
Development and Optimization of a SPME-GC-FID Method for Ethanol Detection

 , ,
, ,  ,
,  and
and
Abstract

1. Introduction
- Alternative Chromatographic Techniques: In addition to gas chromatography, liquid chromatography techniques, such as high-performance liquid chromatography (HPLC), could be explored. Each technique has its advantages, and the choice depends on factors such as sensitivity, selectivity, and the nature of the samples. On the other hand, liquid chromatography (LC) techniques, such as high-performance liquid chromatography (HPLC), may also be used for ethanol analysis. However, the choice between SPME-GC-FID and LC depends on the specific requirements of the analysis and the properties of the target analyte. Liquid chromatography generally excels in separating and analyzing a broader range of compounds, including those with higher molecular weights or polar characteristics. However, some studies reveal that for volatile compounds like ethanol, SPME-GC-FID often provides better sensitivity and faster analysis times [1,2,3,4];
- Spectroscopic Techniques: Techniques like NIR, NMR, and MS could be considered for the direct detection of ethanol. These methods might provide alternative approaches with different detection sensitivities and specificities; however, studies reveal that the direct injection of ethanol in aqueous matrices reveals lower sensitivity [8,9,10,11];
- Sensor Technologies: The use of sensors, such as electrochemical or optical sensors, could provide real-time detection and potentially offer advantages in terms of simplicity and rapidity. There are several available commercial sensors capable of transdermal monitoring of alcohol consumption using insensible sweat, such as the SCRAM™ and the WrisTAS™ biosensors. These sensors can provide information about days of abstinence, and even distinguish between heavy or sporadic alcohol ingestions. However, the reported devices have disadvantages, such as limited specificity and time delay after alcohol intake. Recent reports have demonstrated wearable alcohol biosensors capable of greater specificity and near real-time monitoring through amperometric detection in sweat [18].
- (i)
- Validation of a SPME injection gas chromatography with flame ionization detection method (GC-FID) using a capillary column for the accurate detection of ethanol, exploring different factors that can influence its sensitivity and reproducibility;
- (ii)
- Application of the method for the determination of ethanol in fluids with biomedical, clinical, and forensic importance, including water, phosphate-buffered saline (PBS), and artificial sweat;
- (iii)
- Establishment of a strategy that yields good peak resolution, demonstrating a linear correlation between concentration and peak areas for ethanol in all matrices. The method exhibits inter- and intra-day precisions below 15.5% and 6.5%, respectively, with detection limits below 1.3 mg/L across matrices. Notably, this approach allows for ethanol detection at lower concentrations using a lower temperature and time of extraction, distinguishing it from previous studies.
2. Materials and Methods
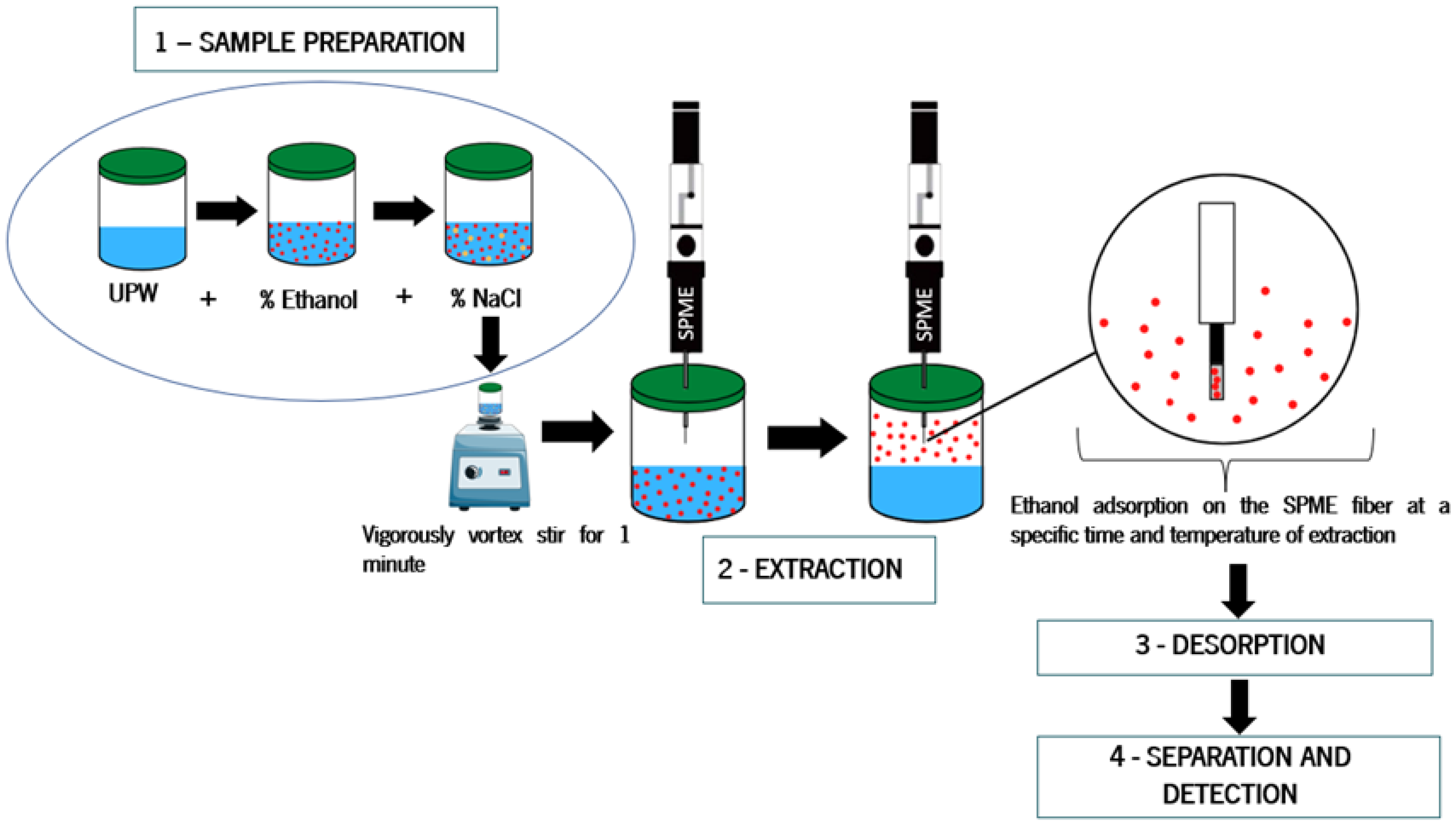
2.1. Samples Preparation and GC-FID Procedure
- Sample preparation: the samples were prepared as mentioned before;
- Extraction: the SPME fiber was exposed to the sample matrix headspace and the volatile compounds adsorbed into the fiber coating for 20 min at 30 °C;
- Desorption: after extraction, the fiber was inserted into the GC injection port, where the compounds were thermally desorbed from the fiber coating and transferred to the GC column;
- Separation and detection: the separated compounds were detected by a flame ionization detector (FID).
2.2. Artificial Sweat and PBS Sample Solutions
2.3. GC Conditions
2.4. Statistical Analysis
3. Results
3.1. SPME Fiber Selection

3.2. Salt Concentration

3.3. Extraction Time

3.4. Extraction Temperature

3.5. Sample Matrix

3.6. Results Validation
Linearity, Precision (CV), Limit of Detection (LOD), and Limit of Quantification (LOQ)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Lin, Z.; Wang, H.; Jones, A.W.; Wang, F.; Zhang, Y.; Rao, Y. Evaluation and review of ways to differentiate sources of ethanol in postmortem blood. Int. J. Leg. Med. 2020, 134, 2081–2093. [Google Scholar] [CrossRef] [PubMed]
- van den Broek, J.; Abegg, S.; Pratsinis, S.E.; Güntner, A.T. Highly selective detection of methanol over ethanol by a handheld gas sensor. Nat. Commun. 2019, 10, 4220. [Google Scholar] [CrossRef] [PubMed]
- Tiscione, N.B.; Alford, I.; Yeatman, D.T.; Shan, X. Ethanol Analysis by Headspace Gas Chromatography with Simultaneous Flame-Ionization and Mass Spectrometry Detection. J. Anal. Toxicol. 2011, 35, 501–511. [Google Scholar] [CrossRef] [PubMed]
- De Martinis, B.S.; Ruzzene, M.A.M.; Martin, C.C.S. Determination of ethanol in human blood and urine by automated headspace solid-phase microextraction and capillary gas chromatography. Anal. Chim. Acta 2004, 522, 163–168. [Google Scholar] [CrossRef]
- Wasfi, I.A.; Al-Awadhi, A.H.; Al-Hatali, Z.N.; Al-Rayami, F.J.; Al Katheeri, N.A. Rapid and sensitive static headspace gas chromatography–mass spectrometry method for the analysis of ethanol and abused inhalants in blood. J. Chromatogr. B 2003, 799, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Pragst, F.; Balikova, M.A. State of the art in hair analysis for detection of drug and alcohol abuse. Clin. Chim. Acta 2006, 370, 17–49. [Google Scholar] [CrossRef] [PubMed]
- Hameedat, F.; Hawamdeh, S.; Alnabulsi, S.; Zayed, A. High Performance Liquid Chromatography (HPLC) with Fluorescence Detection for Quantification of Steroids in Clinical, Pharmaceutical, and Environmental Samples: A Review. Molecules 2022, 27, 1807. [Google Scholar] [CrossRef] [PubMed]
- Spiehler, V.; Cooper, G. Drugs-of-Abuse Testing in Saliva or Oral Fluid. In Drug Testing in Alternate Biological Specimens; Springer: Berlin/Heidelberg, Germany, 2008; pp. 83–99. [Google Scholar] [CrossRef]
- Jessop, D.S.; Turner-Cobb, J.M. Measurement and meaning of salivary cortisol: A focus on health and disease in children. Stress Int. J. Biol. Stress 2008, 11, 1–14. [Google Scholar] [CrossRef]
- Fulgêncio, A.C.d.C.; Resende, G.A.P.; Teixeira, M.C.F.; Botelho, B.G.; Sena, M.M. Combining portable NIR spectroscopy and multivariate calibration for the determination of ethanol in fermented alcoholic beverages by a multi-product model. Talanta Open 2023, 7, 100180. [Google Scholar] [CrossRef]
- Naqvi, R.A.; Arsalan, M.; Batchuluun, G.; Yoon, H.S.; Park, K.R. Deep Learning-Based Gaze Detection System for Automobile Drivers Using a NIR Camera Sensor. Sensors 2018, 18, 456. [Google Scholar] [CrossRef]
- Fiorini, D.; Caprioli, G.; Sagratini, G.; Maggi, F.; Vittori, S.; Marcantoni, E.; Ballini, R. Quantitative Profiling of Volatile and Phenolic Substances in the Wine Vernaccia di Serrapetrona by Development of an HS-SPME-GC-FID/MS Method and HPLC-MS. Food Anal. Methods 2014, 7, 1651–1660. [Google Scholar] [CrossRef]
- Dziekońska-Kubczak, U.; Pielech-Przybylska, K.; Patelski, P.; Balcerek, M. Development of the Method for Determination of Volatile Sulfur Compounds (VSCs) in Fruit Brandy with the Use of HS–SPME/GC–MS. Molecules 2020, 25, 1232. [Google Scholar] [CrossRef] [PubMed]
- Charry-Parra, G.; DeJesus-Echevarria, M.; Perez, F.J. Beer Volatile Analysis: Optimization of HS/SPME Coupled to GC/MS/FID. J. Food Sci. 2011, 76, C205–C211. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Lu, X.; Tian, B.; Wang, C.; Shi, H.; Luo, C.; Li, X. A gas chromatography-flame ionization detection method for direct and rapid determination of small molecule volatile organic compounds in aqueous phase. 3 Biotech 2020, 10, 520. [Google Scholar] [CrossRef] [PubMed]
- Žnideršič, L.; Mlakar, A.; Prosen, H. Development of a SPME-GC-MS/MS method for the determination of some contaminants from food contact material in beverages. Food Chem. Toxicol. 2019, 134, 110829. [Google Scholar] [CrossRef] [PubMed]
- Kristoffersen, L.; Stormyhr, L.-E.; Smith-Kielland, A. Headspace gas chromatographic determination of ethanol: The use of factorial design to study effects of blood storage and headspace conditions on ethanol stability and acetaldehyde formation in whole blood and plasma. Forensic Sci. Int. 2006, 161, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Costa, N.G.; Antunes, J.C.; Paleo, A.J.; Rocha, A.M. A Review on Flexible Electrochemical Biosensors to Monitor Alcohol in Sweat. Biosensors 2022, 12, 252. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.P.; Silva, L.R.; Andrade, P.B.; Valentão, P.; Silva, B.M.; Pereira, J.A.; de Pinho, P.G. Determination of low molecular weight volatiles in Ficus carica using HS-SPME and GC/FID. Food Chem. 2010, 121, 1289–1295. [Google Scholar] [CrossRef]
- Poole, C.F. Ionization-based detectors for gas chromatography. J. Chromatogr. A 2015, 1421, 137–153. [Google Scholar] [CrossRef]
- Joseph, J.A.; Joseph, J.A.; Akkermans, S.; Akkermans, S.; Van Impe, J.F.M.; Van Impe, J.F.M. Processing Method for the Quantification of Methanol and Ethanol from Bioreactor Samples Using Gas Chromatography–Flame Ionization Detection. ACS Omega 2022, 7, 24121–24133. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chai, X.-S.; Deng, Y.; Zhan, H.; Fu, S. Rapid determination of ethanol in fermentation liquor by full evaporation headspace gas chromatography. J. Chromatogr. A 2009, 1216, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-L.; Choong, Y.-M.; Su, N.-W.; Lee, M.-S. A rapid method for determination of ethanol in alcoholic beverages using capillary gas chromatography. J. Food Drug Anal. 2003, 11, 3. [Google Scholar] [CrossRef]
- Savini, F.; Tartaglia, A.; Coccia, L.; Palestini, D.; D’Ovidio, C.; de Grazia, U.; Merone, G.M.; Bassotti, E.; Locatelli, M. Ethanol Determination in Post-Mortem Samples: Correlation between Blood and Vitreous Humor Concentration. Molecules 2020, 25, 2724. [Google Scholar] [CrossRef] [PubMed]
- Kilic, T.; Brunner, V.; Audoly, L.; Carrara, S. Smart e-Patch for drugs monitoring in schizophrenia. In Proceedings of the 2016 IEEE International Conference on Electronics, Circuits and Systems (ICECS), Monte Carlo, Monaco, 11–14 December 2016; pp. 57–60. [Google Scholar] [CrossRef]
- Mihretu, L.D.; Gebru, A.G.; Mekonnen, K.N.; Asgedom, A.G.; Desta, Y.H. Determination of ethanol in blood using headspace gas chromatography with flameionization detector (HS-GC-FID): Validation of a method. Cogent Chem. 2020, 6, 1760187. [Google Scholar] [CrossRef]
- Lo, M.-M.; Benfodda, Z.; Bénimélis, D.; Fontaine, J.-X.; Molinié, R.; Meffre, P. Development of a HS-SPME/GC-MS Method for the Extraction and Identification of the Volatile Compounds Emitted by Flowers of Tillandsia xiphioides. ACS Omega 2021, 6, 12691–12698. [Google Scholar] [CrossRef] [PubMed]
- Bariya, M.; Nyein, H.Y.Y.; Javey, A. Wearable sweat sensors. Nat. Electron. 2018, 1, 160–171. [Google Scholar] [CrossRef]
- Bruce, P.; Minkkinen, P.; Riekkola, M.-L. Practical method validation: Validation sufficient for an analysis method. Microchim. Acta 1998, 128, 93–106. [Google Scholar] [CrossRef]
- ICH. Validation of Analytical Procedures: Text and Methodology Q2(R1). 2014. Available online: www.somatek.com (accessed on 27 January 2022).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Concentration | ||
|---|---|---|---|
| Artificial Sweat | PBS | ||
| NaCl | Sodium Chloride | 20 g/L | 80 g/L |
| NH4Cl | Amonium Chloride | 17.5 g/L | - |
| CH3COOH | Acetic Acid | 5 g/L | - |
| C3H6O3 | Lactc Acid | 15 g/L | - |
| C6H8O6 | Ascorbic Acid | 0.0018 g/L | - |
| C6H12O6 | Glucose | 0.030 g/L | - |
| C5H9NO4 | Glutamic Acid | 0.054 g/L | - |
| CH4N2O | Urea | 0.006 g/L | - |
| KCL | Potassium Chloride | - | 2 g/L |
| NH2HPO4 | Disodium phosphate | - | 14.4 g/L |
| KH2PO4 | Monopotassium phosphate | - | 2.4 g/L |
| GC-Conditions | Equation | R2 | LOD (mg/L) | LOQ (mg/mL) |
|---|---|---|---|---|
| 30 °C_20min_30%NaCl (Matrix A) | y = 12520.6x + 6790.3 | 0.99875 | 0.22 | 0.68 |
| Matrix | Equation | R2 | LOD (mg/L) | LOQ (mg/L) | Reproducibility | |
|---|---|---|---|---|---|---|
| Intra Day n = 5 | Inter Day n = 5 | |||||
| A | y = 12,710.7x + 6534.4 | 0.99875 | 0.22 | 0.68 | 3.8% | 5.1% |
| B | y = 1999.8x + 1124.8 | 0.98800 | 0.36 | 0.96 | 6.4% | 14.5% |
| C | y = 2130.2x + 1581.9 | 0.99789 | 0.43 | 1.29 | 3.8% | 15.2% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costa, N.G.; Freitas, D.S.; Barros, A.; Silva, C.; Antunes, J.C.; Rocha, A.M. Development and Optimization of a SPME-GC-FID Method for Ethanol Detection. Processes 2024, 12, 247. https://doi.org/10.3390/pr12020247
Costa NG, Freitas DS, Barros A, Silva C, Antunes JC, Rocha AM. Development and Optimization of a SPME-GC-FID Method for Ethanol Detection. Processes. 2024; 12(2):247. https://doi.org/10.3390/pr12020247
Chicago/Turabian StyleCosta, Nuna G., David S. Freitas, Aline Barros, Carla Silva, Joana C. Antunes, and Ana M. Rocha. 2024. "Development and Optimization of a SPME-GC-FID Method for Ethanol Detection" Processes 12, no. 2: 247. https://doi.org/10.3390/pr12020247
APA StyleCosta, N. G., Freitas, D. S., Barros, A., Silva, C., Antunes, J. C., & Rocha, A. M. (2024). Development and Optimization of a SPME-GC-FID Method for Ethanol Detection. Processes, 12(2), 247. https://doi.org/10.3390/pr12020247