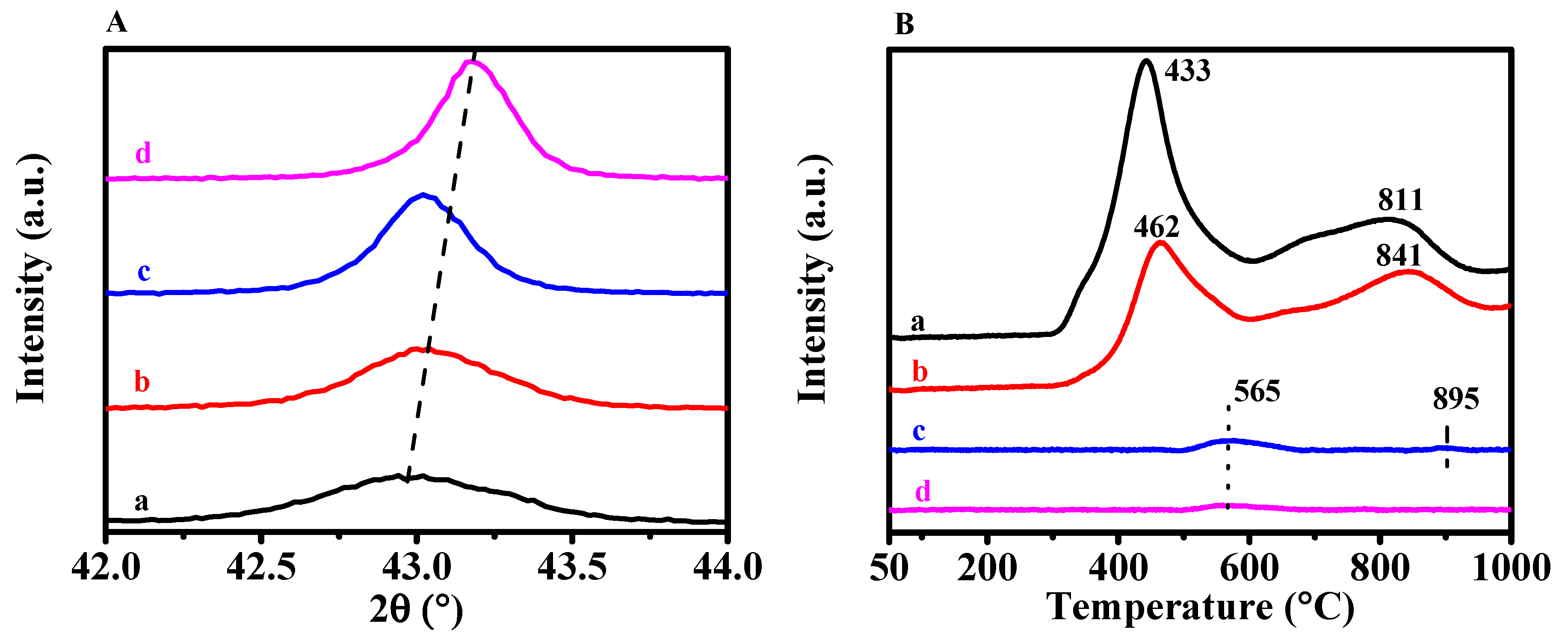
3.1. Effects of Complexing Agents
3.2. CRM Performance
3.3. Characteristics of the Spent Catalysts
3.4. Impact of Preparation Parameters of NiO-MgO on CRM Performance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, M.J.; Sun, Z.X.; Hu, Y.H. Catalysts for CO2 reforming of CH4: A review. J. Mater. Chem. A 2021, 9, 12495. [Google Scholar] [CrossRef]
- Abdulrasheed, A.; Jalil, A.A.; Gambo, Y.; Ibrahim, M.; Hambali, H.U.; Shahul Hamid, M.Y. A review on catalyst development for dry reforming of methane to syngas: Recent advances. Renew. Sustain. Energy Rev. 2019, 108, 175–193. [Google Scholar] [CrossRef]
- Sinh, R.; Dhir, A.; Mohapata, S.K.; Mahla, S.K. Dry reforming of methane using various catalysts in the process: Review. Biomass. Conv. Bioref. 2019, 10, 567–587. [Google Scholar] [CrossRef]
- Li, Z.W.; Lin, Q.; Li, M.; Cao, J.X.; Liu, F.; Pan, H.Y.; Wang, Z.G.; Kawi, S. Recent advances in process and catalyst for CO2 reforming of methane. Renew. Sustain. Energy Rev. 2020, 134, 110312. [Google Scholar] [CrossRef]
- Bacariza, C.; Karam, L.; Hassan, N.E.; Lopes, J.M.; Henriques, C. Carbon dioxide reforming of methane over nickel-supported zeolites: A screening study. Processes 2022, 10, 1331. [Google Scholar] [CrossRef]
- Azizi, Z.; Rezaeimanesh, M.; Tohidian, T.; Rahimpour, M.R. Dimethyl ether: A review of technologies and production challenges. Chem. Eng. Process. 2014, 82, 150–172. [Google Scholar] [CrossRef]
- San-José-Alonso, D.; Juan-Juan, J.; Illán-Gómez, M.J.; Román-Martínez, M.C. Ni, Co and bimetallic Ni–Co catalysts for the dry reforming of methane. Appl. Catal. A Gen. 2009, 371, 54–59. [Google Scholar] [CrossRef]
- Wang, N.; Yu, X.; Wang, Y.; Chu, W.; Liu, M. A comparison study on methane dry reforming with carbon dioxide over LaNiO3 perovskite catalysts supported on mesoporous SBA-15, MCM-41 and silica carrier. Catal. Today 2013, 212, 98–107. [Google Scholar] [CrossRef]
- Sajjadi, S.M.; Haghighi, M.; Eshghi, J. Synergic influence of potassium loading and plasma-treatment on anti-coke property of K-promoted bimetallic NiCo-NiAl2O4 nanocatalyst applied in O2-enhanced dry reforming of CH4. Int. J. Hydrogen Energy 2019, 44, 13397–13414. [Google Scholar] [CrossRef]
- Liu, C.J.; Ye, J.; Jiang, J.; Pan, Y. Progresses in the Preparation of Coke Resistant Ni-based Catalyst for Steam and CO2 Reforming of Methane. ChemCatChem 2011, 3, 529–541. [Google Scholar] [CrossRef]
- Sajjadi, S.M.; Haghighi, M.; Rahmani, F.; Eshghi, J. Plasma-enhanced sol-gel fabrication of CoWNiAl2O4 nanocatalyst used in oxidative conversion of greenhouse CH4/CO2 gas mixture to H2/CO. J. CO2 Util. 2022, 61, 102037. [Google Scholar] [CrossRef]
- Abdullah, B.; Abd Ghani, N.A.; Vo, D.V.N. Recent advances in dry reforming of methane over Ni-based catalysts. J. Clean. Prod. 2017, 162, 170–185. [Google Scholar] [CrossRef] [Green Version]
- Salaev, M.A.; Liotta, L.F.; Vodyankina, O.V. Lanthanoid-containing Ni-based catalysts for dry reforming of methane: A review. Int. J. Hydrogen Energy 2022, 47, 4489–4535. [Google Scholar] [CrossRef]
- Ren, H.-P.; Song, Y.-H.; Wang, W.; Chen, J.-G.; Cheng, J.; Jiang, J.; Liu, Z.-T.; Liu, Z.-W.; Hao, Z.; Lu, J. Insights into CeO2-modified Ni–Mg–Al oxides for pressurized carbon dioxide reforming of methane. Chem. Eng. J. 2015, 259, 581–593. [Google Scholar] [CrossRef]
- Xu, L.; Song, H.; Chou, L. One-Pot Synthesis of Ordered Mesoporous NiO–CaO–Al2O3 Composite Oxides for Catalyzing CO2 Reforming of CH4. ACS Catal. 2012, 2, 1331–1342. [Google Scholar] [CrossRef]
- Yang, R.; Xing, C.; Lv, C.; Shi, L.; Tsubaki, N. Promotional effect of La2O3 and CeO2 on Ni/γ-Al2O3 catalysts for CO2 reforming of CH4. Appl. Catal. A Gen. 2010, 385, 92–100. [Google Scholar] [CrossRef]
- Akri, M.; Zhao, S.; Li, X.; Zang, K.; Lee, A.F.; Isaacs, M.A.; Xi, W.; Gangarajula, Y.; Luo, J.; Ren, Y.; et al. Atomically dispersed nickel as coke-resistant active sites for methane dry reforming. Nat. Commun. 2019, 10, 5181. [Google Scholar] [CrossRef] [Green Version]
- Zuo, Z.; Liu, S.; Wang, Z.; Liu, C.; Huang, W.; Huang, J.; Liu, P. Dry Reforming of Methane on Single-Site Ni/MgO Catalysts: Importance of Site Confinement. ACS Catal. 2018, 8, 9821–9835. [Google Scholar] [CrossRef]
- Song, Y.D.; Ramesh, S.; Adishev, A.; Subramanian, S.; Harale, A.; Albuali, M.; Fadhel, B.A.; Jamal, A.; Moon, D.; Choi, S.H.; et al. Dry reforming of methane by stable Ni–Mo nanocatalysts on single-crystalline MgO. Science 2020, 367, 777–781. [Google Scholar] [CrossRef]
- Ruckenstein, E.; Hu, Y.H. Carbon dioxide reforming of methane over nickel/alkaline earth metal oxide catalysts. Appl. Catal. A Gen. 1995, 133, 149–161. [Google Scholar] [CrossRef]
- Kuzmin, A.; Mironova, N. Composition dependence of the lattice parameter in NicMg1−cO solid solutions. J. Phys. Condens. Mat. 1998, 10, 7937–7944. [Google Scholar] [CrossRef]
- Ren, H.-P.; Hao, Q.-Q.; Wang, W.; Song, Y.-H.; Cheng, J.; Liu, Z.-W.; Liu, Z.-T.; Lu, J.; Hao, Z. High-performance Ni–SiO2 for pressurized carbon dioxide reforming of methane. Int. J. Hydrogen Energy 2014, 39, 11592–11605. [Google Scholar] [CrossRef]
- Qin, P.; Xu, H.; Long, H.; Ran, Y.; Shang, S.; Yin, Y.; Dai, X. Ni/MgO catalyst prepared using atmospheric high-frequency discharge plasma for CO2 reforming of methane. J. Nat. Gas Chem. 2011, 20, 487–492. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Liu, H.-M.; Xu, B.-Q. Durable Ni/MgO catalysts for CO2 reforming of methane: Activity and metal—Support interaction. J. Mol. Catal. A Chem. 2009, 299, 44–52. [Google Scholar] [CrossRef]
- Zhang, Z.-F.; Liu, Z.-T.; Liu, Z.-W.; Lu, J. DMC Formation over Ce0.5Zr0.5O2 Prepared by Complex-decomposition Method. Catal. Lett. 2008, 129, 428–436. [Google Scholar] [CrossRef]
- Arena, F.; Licciardello, A.; Parmalian, A. The role of Ni2+ diffusion on the reducibility of NiO/MgO system: A combined TPR-XPS study. Catal. Lett. 1990, 6, 139–1501. [Google Scholar] [CrossRef]
- Liu, Y.J.; Qing, S.J.; Hou, X.N.; Qin, F.J.; Wang, X.; Gao, Z.X.; Xiang, H.W. Temperature dependence of Cu–Al spinel formation and its catalytic performance in methanol steam reforming. Catal. Sci. Technol. 2017, 7, 5069–5078. [Google Scholar] [CrossRef]
- Taherian, Z.; Khataee, A.; Han, N.; Orooji, Y. Hydrogen production through methane reforming processes using promoted-Ni/mesoporous silica: A review. J. Ind. Eng. Chem. 2022, 107, 20–30. [Google Scholar] [CrossRef]
- Chen, D.; Christensen, K.; Ochoafernandez, E.; Yu, Z.; Totdal, B.; Latorre, N.; Monzon, A.; Holmen, A. Synthesis of carbon nanofibers: Effects of Ni crystal size during methane decomposition. J. Catal. 2005, 229, 82–96. [Google Scholar] [CrossRef]
- Park, K.S.; Cho, J.M.; Park, Y.M.; Kwon, J.H.; Yu, J.S.; Jeong, H.E.; Choung, J.W.; Bae, J.W. Enhanced thermal stability of Ni nanoparticles in ordered mesoporous supports for dry reforming of methane with CO2. Catal. Today 2022, 388–389, 224–230. [Google Scholar] [CrossRef]
- Seo, J.-C.; Kim, H.; Lee, Y.-L.; Nam, S.; Roh, H.-S.; Lee, K.; Park, S.B. One-pot synthesis of full-featured mesoporous Ni/Al2O3 catalysts via pyrolysis-assisted evaporation-induced self-assembly method for dry reforming of methane. ACS Sustain. Chem. Eng. 2021, 9, 894–904. [Google Scholar] [CrossRef]
- Niu, J.; Wang, Y.; Liland, S.E.; Regli, S.K.; Yang, J.; Rout, K.R.; Luo, J.; Ronning, M.; Ran, J.; Chen, D. Unraveling enhanced activity, selectivity, and coke resistance of Pt-Ni bimetallic clusters in dry reforming. ACS Catal. 2021, 11, 2398–2411. [Google Scholar] [CrossRef]
- Li, K.; Pei, C.; Li, X.; Chen, S.; Zhang, X.; Liu, R.; Gong, J. Dry reforming of methane over La2O2CO3-modified Ni/Al2O3 catalysts with moderate metal support interaction. Appl. Catal. B Environ. 2020, 264, 118448. [Google Scholar] [CrossRef]
- Ma, Q.; Han, Y.; Wei, Q.; Makpal, S.; Gao, X.; Zhang, J.; Zhao, T.-S. Stabilizing Ni on bimodal mesoporous-macroporous alumina with enhanced coke tolerance in dry reforming of methane to syngas. J. CO2 Util. 2020, 35, 288–297. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | BET Surface Area (m2·g−1) | Pore Volume (cm3·g−1) | Average Pore Size (nm) |
|---|---|---|---|
| Ni0.1Mg0.9O-800-Gly | 25.5 | 0.15 | 23.3 |
| Ni0.1Mg0.9O-800-Gla | 27.4 | 0.13 | 21.2 |
| Ni0.1Mg0.9O-800-Oxa | 34.6 | 0.39 | 35.1 |
| Ni0.1Mg0.9O-800-Pro | 26.0 | 0.31 | 20.5 |
| Ni0.1Mg0.9O-800-Ser | 34.5 | 0.24 | 34.1 |
| Ni0.1Mg0.9O-800-Urea | 27.7 | 0.17 | 21.3 |
| Ni0.1Mg0.9O-800-Ala | 28.1 | 0.24 | 34.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Li, B.; Xiao, Y.-S.; Liu, Z.-W. NiO-MgO Prepared by the Complex-Decomposition Method as a Catalyst for Carbon Dioxide Reforming of Methane. Processes 2023, 11, 596. https://doi.org/10.3390/pr11020596
Wang Y, Li B, Xiao Y-S, Liu Z-W. NiO-MgO Prepared by the Complex-Decomposition Method as a Catalyst for Carbon Dioxide Reforming of Methane. Processes. 2023; 11(2):596. https://doi.org/10.3390/pr11020596
Chicago/Turabian StyleWang, Ying, Bin Li, Yong-Shan Xiao, and Zhong-Wen Liu. 2023. "NiO-MgO Prepared by the Complex-Decomposition Method as a Catalyst for Carbon Dioxide Reforming of Methane" Processes 11, no. 2: 596. https://doi.org/10.3390/pr11020596
APA StyleWang, Y., Li, B., Xiao, Y. -S., & Liu, Z. -W. (2023). NiO-MgO Prepared by the Complex-Decomposition Method as a Catalyst for Carbon Dioxide Reforming of Methane. Processes, 11(2), 596. https://doi.org/10.3390/pr11020596