Abstract
In this study, we investigate the structural changes, electronic properties, and charge redistribution within azo-bithiophene (Azo-BT)-chemisorbed monolayers under different light stimuli using the density functional theory and molecular dynamics simulations. We consider two types of switches, Azo-BT and BT-Azo, with different arrangements of the Azo and BT blocks counting from the anchor thiol group. The chemisorbed monolayers of pure cis- and trans-isomers with a surface concentration of approximately 2.7 molecules per nm are modeled on a gold surface using the classical all-atom molecular dynamics. Our results reveal a significant shrinkage of the BT-Azo layer under UV illumination, whereas the thicknesses of the Azo-BT layer remain comparable for both isomers. This difference in behavior is attributed to the ordering of the trans-molecules in the layers, which is more pronounced for Azo-BT, leading to a narrow distribution of the inclination angle to the gold surface. Conversely, both layers consisting of cis-switches exhibit disorder, resulting in similar brush heights. To study charge transfer within the immobilized layers, we analyze each snapshot of the layer and calculate the mean charge transfer integrals using Nelsen’s algorithm for a number of interacting neighboring molecules. Combining these integrals with reorganization energies defined for the isolated molecules, we evaluate the charge transfer rates and mobilities for electron and hole hopping within the layers at room temperature based on Marcus’ theory. This research offers new perspectives for the innovative design of electrode surface modifications and provides insights into controlling charge transfer within immobilized layers using light triggers. Additionally, we identify molecular properties that are enhanced via specific molecular design, which contributes to the development of more efficient molecular switches for various electronic applications.
1. Introduction
Molecular switches (MSs) represent the molecules that respond to stimuli by changing between two or more conformational or configurational states. In the former case, the switch involves a physical transformation in the geometry of the molecule, while configurational switches are related to bond-breaking and bond-forming reactions [1]. An example of a configurational molecular switch is one with an azobenzene (Azo) group, which undergoes reversible photoisomerization depending on the wavelength of the light trigger. In the dark or upon irradiation with visible (usually blue) light, azobenzene adopts a trans-state, which represents a global minimum on the potential energy surface. Under UV light, a cis-state is formed as a product of isomerization. The process is fully reversible, and under visible light or upon heating, the metastable cis-isomers change their configuration back to the trans-state. Responsive monolayers of Azo-MS [2] are the key building blocks for advanced applications ranging from smart surfaces with switchable wettability for textile industry [3] and biotechnology, where the binding and release of ions [4], biomacromolecules [5] or both gene and drug delivery [6,7,8] can be controlled, to modern elements of organic electronics, such as molecular junctions (MJ), where MS are bound to metal or semiconductive electrodes [9,10], and sensors [11].
Embedding MS into semiconductive material is a straightforward method for implementing the versatility of optoelectronic devices. The organic semiconductor (OSC)/ electrode interfaces can also act as places where MS can function [12]. According to the recently published review by Chung et al. [12], MJ engineering can be divided into two categories: photo-programmable tunneling injection control and work function tuning of the electrode.
Literature surveys have demonstrated that azobenzene-based molecules organized as molecular junctions on electrode surfaces can work as photoswitches of (i) conductance [13,14,15,16,17,18], (ii) electrode work function [16,19,20,21], and (iii) magnetization/magnetic transitions [22,23]. Changes in conductance are mainly related to the tunneling barrier length between the top probe/electrode (such as Hg droplet and C-AFM or STM tips) and the bottom electrode, usually a gold surface, Au nanoparticle (Au–NP) [24,25], or cobalt [18]. In other words, they are related to the configurational changes that directly affect the molecule–probe intervals. The height of the MS layer between two electrodes for trans-isomers is larger, and in the majority of the experimental papers, this isomer is attributed as the OFF state with a higher resistance, whereas the shorter cis-form is the ON state. The ON/OFF conductance ratio sometimes reaches dramatic values [14,15,17,25,26,27]. However, the identification of the cis-state as the conducting one is not unambiguous. For example, Kumar and colleagues [28] have shown that the cis-azobenzenes incorporated in 1-decanethiolate SAM (self-assembled monolayer) films, i.e., in two-component SAMs, have the properties of the OFF state. Here, the azobenzene-containing molecules work as approximately 2.1 ± 0.3 Å and ca. 0.7 ± 0.2 Å apparent protrusions from the 1-decanethiolate matrix for trans- and cis-isomers, respectively, with a longer protrusion providing contact with the top electrode. Another example by Del Valle et al. [29] illustrates via first-principle calculations that the conductance of azobenzenes is possibly electrode sensitive, with the cis-isomer being the OFF state for carbon nanotube electrodes and the cis-isomer being the ON state for silicon contacts.
The photo-programmable electrode work function tuning is attributed to differences originating from the significant metal-to-molecule electron charge transfer or to the role of the dipole moment of the MS. The latter effect is especially intriguing in light of the fact that azobenzene isomers vary significantly in the magnitude of the dipole moment and its direction [30,31,32] and can lead to a more effective and controlled tuning of the electrical characteristics in biosensing device applications [21].
In general, the photo-programmable MS layer in MJ is situated on the upper surface of the electrode, and ideally, a thin and uniform semiconductor 2D structure is formed on the surface of the MS layer to maximize the switching effect of the device [12]. However, this upper layer of the OSC on MS is difficult to achieve for large-scale solution processing; additionally, its thickness and uniformity are issues in incident light quenching. The new, interesting, and, at the same time, more complex systems to circumvent the aforementioned challenges are the MJ layers consisting of azobenzene molecules comprising the Azo group conjugated with aromatic units or semiconducting oligomers. For example, additional benzene [17,18,19,33,34,35,36,37,38,39,40,41] or fluorinated benzene rings [20,42,43,44], the second covalently attached azobenzene [45], biphenyl [46], azothiophene [47], bithiophene [13,14,18,25,48,49] were included into the chemical structures of one-component Azo-MS. The newly introduced blocks have
- Influenced the extent of conjugation;
- Affected dipole moment;
- Changed backbone rigidity,
- Brought new functionalities such as strong stacking between the aromatic units,
- Altered the system’s response to light stimulus (i.e., accelerated collective switching behavior or caused significant broadening of the optical absorption spectra);
- Emphasized the role of spacer length [14]: a shorter spacer should facilitate the charge transfer rate through the junction and increase conductance, while a longer spacer is prone to decoupling of the Azo from the electrode;
- Allowed larger dynamics of the switching event;
- Allowed a larger ON/OFF conductance ratio.
The findings listed above have important implications for understanding the charge transport mechanisms in MS devices. Developing correlations between structure and function is necessary for fully understanding the properties of molecular junction platforms. Therefore, it is important to account for molecular-level variations that can occur in the bound layers. In this context, optical and transport experimental and theoretical characterizations can provide insights into the structural aspects of MS layers.
Here, we report the structural, optical, and transport properties of a molecular switch in which the azobenzene moiety is linked to a bithiophene spacer and a short alkanethiol (Figure 1). This design (Figure 1b) tethered to a gold electrode or Au–NP has been previously developed by Lenfant and others and studied in several works [13,14,25,48,49,50]. The incorporation of the bithiophene segment, which is redox active at moderately positive potentials, allowed for the preliminary characterization of the immobilization process using simple electrochemical experiments [13]. The bithiophene unit also helps maintain a high level of current in the BT-Azo molecular junction compared to a fully saturated n-alkyl spacer with the same length [14]. In addition, the bithiophene segment promotes possible stacking interactions in the molecular junction layer and endows the molecule with more conformational rigidity [51], as suggested by Lenfant and colleagues.

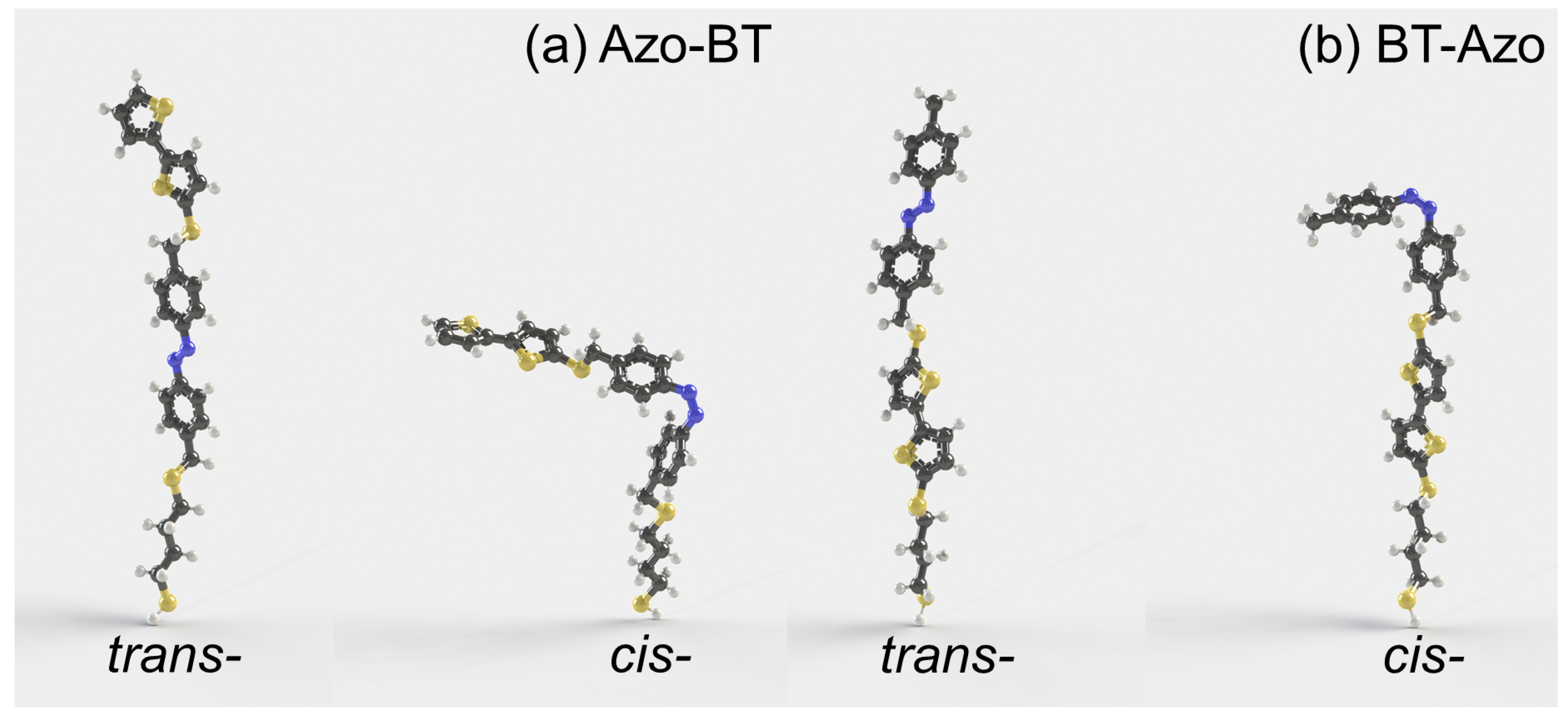
Figure 1.
The optimized geometries of Azo-BT (a) and BT-Azo (b) in trans- and cis-states with a common formula CHNS. The abbreviations Azo and BT stand for azobenzene and bithiophene, respectively. The molecules (a,b) differ in the sequence of Azo and BT blocks counting from the spacer with thiol group, i.e., they are structural isomers. For example, this group anchors BT-Azo (b) molecules to the surface of an electrode [13,14,50] or a nanoparticle [25,48,49]. We use Azo-BT and BT-Azo nomenclature for the sake of convenience only. The carbon, nitrogen, sulfur, and hydrogen atoms are colored in gray, blue, yellow, and white. This color scheme is used throughout this paper. The static dipole moments are 1.67 and 3.52D for (a) trans- and cis-isomers, and 1.34 and 3.22 D for (b) trans- and cis-states, respectively, as obtained in our recent publication [32].
In addition to the synthesis and experimental characterization of the BT-Azo molecular junctions, Lenfant et al. [25] have simulated the isolated trans- and cis-isomers of this switch applying density functional theory (DFT) with B3LYP/6-311G options and calculated the optical absorption spectra for the optimized isomers in vacuum using time-dependent DFT. The same BT-Azo states in contact with the top electrode distanced at 0.2 nm above the molecule and modeled as Au cluster were described as employing the DFT with B3LYP functional and 6-31G and LANL2DZ basis sets for the organic and inorganic parts of the system, respectively [14]. The main theoretical output was that the high ON/OFF cis/trans conductance ratio is primarily due to the changes in both the molecule–electrode coupling and the electronic structure of the metal–molecules–metal junction rather than due to the variation in the SAM thickness. In particular, a top mechanical electrode contacts the “insulating” methyl group in trans-SAM (here, –CH is acting as a tunnel barrier) and the Azo bond or aromatic benzene ring in cis-SAM under UV light (these groups are strongly coupled electronically to the Au cluster). Moreover, the electronic density delocalization and the charge transfer from the switch to the top electrode was found to be more pronounced for the cis-isomer.
Five years later, Lenfant, Cleri et al. conducted a classical molecular dynamics (MD) simulation of realistic SAMs utilizing the MM3 empirical force field in order to emulate the dynamical structure of the interfaces. The focus was the evaluation of the intermolecular interactions and mutual orientations of two contacting flat or curved layers of interdigitating densely packed BT-Azo switches (Figure 1b) from adjacent spherical or icosahedral Au–NPs forming the so-called nanoparticle self-assembled networks (NPSANs) [25]. The main conclusions were that switching between configurations changes the overlap (here, the physical touch of the molecules, their interdigitation, and the stacking) between the molecules, which causes variations in conductance. Interestingly, the overlap between the cis-switches at the NP interface in NPSANs is drastically reduced already at 100 K and vanishes at 300 K, whereas the trans-switches keep the overlap at all temperatures tested, and electrons have a higher tunneling probability to jump between NPs. Nevertheless, the cis-isomer, as measured in the experiments, still plays a role of the ON state, and the mentioned effect from a more efficient molecular overlap does not prevail over the tunneling barrier length, i.e., the SAM thickness, which is larger for the trans-switch (OFF–state). As a result, a smaller ON/OFF conductance ratio of up to 620 has been detected compared to the previously observed value of 7 × 10 for the planar SAMs in contact with a mechanical top electrode [13,14]. In the latest published work dedicated to BT-Azo MJ [18], the MS effects on the cobalt electrode work function have been characterized. The DFT model included both electrodes (Co and the gold top electrode) and a single BT-Azo molecule in different configurational and conformational states between them. We should note that in contrast to the previously studied systems [14,25], where the molecules were standing almost upright on the surface, the MS molecules on Co are tilted with respect to the substrate with tilting angle of 73 ± 4. In this system, the trans-isomer has been identified as an ON state, while the cis-isomer is responsible for lower conductance.
To the best of our knowledge, only three publications [14,18,25] have theoretically described the azobenzene switches with bithiophene segments coupled in one architecture. The focus, as mentioned, is on the explanation of the conductance changes happening under the light stimulus and the high or moderate ON/OFF cis/trans conductance ratio for the studied samples. The motivation is clear: the authors have pursued the goal of elucidating their own experimental findings. Meanwhile, in the conclusions to their published papers [13,14,25,48,49,50], they have indicated several interesting directions for the future development of research, including purely theoretical approaches, which could uncover the phenomena and issues that have not been gauged in the mentioned works. For instance, the overlap of the molecules found in molecular dynamics (MD) simulations depends not so much on the interdigitation and the mutual molecular orientations, as seen from the analysis of the orientation dihedral angle between the conjugated planes of trans-azobenzenes [14,25]. The reason for this is unknown. Another factor that may come into play is the intermolecular stacking distance, d, which has not been altered in the experiments. Besides the variations of d, which have a strong impact on the electron transport properties, the electronic coupling between the neighbouring molecules and the charge transfer within the MJ layer are also interesting tasks for computer modeling. We would like to address these and other questions related to the rational design of azobenzene–bithiophene molecular switches (Figure 1a) in this present paper.
2. Models and Methods
Optimized isolated molecules have been taken from our previous study [32]. Further calculations with B3LYP/6-311G(d,p) functional and basis set concern the isomers geometries in implicit dichloromethane (the solvent used in experiments on BT-Azo [13]) modeled as polarizable continuum using the integral equation formalism [52]. This functional set has been widely used to optimize geometries of various azobenzene-containing molecules assembled in layers [14,25,41,53,54]. Later, the isomers optimized in dichloromethane are subjected to time-dependent DFT calculations of absorption spectra with CAM-B3LYP/6-311G(d,p). Here, the vertical transition energies to the first hundred excited states are evaluated. The electronic spectra are simulated employing Gaussian functions with the half-width at half height of 0.333 eV to build a continuous spectrum from a collection of transitions corresponding to the time-dependent DFT transition energies and oscillator strengths.
Another property defined for isolated MS is the reorganization energy . The reorganization energy of the parameters for calculation of the charge transfer rate follows the Marcus theory:
where V (the charge transfer integral) is the second variable of . The value of = 0 for the self-exchange reactions, i.e., where the electron or hole is transferred between chemically identical molecules; therefore, [55]
The reorganization energy is calculated following the approach described by Nelsen, Blackstock, and Kim [56], and the V values for interacting neighboring molecules are evaluated by applying the so-called energy splitting in dimers [57] for various intermolecular stacking distances d. These approaches have been utilized in some recent publications [58,59,60]. The values of charge transfer rate later are used for the calculation of electron/hole mobility in one-dimensional molecular stacks via the Einstein–Smoluchowski equation for diffusion of charged particles:
where D is diffusion coefficient, T is temperature (is set at 298K), d is either the intermolecular stacking distance for dimers or the separation between centers of masses of the molecules in interacting dimer (the details are provided below). All DFT and time-dependent DFT calculations are conducted in Gaussian 09, Revision A.01 [61].
The collective behavior chemisorbed layers is modeled in MD runs using Materials Studio [62]. The simulations are carried out in the slab models, which are in the size of 53 × 57 × 103 Å, with 81 Azo-BT or BT-Azo pure cis- or pure trans-layers anchored to the Au(111) with surface concentration of ca. 2.7 molecules per nm, which is close to the experimentally evaluated ligand density of 3.2 molecules per nm for gold nanoparticles [25]. We should note that this relatively high concentration leads to quite dense layers of azobenzenes anchored to the surface. The starting configurations of the layers are constructed using an algorithm developed by us, which is stored at bitbucket, t.ly/81cWk. In short, the optimized geometries (rigid bodies) of isomers have been placed vertically as a grid of 9 × 9 without additional rotations with a distance between any neighboring molecules (atoms) in the rows of 6.8–12 Å. Next, the molecules are moved in x, y, or a diagonal direction with a certain step. The algorithm stops when the minimal chosen distance between the molecules is reached. As a result, denser SAMs of vertically oriented molecules are prepared. Built layers are placed in the simulation box with their thiol groups next to the frozen gold surface modeled as two layers. Further, the thiol–SH groups are constrained. This is needed to mimic chemisorbed layers of MS. We omitted a step of real chemical attachment of the molecules to the gold with dissociation of the thiol proton since it needs a parameterization of newly formed S–Au bonds, we should not check their initial lengths (since molecules are not on the grid on the surface, the distances between S and the neighboring Au vary), and at the same time, chosen simplification is not limiting the model of MJ; we are mostly interested in the phenomena happening at the rim of the layer distant from the surface. The intra- and intermolecular parameters, as well as the interactions with gold, are parameterized using the polymer consistent force field (PCFF) previously applied for the simulations of the assembled Azo-layers [34,63].
Each simulation box containing pure trans- or cis-Azo-BT or BT-Azo is subjected to preliminary equilibration stage using Smart algorithm, followed by initial NVT-MD run with the following parameters: T = 298 K (Nose thermostat, Q ratio is 0.01), 500 ps with a time step of 1 fs, PCFF partial charges (Ewald summation method with accuracy of 0.001 kcal mol and a buffer width of 0.5 Å), atom-based summation method for the calculation of the van der Waals interactions (9-6 Lennard-Jones potential with a cutoff distance of 12.5 Å). The NVT-MD production runs are performed under the same settings with trajectories of 10 ns. Later, these snapshots are used for the analysis of the structural properties of the layers, like the inclination angles of the molecules, the thickness, etc. Moreover, from MD data, the interacting dimers are taken for calculation of the averaged values of transfer integrals, as described above.
As an auxiliary property characterizing changes in the wettability of the monolayer upon light stimulus, the solvation free energies () are evaluated for the molecules dissolved in dichloromethane or water. Here, trans- or cis-isomer is placed in a cubic simulation box with 1500 CHCl or 1700 HO molecules equilibrated in NPT-MD (P = 1 atm, Berendsen barostat) for 500 ps until the solution density reaches the value close to the experimental one: 1.071 g cm (dichloromethane) and 1.018 g cm (water); pure solvents at 25 C have densities of 1.316 g cm and 0.997 g cm, respectively [64]. The prepared solutions are calculated later using thermodynamic integration technique (NPT-MD). Other settings for this calculation are used as described elsewhere [30,31,65]. The averaging is calculated for three independent calculations for each modeled system.
The obtained data are compared with experimentally derived layer thicknesses [25,32] and inclination angles [18] and measured [13,25] and calculated [25] optical absorption spectra and wetting behavior of the layers [14].
3. Results
3.1. Properties of Isolated Molecules
As mentioned above, we study the intermolecular interactions of the two azobenzene switches by combining in their structure bithiophene with a short alkanethiol tail (Figure 1). The latter one is the part of the molecule, which is chemisorbed via the thiol group to the bottom electrode in experiments with BT-Azo [13,14,25,48,49,50]. The structural properties of the isomers of Azo-BT and BT-Azo have been described in our previous study [32]. In that publication, we have considered trans- and cis-Azo-BT and BT-Azo states as isolated molecules and in contact with the top golden tip modeled as Au cluster. The most relevant data from Ref. [32] for this present paper are
- The values of the dipole moments (1.34 and 3.22 D for trans- and cis-BT-Azo; 1.67 and 3.52 D for trans- and cis-Azo-BT);
- The differences in the length of trans- and cis-isomers h are 6.05 Å and 14.69 Å for BT-Azo and Azo-BT, respectively; the experimental value of h = 5.1 Å for the layer of BT-Azo [25];
- Even though the molecular volume reduces upon the trans-cis isomerization for both MS, intermolecular sterical clashes may arise for cis-populated layers, especially for Azo-BT MS.
3.1.1. Optical Properties
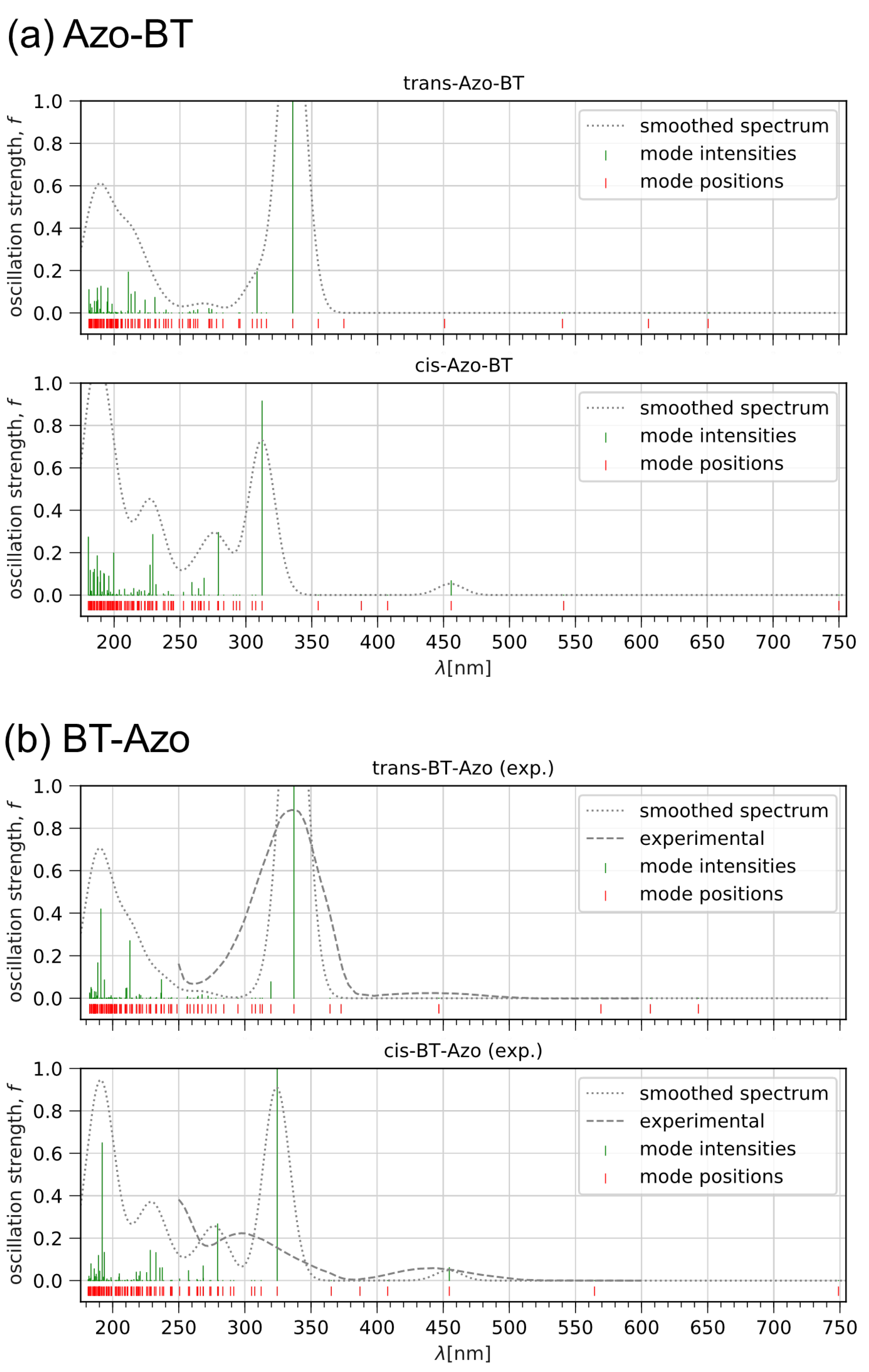
First, let us consider the optical properties predicted for the isolated molecules immersed in implicit dichloromethane. The time-dependent DFT calculated spectra are compared in Figure 2. In the experiment from Ref. [13], it was reported that upon the irradiation of the BT-Azo solution at 360 nm, the band at 342 nm () progressively decreases and broadens, while the absorbance of the n– transition in the 400–450 nm region increases, suggesting the trans-cis isomerization of the molecular switch (MS). The reduction in absorbance close to 340–350 nm during the transition from the trans- to the cis-states was also observed for the chemisorbed BT-Azo on Au nanoparticles in dimethylformamide [25]. The calculated spectrum reported by Viero et al. [25] shows a major active transition at = 353 nm for the trans-isomer, while the cis-state is characterized by three absorption bands located at 300, 337, and 520 nm.

Figure 2.
Absorption spectra (dotted lines) of trans- and cis-Azo-BT (a) and trans- and cis-BT-Azo (b), as received in TD-DFT calculations. The dashed lines on panel (b) are the experimental spectra from Ref. [13] with = 341 nm () and 435 nm ().
Our results predict the position of = 342 nm for the trans-isomers of both MS (Figure 2a,b, top panels), which quantitatively coincides with the experimentally found value. The curves of the spectra are not sensitive to the positions of the Azo and BT blocks in the molecule. In contrast, the data for the cis-isomers are slightly different for Azo-BT and BT-Azo: the band is blue-shifted for Azo-BT at 310 nm (325 nm for BT-Azo) and is characterized by a smaller value of the oscillator strength f. At the same time, the absorbance of the n– transition is seen at 455 nm for both molecules. The latter one for BT-Azo is closer to the experimental value mentioned above compared to the predictions of Viero et al. [25]. The bands around 310–325 nm include excitations coming from the bithiophene fragment linked to the azobenzene moiety via the sulfur bridge, and they are classified as transitions, which is in agreement with experimental findings [13]. Since the bithiophene fragment in Azo-BT and BT-Azo has different neighboring connections (one –S– and two –S– bridges, respectively) and participation in the conjugation due to lone electron pairs, this explains the observed shifts in the absorption spectra.
3.1.2. Reorganization Energies
Table 1 collects the values of the reorganization energies in eV for the hopping of holes () and electrons () calculated for the isolated molecules in implicit CHCl, as described in the previous section.

Table 1.
The reorganization energies for all the considered molecules.
The larger values of the reorganization energy suggest more substantial conformational changes in the molecule during the hopping process. In the case of the experimentally known BT-Azo molecule, the reorganization energy required for hole transport is up to three times higher compared to electron hopping. For both isomers of the designed Azo-BT molecule, is two times smaller than that of BT-Azo. However, there is no clear dependence on the isomeric state for this molecule. Notably, the restructuring of cis-Azo-BT during electron uptake is less energy favorable. With the exception of cis-Azo-BT, electron transport is preferred in the remaining cases. In the following sections, we will explore how the reorganization energy, together with the hopping distance, determines the charge transfer rate and charge carrier mobility.
3.1.3. Gibbs Free Energy of Solvation and Its Changes upon Light Stimulus
The next property of the studied molecules is the free energy of solvation, modeled for two solvents: dichloromethane (CHCl), which is thermodynamically good and in which BT-Azo has been synthesized [13], and water, which is thermodynamically poor and was used in the wetting experiments [13,14]. Table 2 presents the values of with standard deviations and the mean values of the ideal, van der Waals, and electrostatic terms.
Table 2.
The Gibbs free energy of solvation in dichloromethane (a) and water (b) and its averaged terms: ideal, van der Waals and electrostatic. The standard deviation is given for the Gibbs free energy of solvation. All values are in kcal mol.
In the experiments, it was observed that under UV stimulus for 90 min (with an intensity of 70–250 W cm) [13,14], the water contact angle decreases from 98 ± 2 (in the trans state) to 93 ± 2 (in the cis state). The process of partially improved wetting is fully reversible, as shown in those experiments, and the contact angle dynamically changes upon blue (trans-rich layer) to UV (cis-rich layer) illumination. In the experiments conducted by Lenfant and co-authors [13,14,50], this behavior was explained by the difference in the static dipole moment of the trans- and cis-isomers of BT-Azo. In our recent publication, we confirmed that the cis isomer is a more polar molecule, characterized by a dipole moment of 3.22 D (while the trans-state value is 1.34 D) [32].
The values presented in Table 2 complement the aforementioned explanation of the wetting experiments and, at the same time, appear more reliable as they are statistically averaged. These values were obtained via classical simulations at room temperature in systems with explicit solvent molecules, ensuring meaningful intermolecular interactions and preferred molecular solvation. It is worth noting that the partial charges for the isomers were determined from electrostatic potentials, as described elsewhere [30,65].
The solvation calculations in dichloromethane serve as a validation of the model, as all the values are negative, indicating the dissolution of the molecules in this solvent. The solvation energies are independent of the isomerization state and the position of BT and Azo in the molecular structure.
In contrast, the hydration process shows positive for both isomers of the BT-Azo molecule, but the value is two times smaller for the cis-isomer compared to the trans-counterpart. This observation rationalizes the experimental findings [13,14,50]. For the Azo-BT molecule, more significant changes in hydration are predicted. The wetting of the layer under the UV light may become complete, as 0. Therefore, dewetting/wetting switching could be observed under blue/UV illumination. Several factors contribute to this phenomenon, such as the larger dipole moment of the molecule, improved accessibility of the molecular groups for water, lesser conformational rigidity of the backbone, and, consequently, the coordination of water near hydrophilic sites of the molecule.
3.2. Properties of Chemisorbed Monolayers
3.2.1. Structural Properties
Now, let us switch from considering the isolated molecules in implicit or explicit solvents to the properties of the chemisorbed layers. The latter ones are visualized in Figure 3. For the chemisorbed layers, we chose a surface concentration of 2.7 molecules per nm, which is less dense compared to the anchoring on the facets of the gold nanoparticles in experiments [25]. We reduced the surface concentration in our model as we are considering flat surfaces, not 3D surfaces, where more molecules can be chemisorbed due to geometrical considerations.

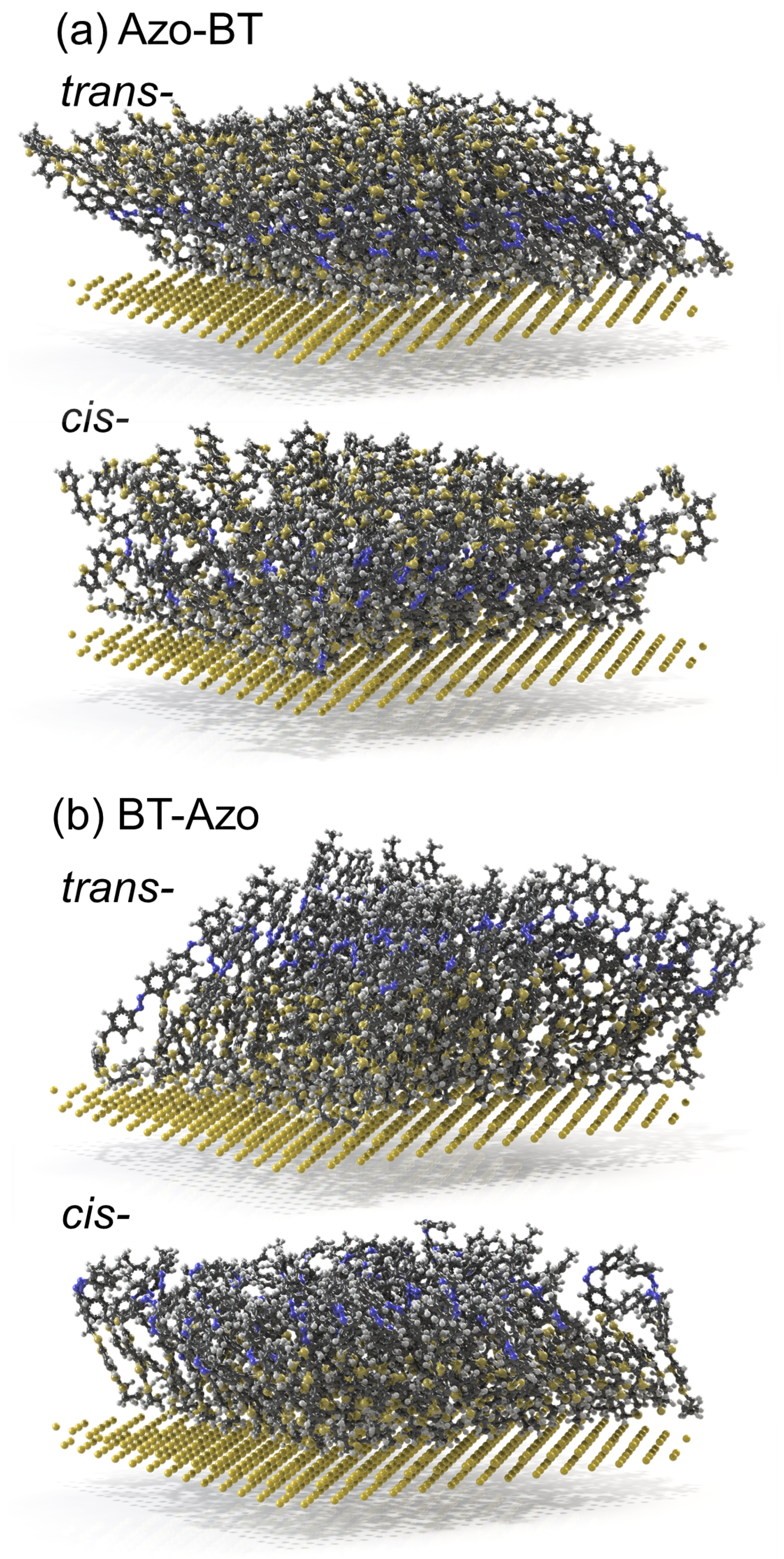
Figure 3.
Chemisorbed monolayers of trans- and cis-Azo-BT (a) and trans- and cis-BT-Azo (b), as received in MD simulations. The gyration radius of the anchored molecules is 3.37 ± 0.20 Å (trans-Azo-BT), 2.38 ± 0.43 Å (cis-Azo-BT), 3.72 ± 0.33 Å (trans-BT-Azo), and 2.84 ± 0.40 Å (cis-BT-Azo).
The snapshots of the systems depict that the trans-layers are rather ordered and densely packed. We note here that the all-trans layer serves as the initial state of the simulations and experiments before any light stimulus is applied [14]. However, based on the isomerization kinetics revealed in the experiments [14], the maximum possible population of the cis isomers under the conditions mentioned by the authors is slightly above 60%. This indicates that the isomerization reaction is quite effective for such densely packed layers, although it does not lead to a complete trans-to-cis conversion. Therefore, the modeled all-cis-layers are merely hypothetical systems. Nonetheless, this approximation does not diminish the value of theoretical investigation; it represents the first logical step towards studying mixed layers, such as 40% trans–60% cis, in future research. Moreover, one effect that may occur on larger scales remains unclear, the so-called cooperativity of isomerization, which could lead to cis-rich or trans-rich domains and their cooperative switching under light.
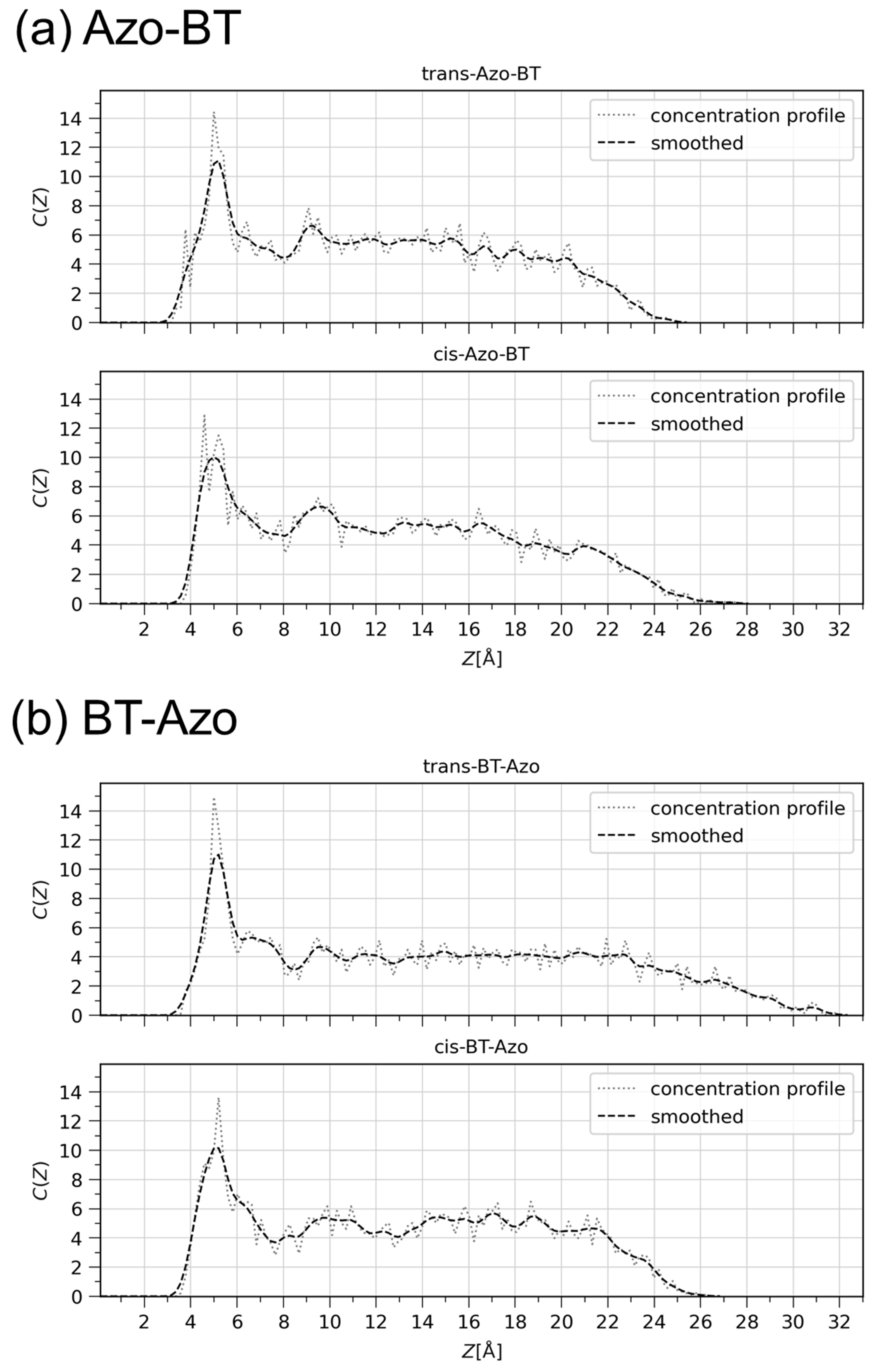
Figure 3 illustrates that the structure of the layers is preserved for all four modeled systems, thanks to the chemisorption of the molecules on gold. The trans-layers appear to be more ordered due to their stretched and planar geometry. Among the cis-layers, the BT-Azo one also exhibits a higher degree of structure due to the positioning of the kink in the molecule away from the anchoring surface. On the contrary, the cis-Azo-BT is a more disorganized system, as the cis-azo bond of each molecule is close to the gold surface, and therefore, no order is expected from this sample. The concentration profiles shown in Figure 4 are consistent with the ellipsometry results, which indicate a shrinkage of the layer height of BT-Azo by approximately 5 ± 1 Å, in agreement with the DFT calculations [14,32]. Such a clear height alteration is explained via the intrinsic ordering of the molecules in the trans- and cis-states. In the literature, there are also examples showing that for relatively dense anchored systems, the all-cis-layers also demonstrate some organization [36].

Figure 4.
Concentration profiles of trans- and cis-Azo-BT (a) and trans- and cis-BT-Azo (b). The concentration profile C(Z) calculates the concentration of particles in a given layer away from the gold surface located at Z = 0. The reported values of C(Z) are relative to a random distribution and are, therefore, unit-less. The values range from 0 (no atoms in the layer), to a maximum value corresponding to the total number of bins if all atoms reside in the same layer. The sum over all layers or stripes is equal to the total number of bins.
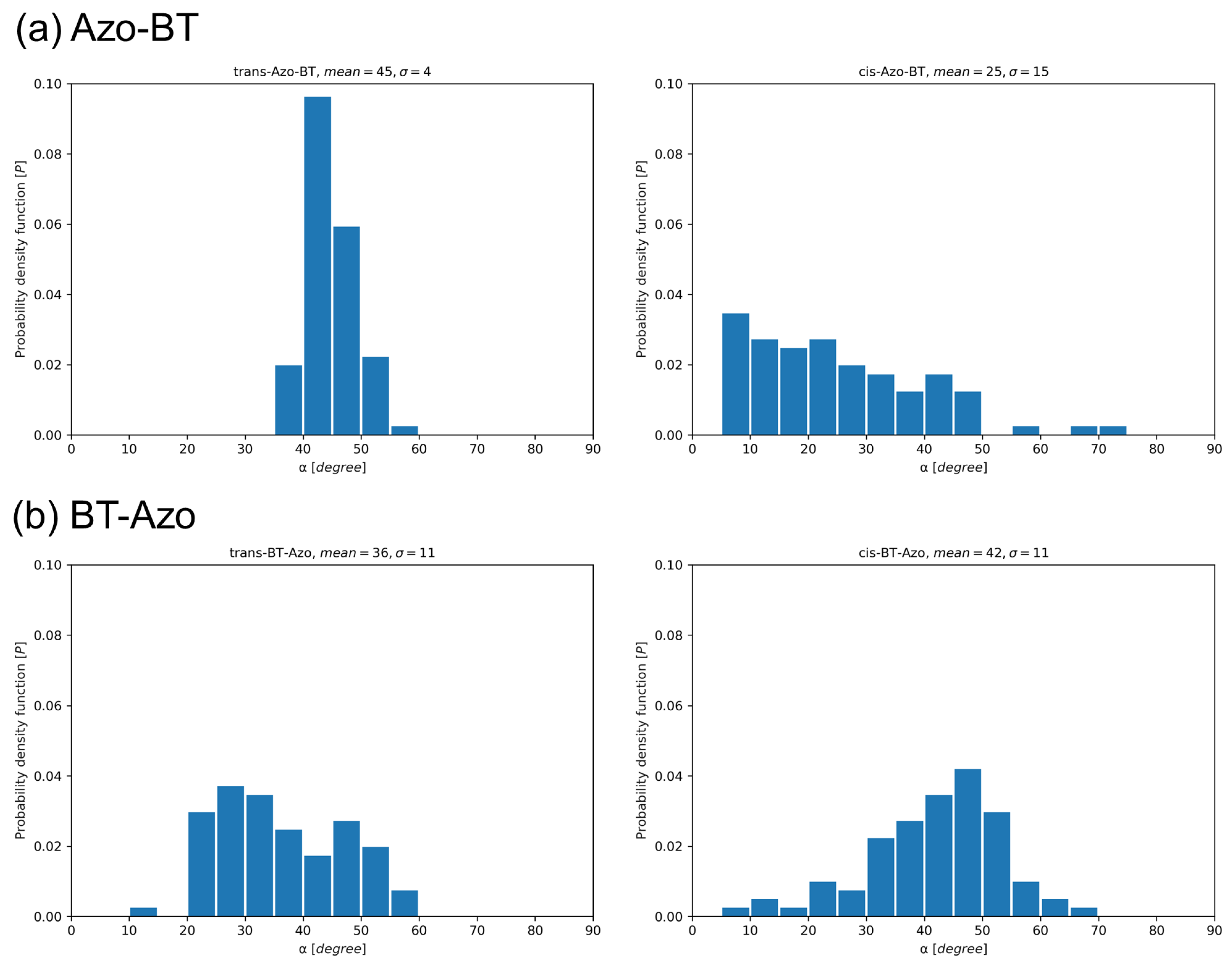
On the other hand, the designed layers of Azo-BT do not exhibit noticeable switching in thickness, as deduced from the comparison of concentration profiles. Firstly, this is caused by the tilting of the molecules in the trans-layer (Figure 5a), which is relatively narrow and centered at an angle of 45 to the surface. Secondly, the cis-Azo-BT layer is the least ordered system, with molecules having the potential to adopt almost any orientation relative to the surface. In fact, the range of tilting angles spans from 7 to 75 degrees.

Figure 5.
Probability density function P showing populations of angles between a normal to the gold surface and a vector connecting thiol sulfur and a middle of the thiophene–thiophene bond in Azo-BT (a) or a middle of the N=N double bond in BT-Azo (b).
The experimentally known molecule BT-Azo also demonstrates a greater extent of tilting to the surface in the trans-state compared to the cis-isomer. The most populated angle is 30 for the trans-isomer and approximately 45 for the cis-isomer. However, due to the differences in the length of the cis-isomer, the resulting layer height is smaller for the layer under UV stimulus.
3.2.2. Photo-Programmable Charge Transfer
Table 3 collects the values of the transfer integrals, the defined com-com distances for dimers, the calculated charge transfer rates, and charge carrier mobilities.
Table 3.
The transfer integrals V, the com–com distances d, as obtained from the analysis of sixty dimers built by the nearest-neighboring molecules taken from chemisorbed layers. The charge transfer rate and the carrier mobility are calculated using Marcus and Einstein–Smoluchowski equations for mean values V and d to demonstrate the order of magnitude.
The transfer integral for the hopping of electrons or holes has similar values for all the studied systems, regardless of their isomerization state or the position of the azobenzene fragment in the backbone. The separations between the center of masses in the considered dimers are also very close to each other, differing by at most 1 angstrom. In this regard, the decisive factor affecting the charge transfer rate is the reorganization energy. Molecules with smaller magnitudes of reorganization energy will facilitate a faster hopping of charge carriers. For example, electron transfer is favorable for the BT-Azo molecule, whereas the hopping of holes is minimal. Upon UV stimulus, the electron transport decreases by a factor of 4. Interestingly, electron hopping for Azo-BT decreases by 52 times under UV light and becomes almost negligible, whereas hole transfer is promoted via the trans-to-cis isomerization by a factor of 2. In absolute terms, the electron mobility in the layers of BT-Azo is rather high. In our opinion, this presents an interesting addition to the properties of the molecular switch BT-Azo, already known from the experiment, which is located between two electrodes.
4. Discussion and Conclusions
In the published papers [13,14,25,48,49,50], the authors have suggested several intriguing avenues for future research, including purely theoretical approaches that may uncover phenomena and issues not yet explored in their previous works. For instance, exploring the electronic coupling between the neighboring molecules and charge transfer within the molecular junction layer are intriguing tasks for computer modeling. In this paper, we addressed these questions pertinent to the rational design of azobenzene–bithiophene molecular switch.
Our primary idea was to investigate how changing the positions of Azo and BT blocks in the molecule impacts the properties of the isolated molecules, as well as the molecular layers attached to the gold surface. Systematically exploring different configurations, we aimed to gain insights into the structural and electronic characteristics of these molecular switches. Additionally, studying the influence of these changes on the charge transfer and solvation energies would allow us to assess the suitability of various configurations for molecular electronics and optoelectronic devices.
Regardless of the block positions, the studied molecules exhibit similar absorption spectra for the trans-isomers, while the cis-state optical spectra show slight shifts. Interestingly, the calculated hydration free energies provide an explanation for the observed dewetting/partial wetting transition in the BT-Azo layers under UV stimulus, as observed in experiments. In the case of the designed Azo-BT layers, the values change sign, indicating more drastic changes in wettability, shifting from dewetting to wetting upon exposure to UV light. These findings suggest that the molecular design and arrangement play crucial roles in dictating the wetting behavior of the layers, making them potential candidates for future applications in surface engineering.
The electron coupling V within the layers is found to be insensitive to the isomerization state or the position of the azobenzene and bithiophene blocks. As a result, the transfer rates and charge carrier mobilities are primarily determined using the reorganization energies and, to a lesser extent, the hopping distance. In our approach, the hopping distance is approximated by the mean distance between the centers of masses in the interacting dimers. Our predictions indicate that the BT-Azo trans-layer exhibits high electron mobility, which can be reduced by 4 times under UV illumination. On the other hand, if the layer consists of Azo-BT molecular switches, the electron mobility can be significantly reduced or even completely turned off. These results highlight the tunability and sensitivity of the electron mobility in these molecular layers, depending on their isomeric states and the specific arrangement of the azobenzene and bithiophene blocks. Such findings are of great interest for potential applications in molecular electronics and provide valuable insights into the design and optimization of molecular switches for various electronic devices.
Author Contributions
Conceptualization, O.G.; methodology, V.S. and A.S.P.; formal analysis, V.S. and M.H.; investigation, V.S., M.H., and A.S.P.; data curation, V.S.; writing—original draft preparation, O.G.; writing—review and editing, all authors; visualization, V.S.; supervision, O.G.; project administration, O.G.; funding acquisition, O.G. All authors have read and agreed to the published version of the manuscript.
Funding
The financial support from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, project GU1510/5-1) is highly appreciated.
Data Availability Statement
On inquiry, the data presented in this study are available from the authors. The multilayer constructor can be found at https://bitbucket.org/gehrmann/multilayer_constructor (accessed on 30 August 2023). The multilayer constructor is based on a fast computational method, provides a possibility for the building of mono- and multilayers for pure ore mixed substances using only coordinate files for the entities having optimized geometries. The following limitations of the protocol are that additional equilibration steps are needed, molecules do not rotate or inclined, and this procedure does not reproduce real physical processes during adsorption and the thin film growth.
Acknowledgments
We would like to acknowledge the high-performance computing support from the Center for Information Services and High Performance Computing (ZIH) provided by Technische Universität Dresden.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| Azo | Azobenzene |
| BT | Bithiophene |
| MS | Molecular switch |
| MJ | Molecular junction |
| DFT | Density functional theory |
| MD | Molecular dynamics simulation |
References
- Fuentes, N.; Martin-Lasanta, A.; de Cienfuegos, L.Á.; Ribagorda, M.; Parra, A.; Cuerva, J.M. Organic-based molecular switches for molecular electronics. Nanoscale 2011, 3, 4003–4014. [Google Scholar] [CrossRef]
- Klajn, R. Immobilized azobenzenes for the construction of photoresponsive materials. Pure Appl. Chem. 2010, 82, 2247–2279. [Google Scholar] [CrossRef]
- Delorme, N.; Bardeau, J.-F.; Bulou, A.; Poncin-Epaillard, F. Azobenzene-containing monolayer with photoswitchable wettability. Langmuir 2005, 21, 12278–12282. [Google Scholar] [CrossRef]
- Takahashi, I.; Honda, Y.; Hirota, S. Regulating copper-binding affinity with photoisomerizable azobenzene ligand by construction of a self-assembled monolayer. Angew. Chem. Int. Ed. 2009, 48, 6065–6068. [Google Scholar] [CrossRef] [PubMed]
- Callari, F.L.; Petralia, S.; Conoci, S.; Sortino, S. Light-triggered DNA release by dynamic monolayer films. New J. Chem. 2008, 32, 1899–1903. [Google Scholar] [CrossRef]
- Kano, K.; Tanaka, Y.; Ogawa, T.; Shimomura, M.; Okahata, Y.; Kunitake, T. Photoresponsive memnranes. Regulation of membrane properties by photoreversible cis-trans isomerization of azobenzenes. Chem. Lett. 1980, 9, 421–424. [Google Scholar] [CrossRef]
- Bisby, R.H.; Mead, C.; Morgan, C.G. Wavelength-programmed solute release from photoresponsive liposomes. Biochem. Biophys. Res. Commun. 2000, 276, 169–173. [Google Scholar] [CrossRef]
- Morgan, C.G.; Thomas, E.W.; Sandhu, S.S.; Yianni, Y.P.; Mitchell, A.C. Light-induced fusion of liposomes with release of trapped marker dyu is sensitised by photochromic phospholipid. Biochim. Biophys. Acta (BBA) Biomembr. 1987, 903, 504–509. [Google Scholar] [CrossRef]
- Huang, X.; Li, T. Recent progress in the development of molecular-scale electronics based on photoswitchable molecules. J. Mater. Chem. C 2020, 8, 821–848. [Google Scholar] [CrossRef]
- Liu, Y.; Qiu, X.; Soni, S.; Chiechi, R.C. Charge transport through molecular ensembles: Recent progress in molecular electronics. Chem. Phys. Rev. 2021, 2, 021303. [Google Scholar] [CrossRef]
- Tang, C.; Shiri, M.; Zhang, H.; Ayinla, R.T.; Wang, K. Light-driven charge transport and optical sensing in molecular junctions. Nanomaterials 2022, 12, 698. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.H.; Hassan, S.Z.; So, C.; Kang, M.; Chung, D.S. Molecular-switch-embedded Solution-processed Semiconductors. Adv. Mater. 2023, 35, 2203401. [Google Scholar] [CrossRef] [PubMed]
- Karpe, S.; Oçafrain, M.; Smaali, K.; Lenfant, S.; Vuillaume, D.; Blanchard, P.; Roncali, J. Oligothiophene-derivatized azobenzene as immobilized photoswitchable conjugated systems. Chem. Commun. 2010, 46, 3657–3659. [Google Scholar] [CrossRef] [PubMed]
- Smaali, K.; Lenfant, S.; Karpe, S.; Oçafrain, M.; Blanchard, P.; Deresmes, D.; Godey, S.; Rochefort, A.; Roncali, J.; Vuillaume, D. High ON-OFF conductance switching ratio in optically-driven self-assembled conjugated molecular systems. ACS Nano 2010, 4, 2411–2421. [Google Scholar] [CrossRef]
- Ferri, V.; Elbing, M.; Pace, G.; Dickey, M.D.; Zharnikov, M.; Samorì, P.; Mayor, M.; Rampi, M.A. Light-powered electrical switch based on cargo-lifting azobenzene monolayers. Angew. Chem. Int. Ed. 2008, 18, 3455–3457. [Google Scholar] [CrossRef]
- Schuster, S.; Füser, M.; Asyuda, A.; Cyganik, P.; Terfort, A.; Zharnikov, M. Photoisomerization of azobenzene-substituted alkanethiolates on Au (111) substrates in the context of work function variation: The effect of structure and packing density. Phys. Chem. Chem. Phys. 2019, 21, 9098–9105. [Google Scholar] [CrossRef]
- Mativetsky, J.M.; Pace, G.; Elbing, M.; Rampi, M.A.; Mayor, M.; Samori, P. Azobenzenes as light-controlled molecular electronic switches in nanoscale metal- molecule- metal junctions. J. Am. Chem. Soc. 2008, 130, 9192–9193. [Google Scholar] [CrossRef]
- Thomas, L.; Arbouch, I.; Guérin, D.; Wallart, X.; Van Dyck, C.; Mélin, T.; Cornil, J.; Vuillaume, D.; Lenfant, S. Conductance switching of azobenzene-based self-assembled monolayers on cobalt probed by UHV conductive-AFM. Nanoscale 2021, 13, 6977–6990. [Google Scholar] [CrossRef]
- Crivillers, N.; Liscio, A.; Di Stasio, F.; Van Dyck, C.; Osella, S.; Cornil, D.; Mian, S.; Lazzerini, G.M.; Fenwick, O.; Orgiu, E.; et al. Photoinduced work function changes by isomerization of a densely packed azobenzene-based SAM on Au: A joint experimental and theoretical study. Phys. Chem. Chem. Phys. 2011, 13, 14302–14310. [Google Scholar] [CrossRef]
- Masillamani, A.M.; Osella, S.; Liscio, A.; Fenwick, O.; Reinders, F.; Mayor, M.; Palermo, V.; Cornil, J.; Samorì, P. Light-induced reversible modification of the work function of a new perfluorinated biphenyl azobenzene chemisorbed on Au (111). Nanoscale 2014, 6, 8969–8977. [Google Scholar] [CrossRef]
- Ah, Q.; Lloyd, F.N.; Akiyama, H.; Nagahiro, T.; Tamada, K.; Wee, A.T.S. Reversible work function changes induced by photoisomerization of asymmetric azobenzene dithiol self-assembled monolayers on gold. Appl. Phys. Lett. 2008, 93, 083109. [Google Scholar]
- Yamamoto, T.; Natsui, K.; Einaga, Y. Photo-Modulation of Superconducting and Magnetic Properties. In Photon-Working Switches; Springer: Berlin/Heidelberg, Germany, 2017; pp. 285–299. [Google Scholar]
- Suda, M.; Kameyama, N.; Ikegami, A.; Suzuki, M.; Kawamura, N.; Einaga, Y. Size-reduction induced ferromagnetism and photo-magnetic effects in azobenzene-thiol-passivated gold nanoparticles. Polyhedron 2009, 28, 1868–1874. [Google Scholar] [CrossRef]
- Karthäuser, S.; Peter, S.; Simon, U. Integration of individual functionalized gold nanoparticles into nanoelectrode configurations: Recent advances. Eur. J. Inorg. Chem. 2020, 40, 3798–3810. [Google Scholar] [CrossRef]
- Viero, Y.; Copie, G.; Guerin, D.; Krzeminski, C.; Vuillaume, D.; Lenfant, S.; Cleri, F. High conductance ratio in molecular optical switching of functionalized nanoparticle self-assembled nanodevices. J. Phys. Chem. C 2015, 119, 21173–21183. [Google Scholar] [CrossRef]
- Ahonen, P.; Laaksonen, T.; Schiffrin, D.J.; Kontturi, K. Photoswitching electron transport properties of an azobenzene containing thiol-SAM. Phys. Chem. Chem. Phys. 2007, 9, 4898–4901. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.; Yang, M.; Shin, N.; Hong, S. Mapping reversible photoswitching of molecular resistance fluctuations during the conformational transformation of azobenzene-terminated molecular switches. Nanotechnology 2018, 29, 365704. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.S.; Ye, T.; Takami, T.; Yu, B.-C.; Flatt, A.K.; Tour, J.M.; Weiss, P.S. Reversible photo-switching of single azobenzene molecules in controlled nanoscale environments. Nano Lett. 2008, 8, 1644–1648. [Google Scholar] [CrossRef] [PubMed]
- Del Valle, M.; Gutiérrez, R.; Tejedor, C.; Cuniberti, G. Tuning the conductance of a molecular switch. Nat. Nanotechnol. 2007, 2, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.; Saphiannikova, M.; Santer, S.; Guskova, O. Photoisomers of azobenzene star with a flat core: Theoretical insights into multiple states from DFT and MD perspective. J. Phys. Chem. B 2017, 121, 8854–8867. [Google Scholar] [CrossRef]
- Montagna, M.; Guskova, O. Photosensitive cationic azobenzene surfactants: Thermodynamics of hydration and the complex formation with poly (methacrylic acid). Langmuir 2018, 34, 311–321. [Google Scholar] [CrossRef]
- Savchenko, V.; Guskova, O. Molecular switch based on bithiophene-azobenzene: How to control conductance through the monolayer using light. Her. Tver State Univ. Ser. Chem. 2021, 3, 7–20. [Google Scholar]
- Kaneta, M.; Honda, T.; Onda, K.; Han, M. Repeated photoswitching performance of azobenzenes adsorbed on gold surfaces: A balance between space, intermolecular interactions, and phase separation. New J. Chem. 2017, 41, 1827–1833. [Google Scholar] [CrossRef]
- Tian, Z.; Wen, J.; Ma, J. Dynamic simulations of stimuli-responsive switching of azobenzene derivatives in self-assembled monolayers: Reactive rotation potential and switching functions. Mol. Simul. 2015, 41, 28–42. [Google Scholar] [CrossRef]
- Cantatore, V.; Granucci, G.; Rousseau, G.; Padula, G.; Persico, M. Photoisomerization of self-assembled monolayers of azobiphenyls: Simulations highlight the role of packing and defects. J. Phys. Chem. Lett. 2016, 7, 4027–4031. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Tian, Z.; Ma, J. Light-and electric-field-induced switching of thiolated azobenzene self-assembled monolayer. J. Phys. Chem. C 2013, 117, 19934–19944. [Google Scholar] [CrossRef]
- Liu, C.; Zheng, D.; Hu, W.; Zhu, Q.; Tian, Z.; Zhao, J.; Zhu, Y.; Ma, J. Tuning the collective switching behavior of azobenzene/Au hybrid materials: Flexible versus rigid azobenzene backbones and Au (111) surfaces versus curved Au nanoparticles. Nanoscale 2017, 9, 16700–16710. [Google Scholar] [CrossRef]
- Fast, E.; Schlimm, A.; Lautenschläger, I.; Clausen, K.U.; Strunskus, T.; Spormann, C.; Lindhorst, T.K.; Tuczek, F. Improving the switching capacity of glyco-self-assembled monolayers on Au (111). Chem. Eur. J. 2020, 26, 485–501. [Google Scholar] [CrossRef]
- Rashid, M.A.M.; Hayati, D.; Kwak, K.; Hong, J. Theoretical investigation of azobenzene-based photochromic dyes for dye-sensitized solar cells. Nanomaterials 2020, 10, 914. [Google Scholar] [CrossRef]
- Crivillers, N.; Orgiu, E.; Reinders, F.; Mayor, M.; Samorì, P. Optical modulation of the charge injection in an organic field-effect transistor based on photochromic self-assembled-monolayer-functionalized electrodes. Adv. Mat. 2011, 23, 1447–1452. [Google Scholar] [CrossRef]
- Tirosh, E.; Benassi, E.; Pipolo, S.; Mayor, M.; Valášek, M.; Frydman, V.; Corni, S.; Cohen, S.R. Direct monitoring of opto-mechanical switching of self-assembled monolayer films containing the azobenzene group. Beilstein J. Nanotechnol. 2011, 2, 834–844. [Google Scholar] [CrossRef]
- Yu, S.H.; Hassan, S.Z.; Nam, G.-H.; An, S.; Kang, B.; Chung, D.S. Consideration of azobenzene-based self-assembled monolayer deposition conditions for maximizing optoelectronic switching performances. Chem. Mater. 2021, 33, 5991–6002. [Google Scholar] [CrossRef]
- Crivillers, N.; Osella, S.; Van Dyck, C.; Lazzerini, G.M.; Cornil, D.; Liscio, A.; Di Stasio, F.; Mian, S.; Fenwick, O.; Reinders, F.; et al. Large work function shift of gold induced by a novel perfluorinated azobenzene-based self-assembled monolayer. Adv. Mater. 2013, 25, 432–436. [Google Scholar] [CrossRef] [PubMed]
- Osella, S.; Samori, P.; Cornil, J. Photoswitching azobenzene derivatives in single molecule junctions: A theoretical insight into the I/V characteristics. J. Phys. Chem. C 2014, 118, 18721–18729. [Google Scholar] [CrossRef]
- Van Dyck, C.; Bergren, A.J.; Mukundan, V.; Fereiro, J.A.; DiLabio, G.A. Extent of conjugation in diazonium-derived layers in molecular junction devices determined by experiment and modelling. Phys. Chem. Chem. Phys. 2019, 21, 16762–16770. [Google Scholar] [CrossRef]
- Rego, L.G.C.; Bortolini, G. Modulating the photoisomerization mechanism of semiconductor-bound azobenzene-functionalized compounds. J. Phys. Chem. C 2019, 123, 5692–5698. [Google Scholar] [CrossRef]
- Bang, G.S.; Lee, J.; Baek, H.Y.; Lee, H.; Jun, K.; Shin, S.R. Adsorption of azothiophene dye having an n-bridging bidentate tail group on gold. Langmuir 2009, 25, 10788–10793. [Google Scholar] [CrossRef] [PubMed]
- Viero, Y.; Guérin, D.; Vladyka, A.; Alibart, F.; Lenfant, S.; Calame, M.; Vuillaume, D. Light-stimulatable molecules/nanoparticles networks for switchable logical functions and reservoir computing. Adv. Func. Mater. 2018, 28, 1801506. [Google Scholar] [CrossRef]
- Stiévenard, D.; Guérin, D.; Lenfant, S.; Lévêque, G.; Nijhuis, C.A.; Vuillaume, D. Electrical detection of plasmon-induced isomerization in molecule–nanoparticle network devices. Nanoscale 2018, 10, 23122–23130. [Google Scholar] [CrossRef]
- Lenfant, S. Charge transport in dynamic molecular junctions. In Molecular Electronics: An Experimental and Theoretical Approach; CRC Press: Boca Raton, FL, USA, 2016; pp. 65–99. [Google Scholar]
- Guskova, O.A. On the inter-ring torsion potential of 2, 2’-bithiophene: A review of open problems and current proposals. In Quantum Systems in Physics, Chemistry, and Biology: Advances in Concepts and Applications; Springer: Berlin/Heidelberg, Germany, 2017; pp. 209–230. [Google Scholar]
- Mennucci, B.; Cances, E.; Tomasi, J. Evaluation of solvent effects in isotropic and anisotropic dielectrics and in ionic solutions with a unified integral equation method: Theoretical bases, computational implementation, and numerical applications. J. Phys. Chem. B 1997, 101, 10506–10517. [Google Scholar] [CrossRef]
- Malyar, I.V.; Titov, E.; Lomadze, N.; Saalfrank, P.; Santer, S. Photoswitching of azobenzene-containing self-assembled monolayers as a tool for control over silicon surface electronic properties. J. Chem. Phys. 2017, 146, 104703. [Google Scholar] [CrossRef]
- Pipolo, S.; Benassi, E.; Corni, S. Structural properties of azobenzene self-assembled monolayers by atomistic simulations. Langmuir 2017, 29, 10505–10512. [Google Scholar] [CrossRef] [PubMed]
- Malagoli, M.; Brédas, J.-L. Density functional theory study of the geometric structure and energetics of triphenylamine-based hole-transporting molecules. Chem. Phys. Lett. 2000, 327, 13–17. [Google Scholar] [CrossRef]
- Nelsen, S.F.; Blackstock, S.C.; Kim, Y. Estimation of inner shell Marcus terms for amino nitrogen compounds by molecular orbital calculations. J. Am. Chem. Soc. 1987, 109, 677–682. [Google Scholar] [CrossRef]
- Koh, S.E.; Risko, C.; da Silva Filho, D.A.; Kwon, O.; Facchetti, A.; Brédas, J.-L.; Marks, T.J.; Ratner, M.A. Modeling electron and hole transport in fluoroarene-oligothiopene semiconductors: Investigation of geometric and electronic structure properties. Adv. Funct. Mater. 2008, 18, 332–340. [Google Scholar] [CrossRef]
- Savchenko, V.A.; Guskova, O.A. The effect of alkyl substitutes on the characteristics of charge transfer in stacks of D-π-A-π-D molecules. Rev. Adv. Chem. 2022, 12, 214–221. [Google Scholar] [CrossRef]
- Raychev, D.; Méndez López, R.D.; Kiriy, A.; Seifert, G.; Sommer, J.-U.; Guskova, O. Copolymers of diketopyrrolopyrrole and benzothiadiazole: Design and function from simulations with experimental support. Macromolecules 2019, 52, 904–914. [Google Scholar] [CrossRef]
- Makarova, M.V.; Semenov, S.G.; Guskova, O.A. Computational study of structure, electronic, and microscopic charge transport properties of small conjugated diketopyrrolopyrrole-thiophene molecules. Int. J. Quant. Chem. 2016, 116, 1459–1466. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision A.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- BIOVIA, Dassault Systèmes, Materials Studio 09; Dassault Systèmes: San Diego, CA, USA, 2019.
- Tian, Z.; Wen, J.; Ma, J. Reactive molecular dynamics simulations of switching processes of azobenzene-based monolayer on surface. J. Chem. Phys. 2013, 139, 014706. [Google Scholar] [CrossRef]
- Riddick, J.A.; Bunger, W.B. Organic Solvents: Physical Properties and Methods of Purification, 3rd ed.; Wiley-Interscience: New York, NY, USA, 1986; pp. 66–69. [Google Scholar]
- Savchenko, V.; Lomadze, N.; Santer, S.; Guskova, O. Spiropyran/Merocyanine Amphiphile in Various Solvents: A Joint Experimental–Theoretical Approach to Photophysical Properties and Self-Assembly. Int. J. Mol. Sci. 2022, 23, 11535. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).