
Nirmatrelvir: From Discovery to Modern and Alternative Synthetic Approaches
 , , and
, , and
Abstract
:1. Introduction
2. Development of Nirmatrelvir
2.1. Target Validation: SARS-CoV-2 Mpro
2.2. Lead Discovery and Optimization
3. Synthetic Approaches to Nirmatrelvir
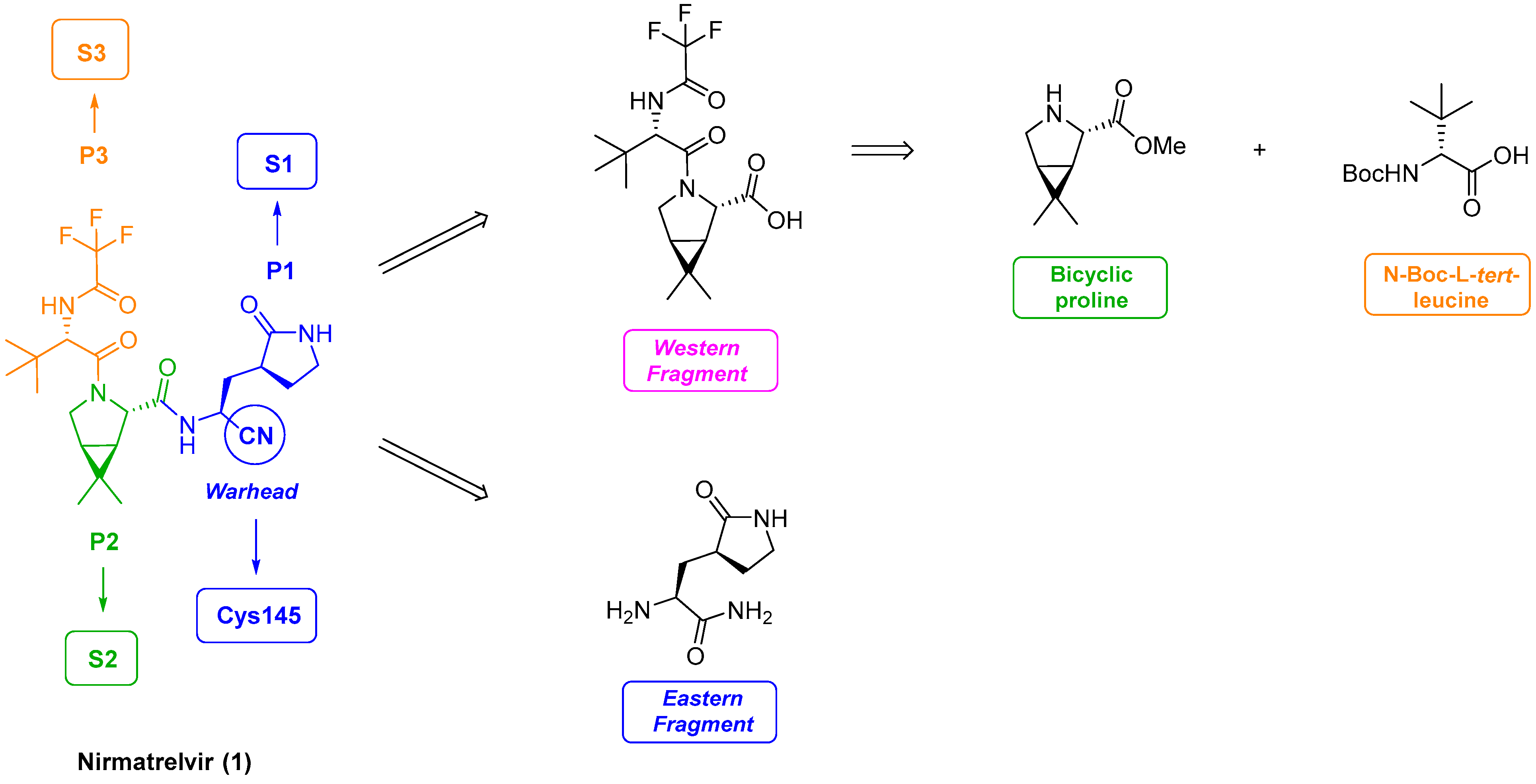
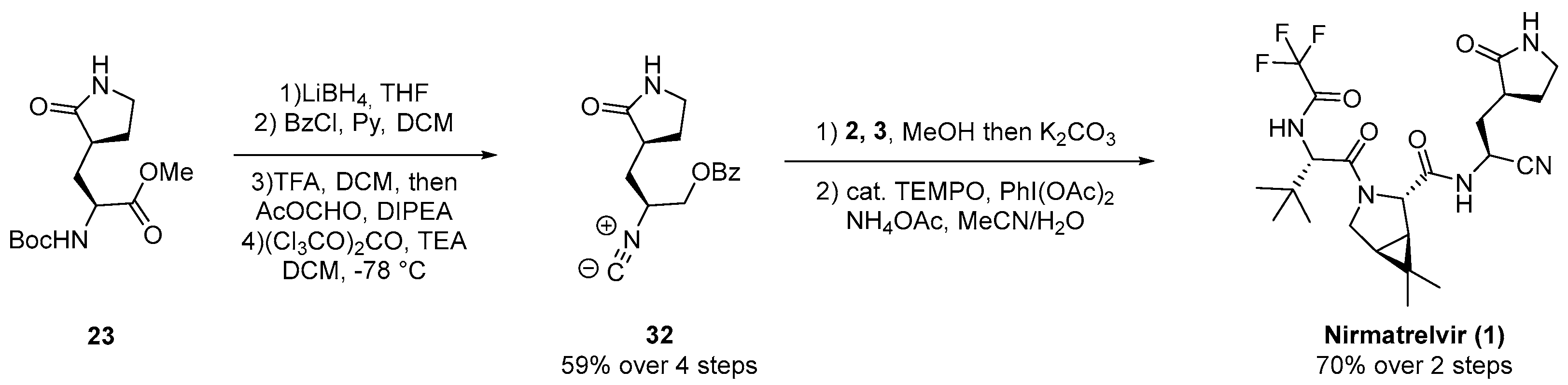
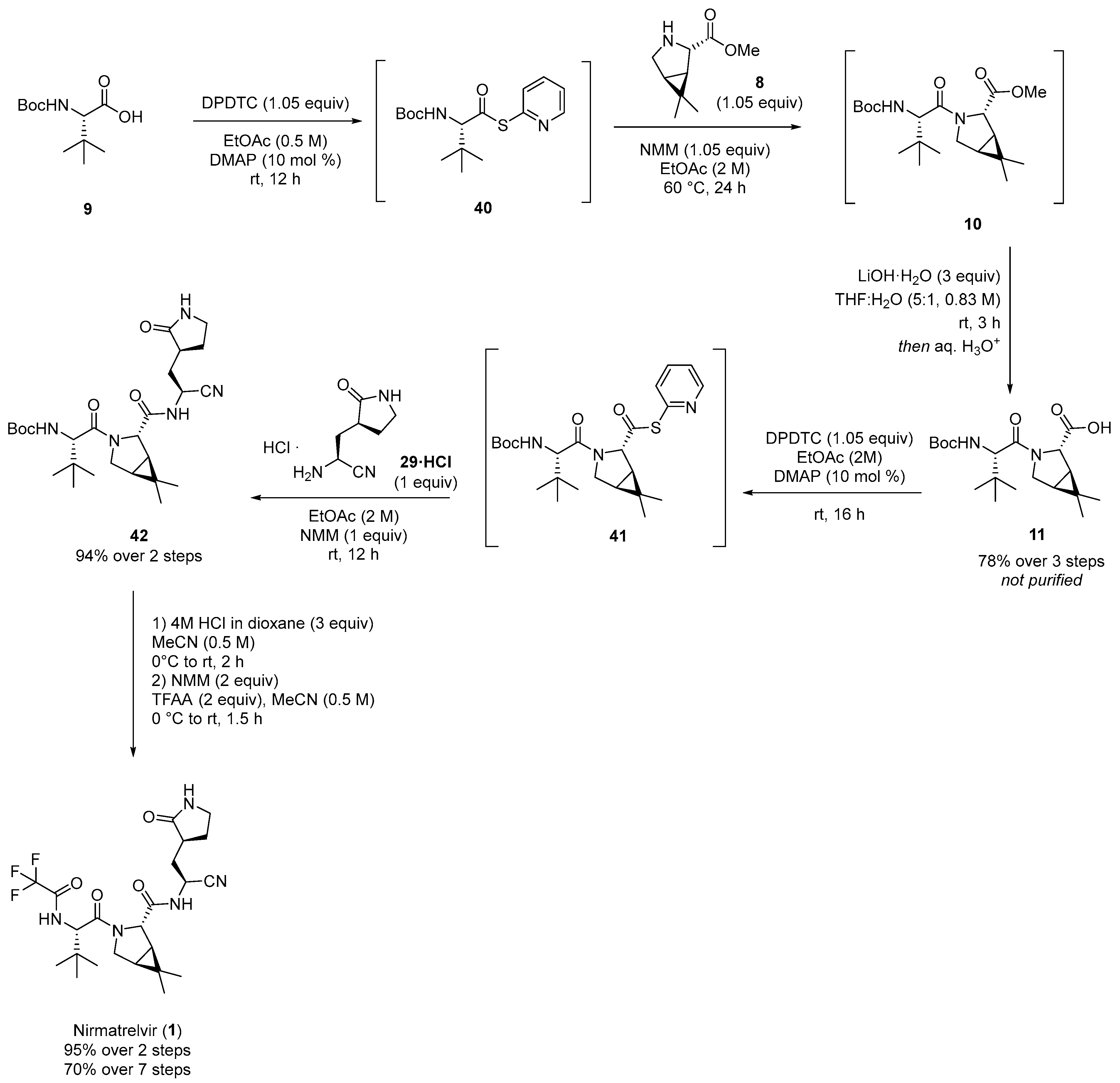
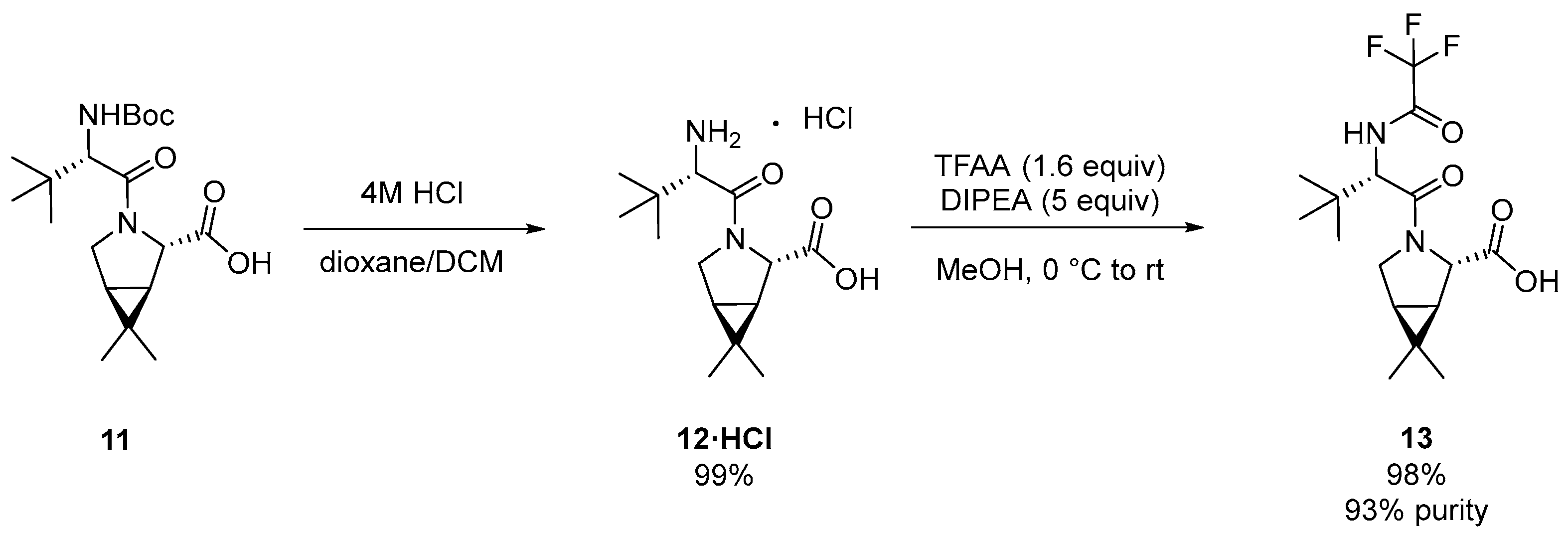
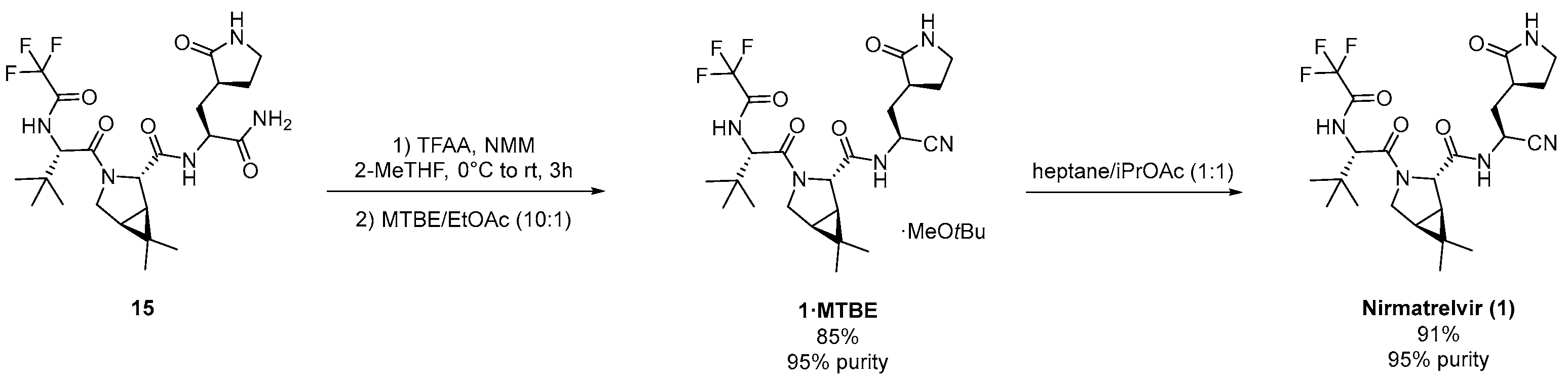
3.1. Pfizer’s Synthesis
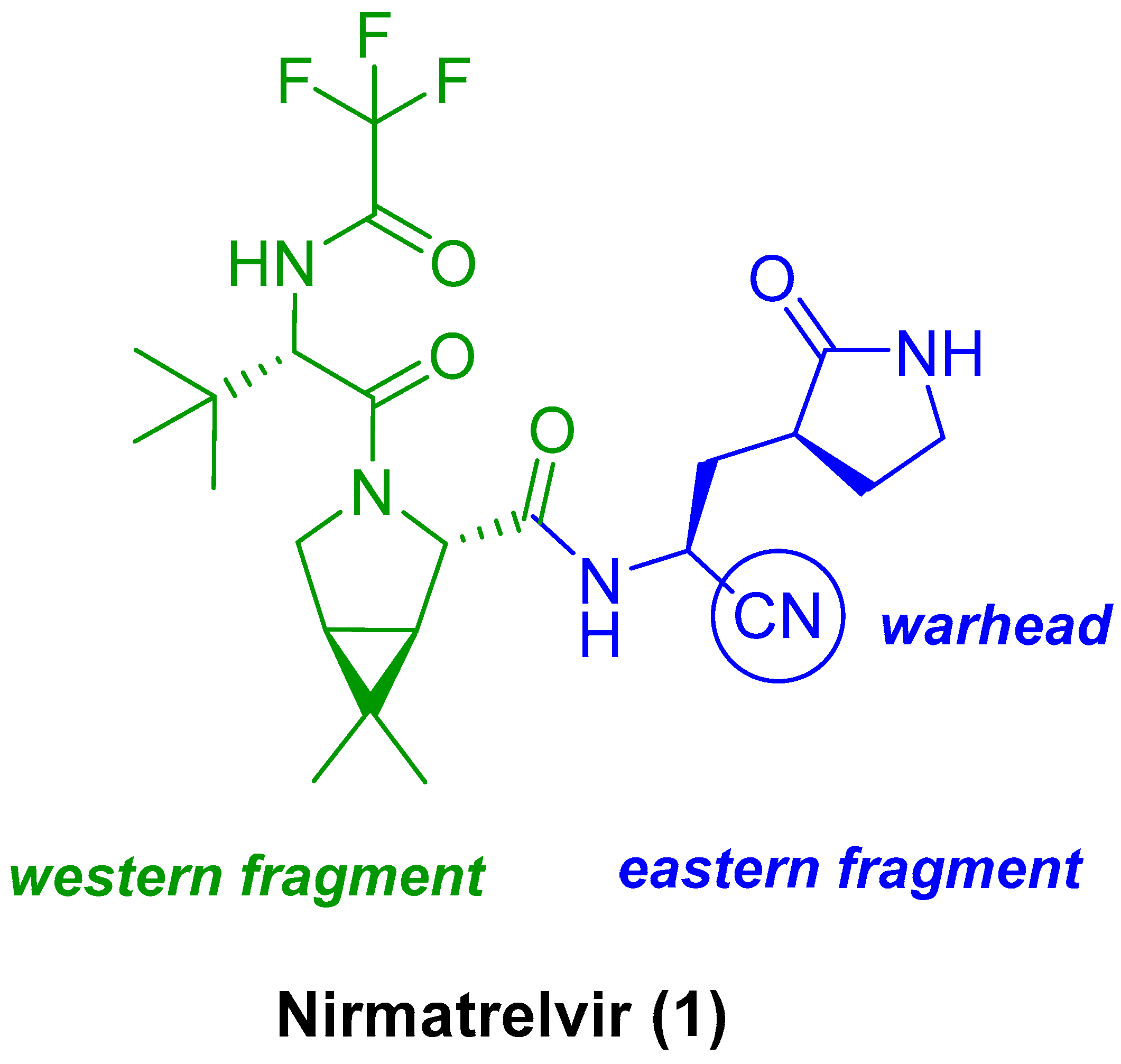
3.2. Structural Components of Nirmatrelvir
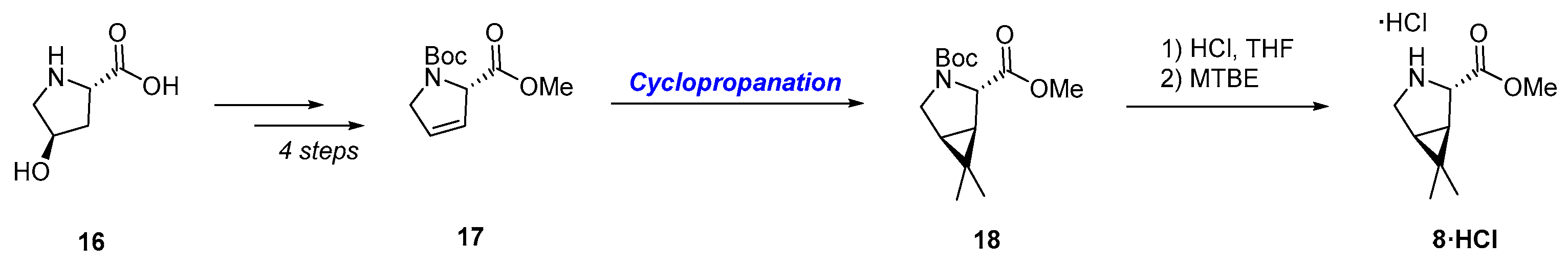
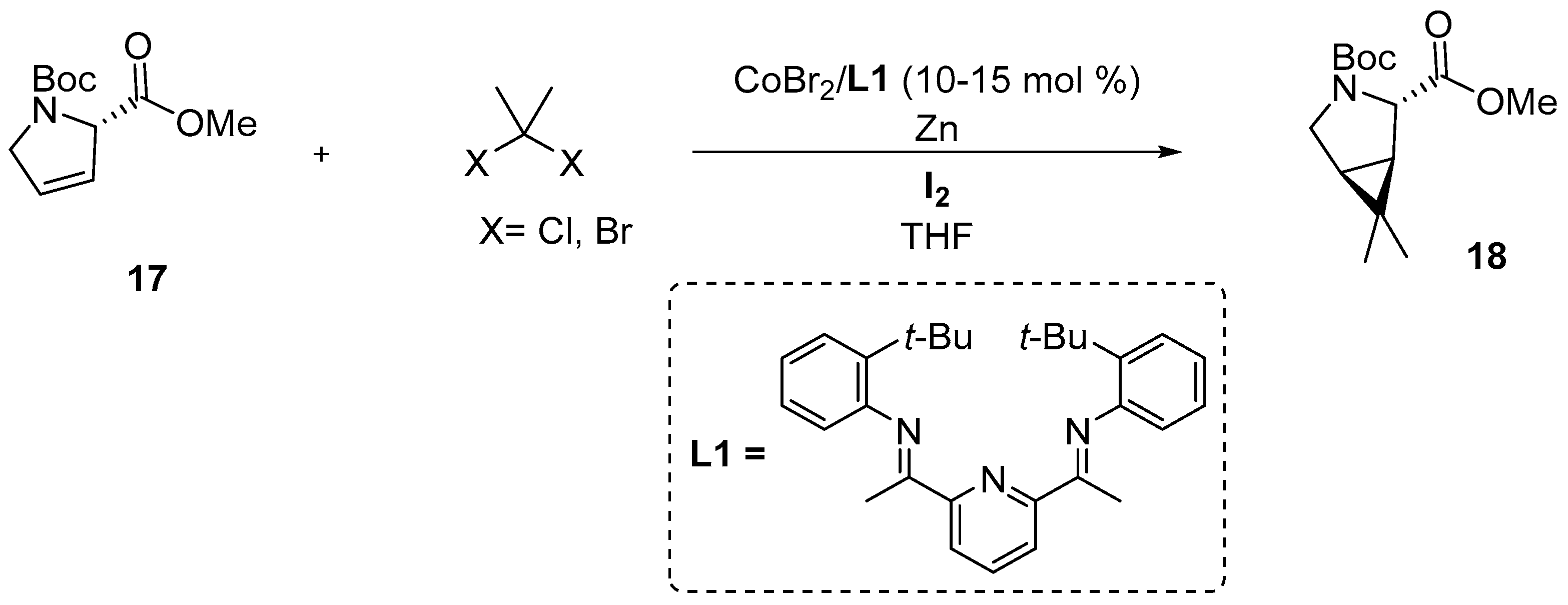
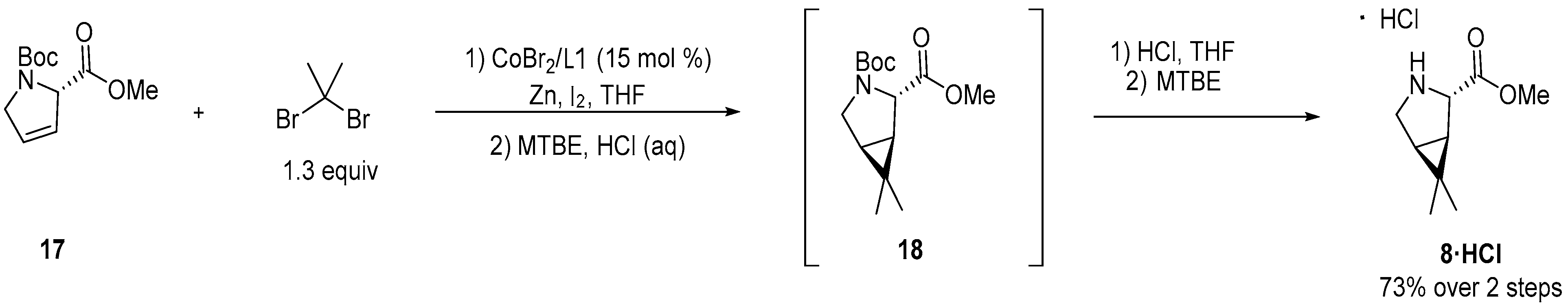
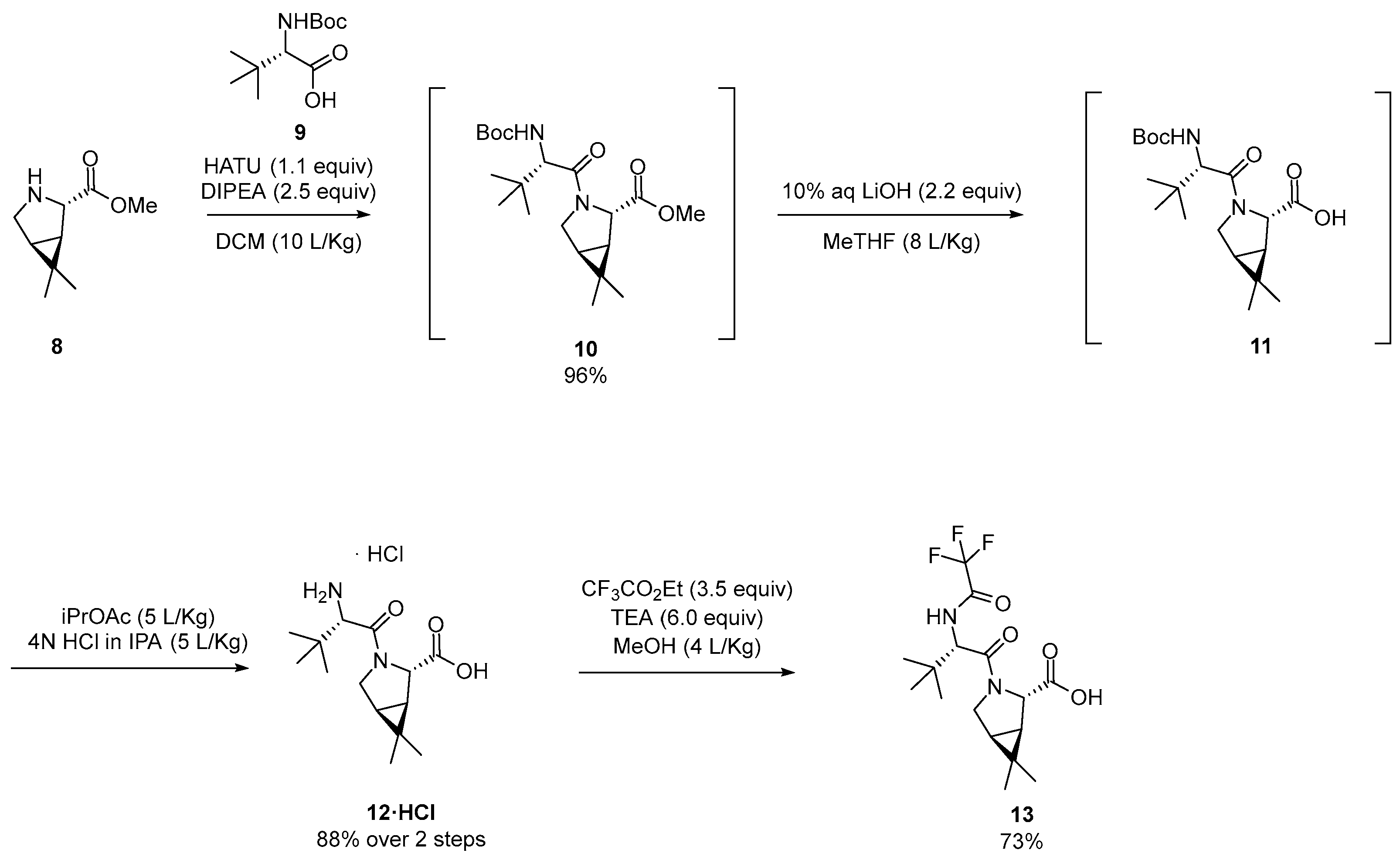
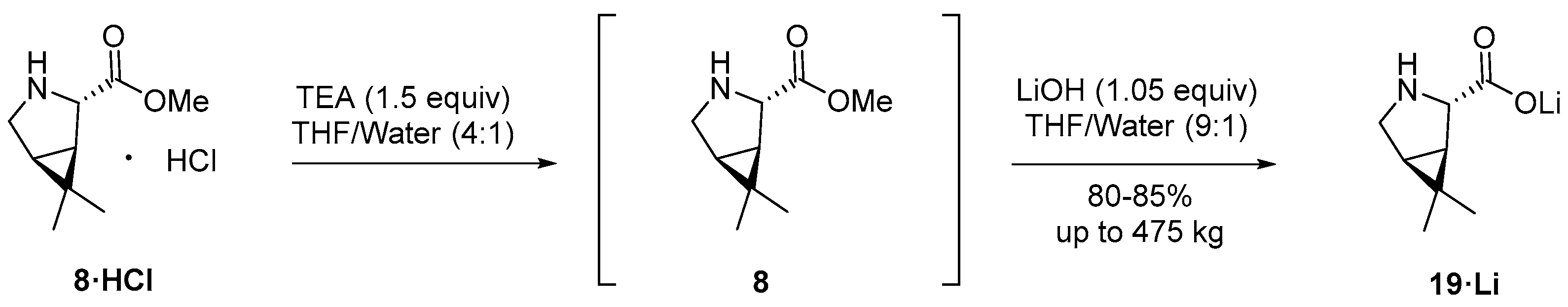
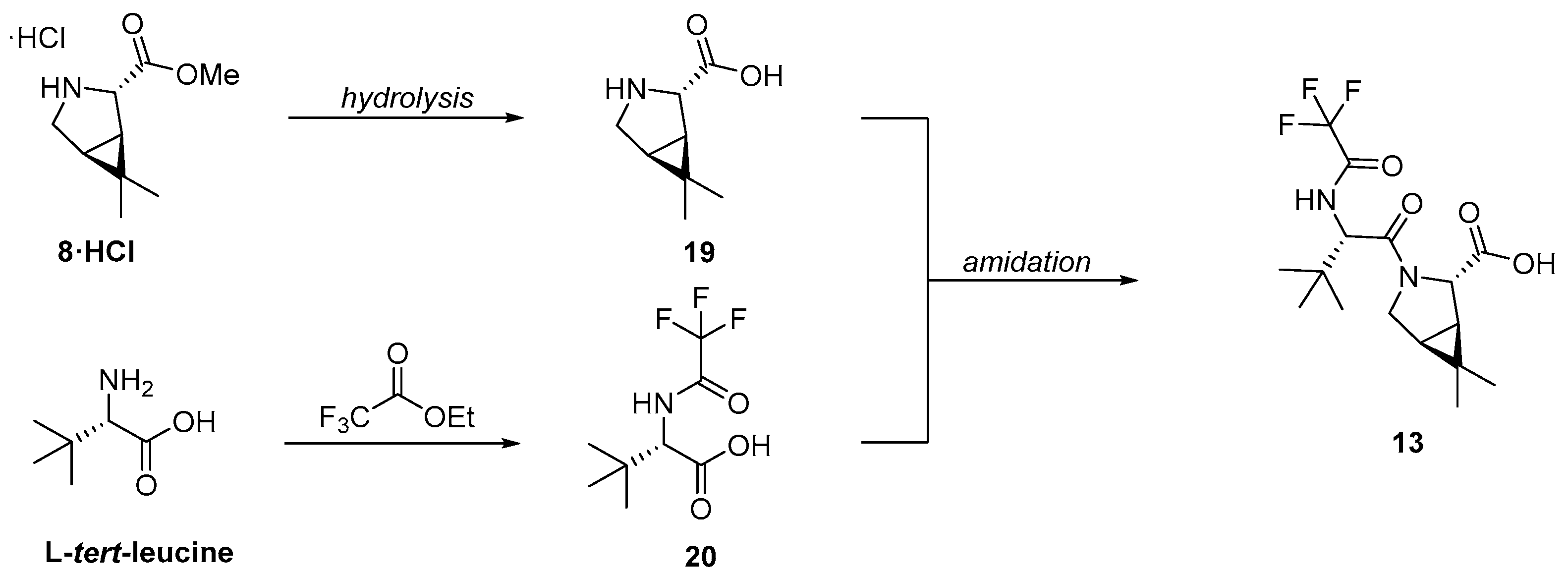
3.2.1. Synthesis of the Bicyclic [3.1.0] Proline Building Block
3.2.2. Synthesis of the Western Fragment
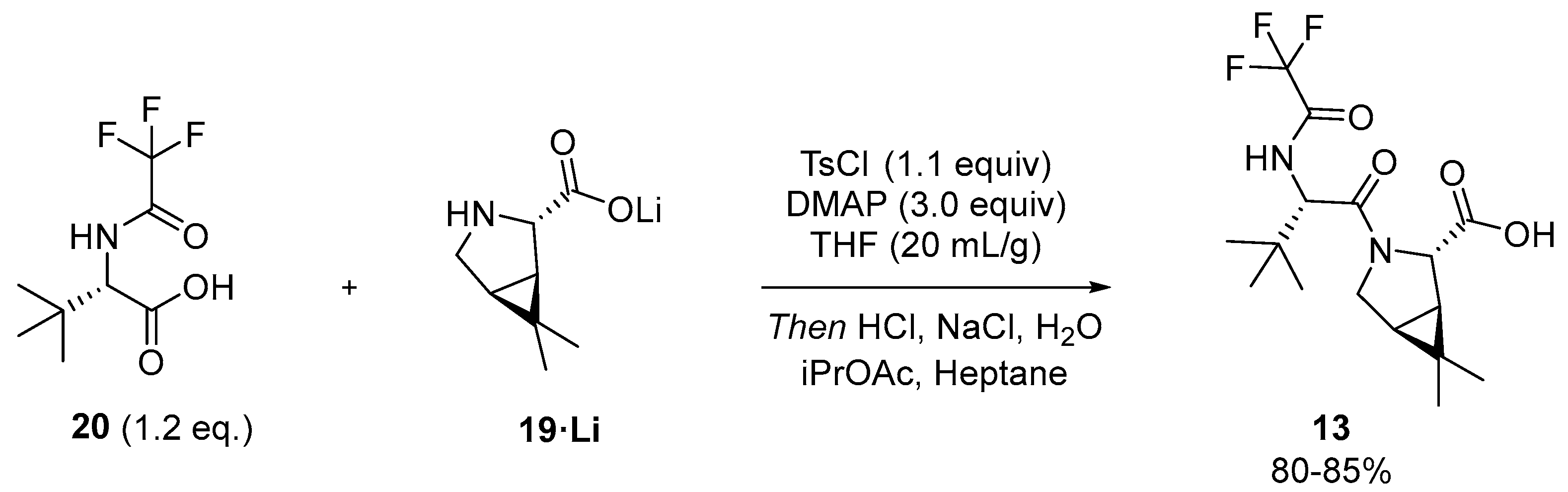
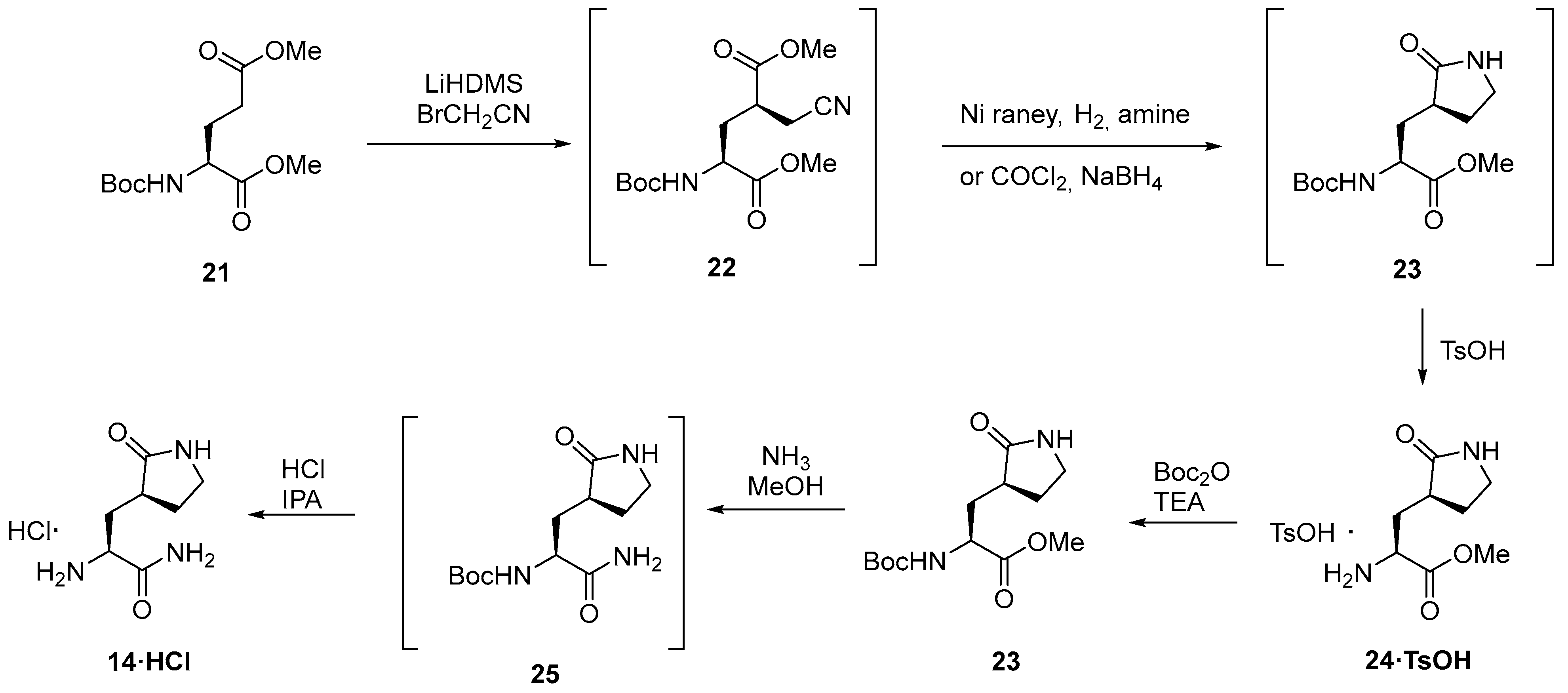
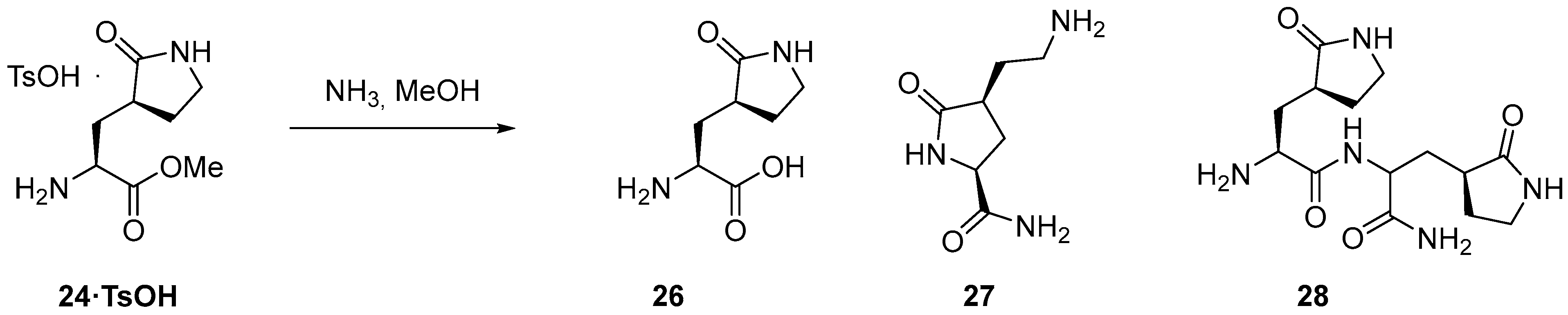
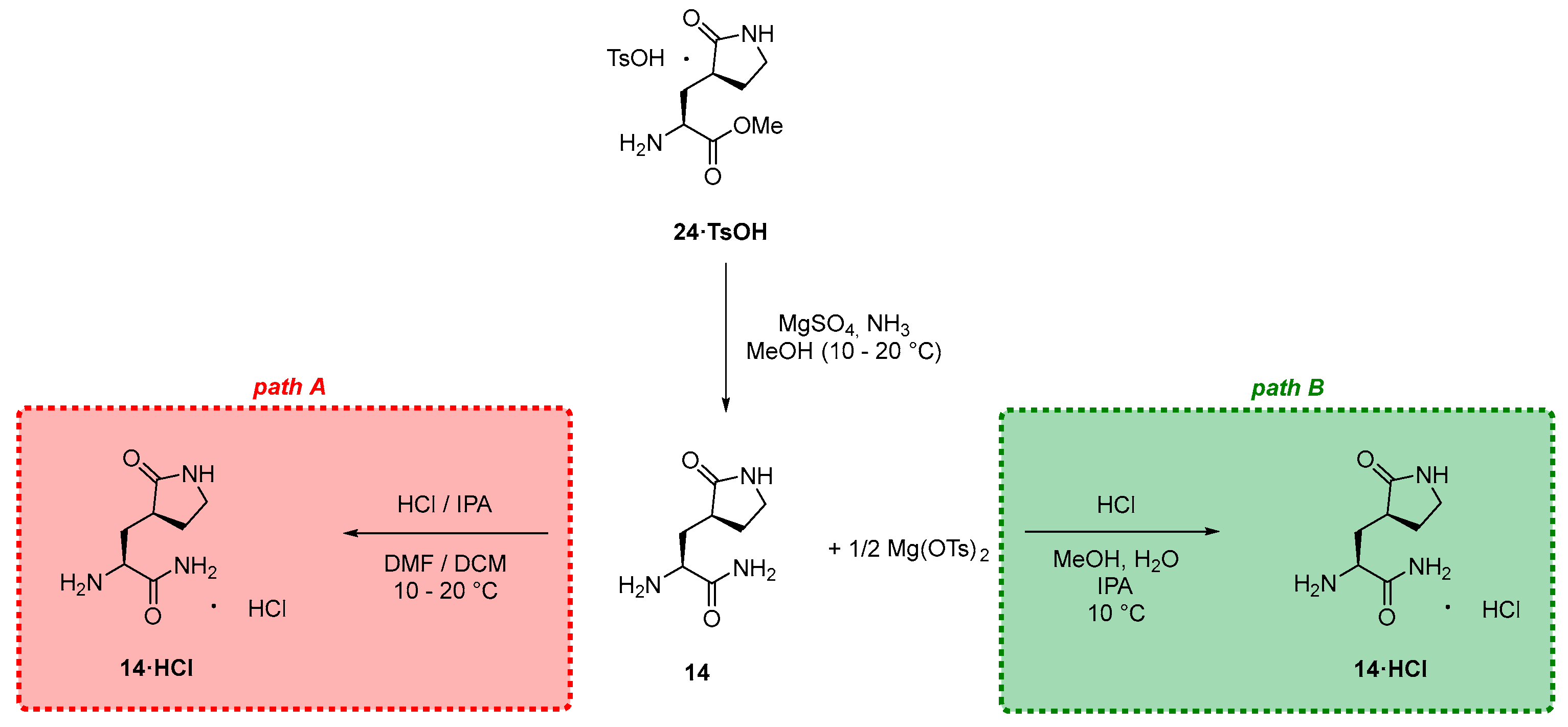
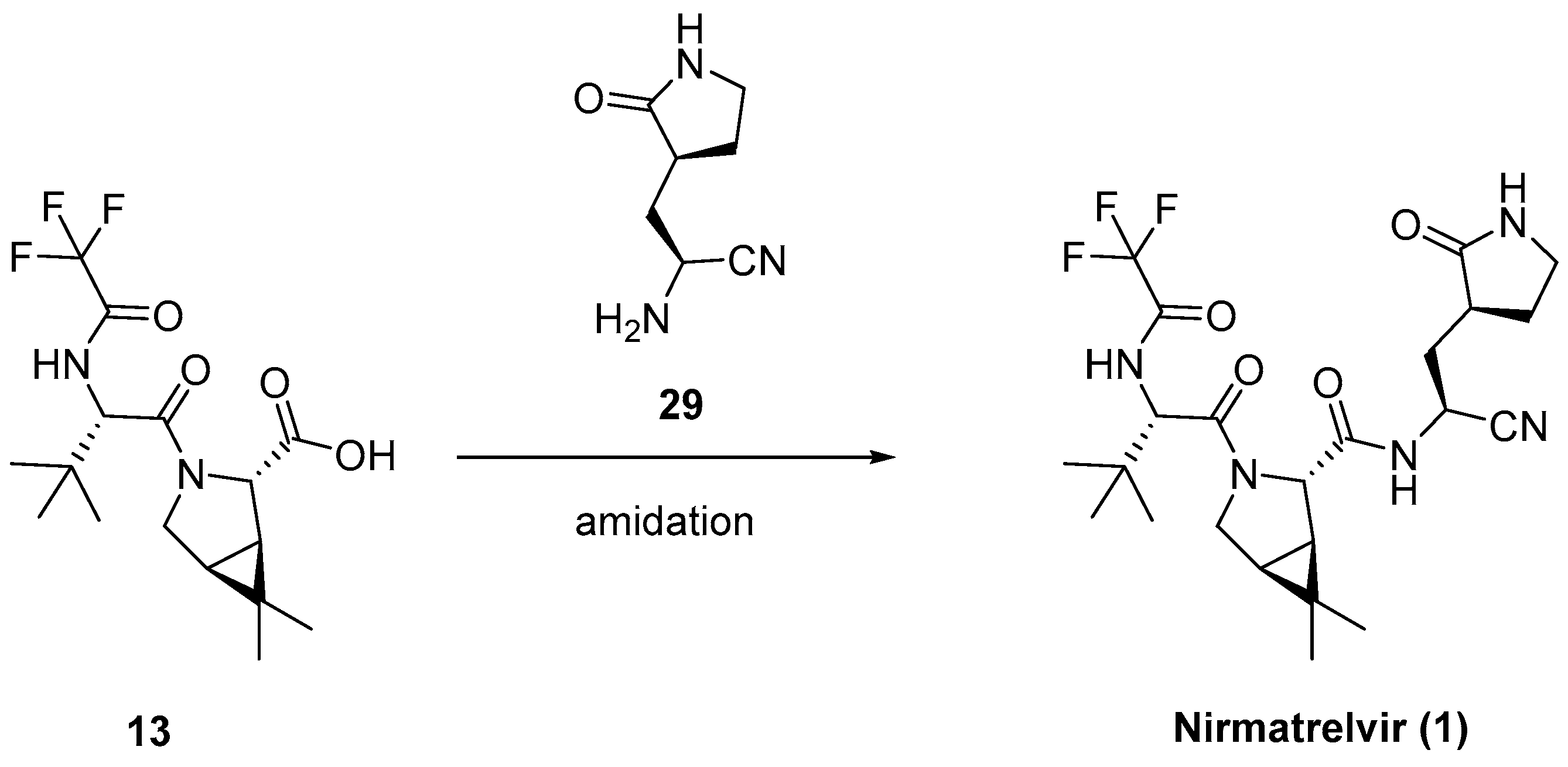
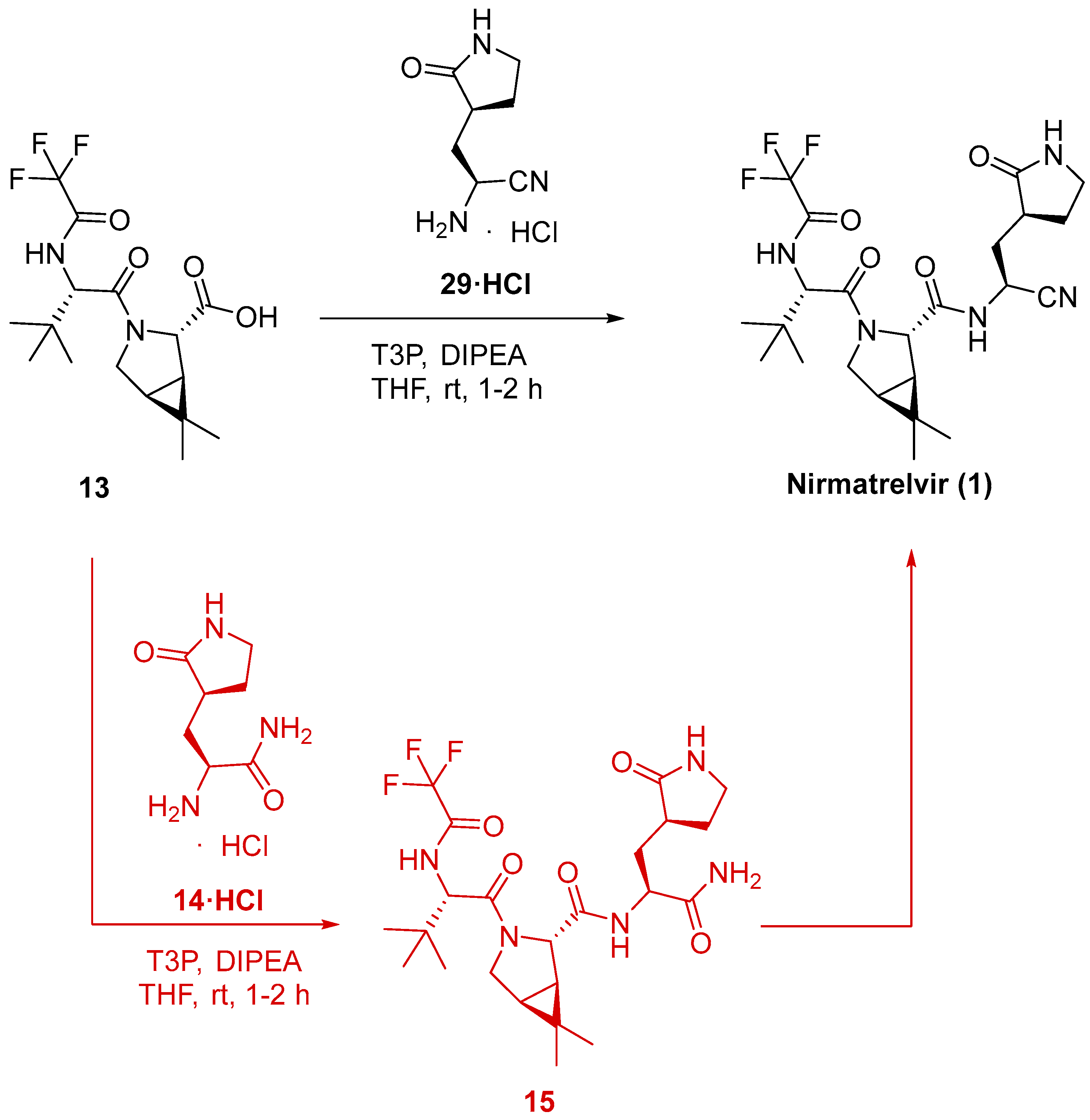
3.2.3. Synthesis of the Eastern Fragment
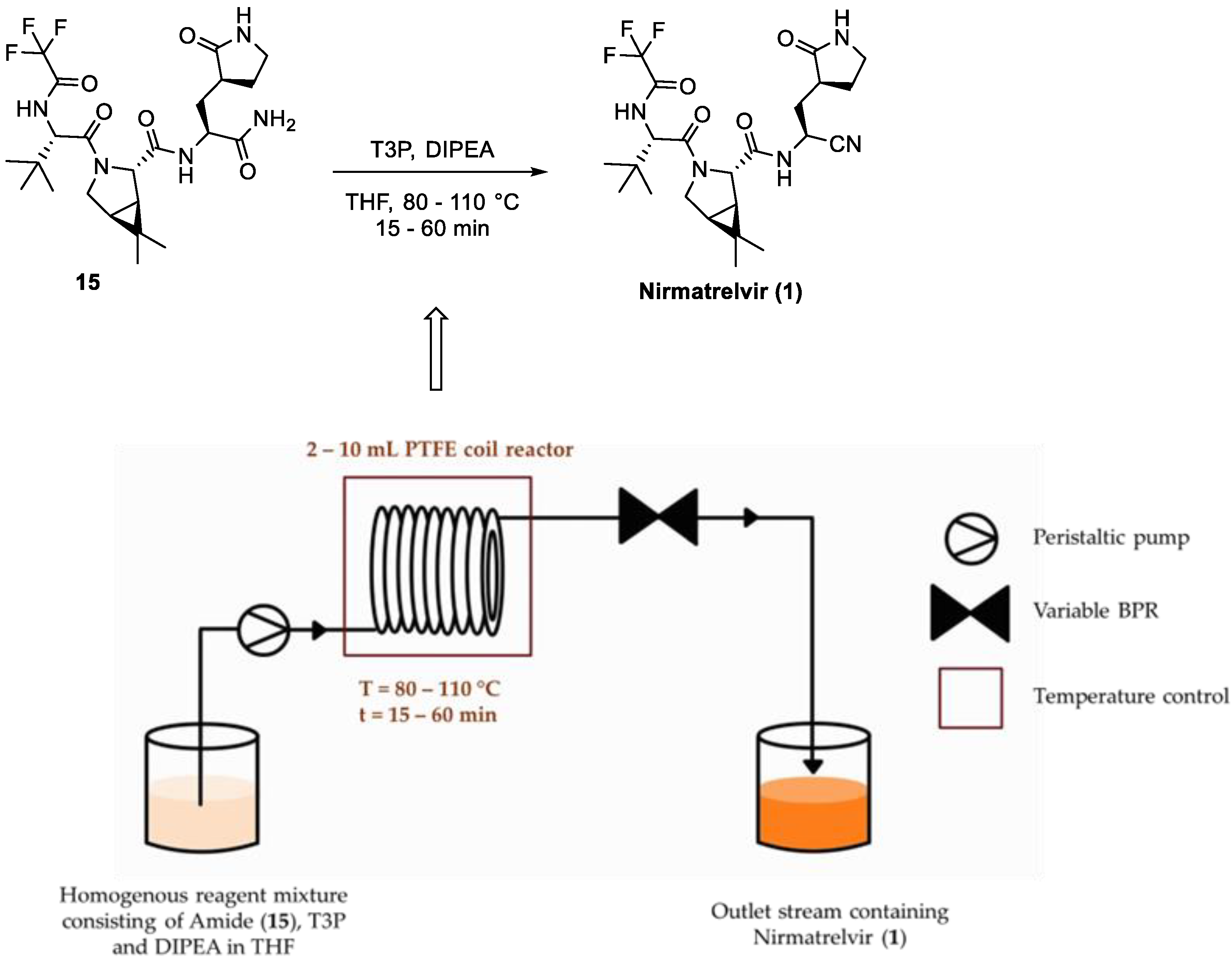
3.3. Application of Flow Chemistry in Nirmatrelvir Synthesis
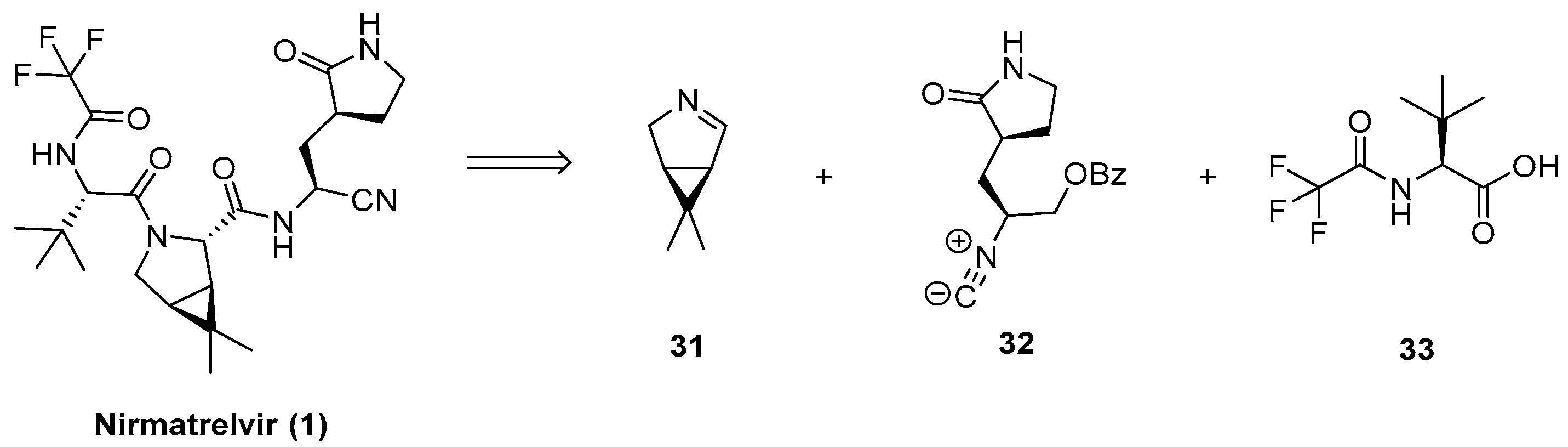
3.4. Multicomponent Approach
3.5. Sustainable Synthesis
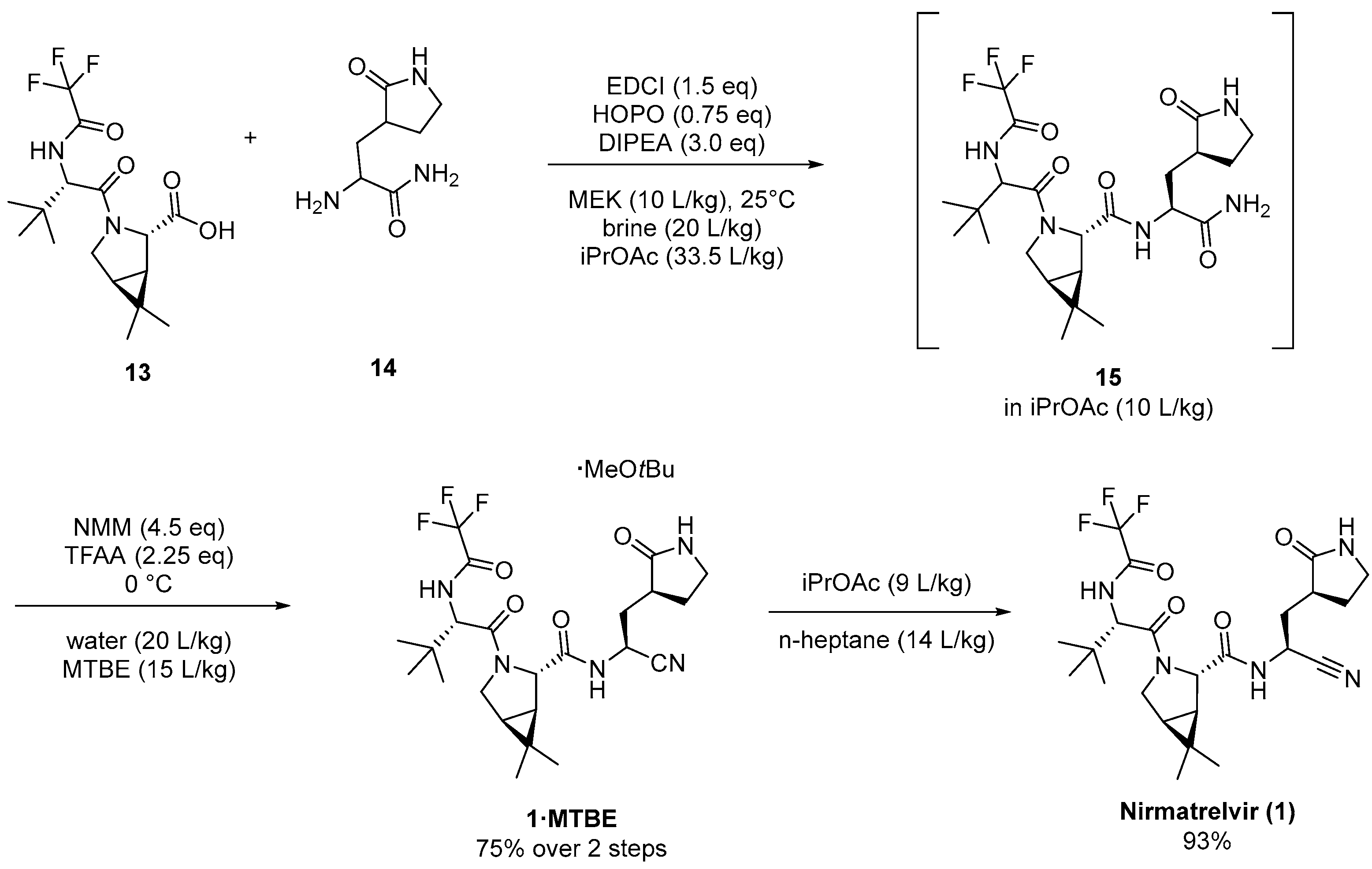
4. Industrial Scale-Up of Nirmatrelvir Synthesis
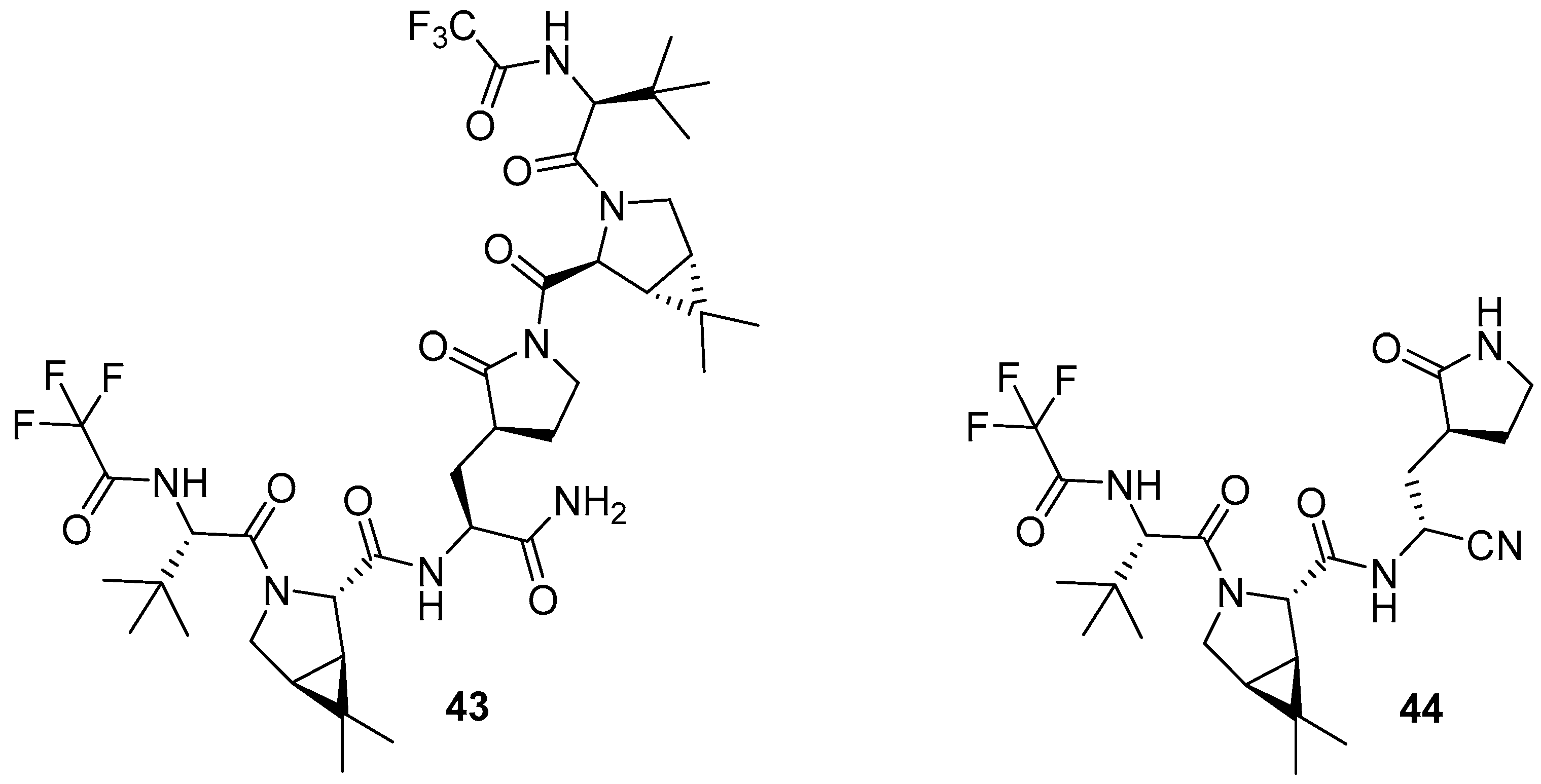
5. Variants and Modifications to Nirmatrelvir
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- De Vries, M.; Mohamed, A.S.; Prescott, R.A.; Valero-Jimenez, A.M.; Desvignes, L.; O’Connor, R.; Steppan, C.; Devlin, J.C.; Ivanova, E.; Herrera, A.; et al. A comparative analysis of SARS-CoV-2 antivirals characterizes 3CL(pro) inhibitor PF-00835231 as a potential new treatment for COVID-19. J. Virol. 2021, 95, e01819-20. [Google Scholar] [CrossRef] [PubMed]
- Lamb, Y.N. Nirmatrelvir Plus Ritonavir: First Approval. Drugs 2022, 82, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Citarella, A.; Dimasi, A.; Moi, D.; Passarella, D.; Scala, A.; Piperno, A.; Micale, N. Recent Advances in SARS-CoV-2 Main Protease Inhibitors: From Nirmatrelvir to Future Perspectives. Biomolecules 2023, 13, 1339. [Google Scholar] [CrossRef] [PubMed]
- Bege, M.; Borbás, A. The Design, Synthesis and Mechanism of Action of Paxlovid, a Protease Inhibitor Drug Combination for the Treatment of COVID-19. Pharmaceutics 2024, 16, 217. [Google Scholar] [CrossRef] [PubMed]
- Karniadakis, I.; Mazonakis, N.; Tsioutis, C.; Papadakis, M.; Markaki, I.; Spernovasilis, N. Oral Molnupiravir and Nirmatrelvir/Ritonavir for the Treatment of COVID-19: A Literature Review with a Focus on Real-World Evidence. Infect. Dis. Rep. 2023, 15, 662–678. [Google Scholar] [CrossRef] [PubMed]
- Akinosoglou, K.; Schinas, G.; Gogos, C. Oral Antiviral Treatment for COVID-19: A Comprehensive Review on Nirmatrelvir/Ritonavir. Viruses 2022, 14, 2540. [Google Scholar] [CrossRef] [PubMed]
- Marzi, M.; Vakil, M.K.; Bahmanyar, M.; Zarenezhad, E. Paxlovid: Mechanism of Action, Synthesis, and In Silico Study. BioMed Res. Int. 2022, 2022, 7341493. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.X.; Du, W.W.; Xia, Y.L.; Zhang, Z.B.; Yu, Z.F.; Fu, Y.X.; Liu, S.Q. Identification of and Mechanistic Insights into SARS-CoV-2 Main Protease Non-Covalent Inhibitors: An In-Silico Study. Int. J. Mol. Sci. 2023, 24, 4237. [Google Scholar] [CrossRef]
- Saied, E.M.; El-Maradny, Y.A.; Osman, A.A.; Darwish, A.M.G.; Abo Nahas, H.H.; Niedbała, G.; Piekutowska, M.; Abdel-Rahman, M.A.; Balbool, B.A.; Abdel-Azeem, A.M. A Comprehensive Review about the Molecular Structure of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): Insights into Natural Products against COVID-19. Pharmaceutics 2021, 13, 1759. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Dai, W.; Zhang, B.; Jiang, X.M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; George, S.; Kusov, Y.; Perbandt, M.; Anemüller, S.; Mesters, J.R.; Norder, H.; Coutard, B.; Lacroix, C.; Leyssen, P.; et al. 3C protease of enterovirus 68: Structure-based design of Michael acceptor inhibitors and their broad-spectrum antiviral effects against picornaviruses. J. Virol. 2013, 87, 4339–4351. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An oral SARS-CoV-2 M(pro) inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef]
- Fu, L.; Ye, F.; Feng, Y.; Yu, F.; Wang, Q.; Wu, Y.; Zhao, C.; Sun, H.; Huang, B.; Niu, P.; et al. Both Boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat. Commun. 2020, 11, 4417. [Google Scholar] [CrossRef] [PubMed]
- Joyce, R.P.; Hu, V.W.; Wang, J. The history, mechanism, and perspectives of nirmatrelvir (PF-07321332): An orally bioavailable main protease inhibitor used in combination with ritonavir to reduce COVID-19-related hospitalizations. Med. Chem. Res. 2022, 31, 1637–1646. [Google Scholar] [CrossRef] [PubMed]
- Bonatto, V.; Lameiro, R.F.; Rocho, F.R.; Lameira, J.; Leitão, A.; Montanari, C.A. Nitriles: An attractive approach to the development of covalent inhibitors. RSC Med. Chem. 2023, 14, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Cotrim, B.A.; Barros, J.C. Development and patent synthesis of nirmatrelvir–the main component of the first oral drug against SARS-CoV-2 Paxlovid®. Aust. J. Chem. 2022, 75, 487–491. [Google Scholar] [CrossRef]
- Jiang, B.; Li, G.; Yu, J.; Xu, X.; Pan, H.; Zhao, C.; Zhong, J.; Zhang, F. Synthesis and crystal characteristics of nirmatrelvir. React. Chem. Eng. 2023, 8, 1747–1759. [Google Scholar] [CrossRef]
- Algera, R.F.; Allais, C.; Baldwin, A.F.; Becirovic, H.; Bowles, P.; Brown, A.R.; Buske, J.M.; Clarke, H.J.; Do, N.M.; Doyle, K.; et al. Synthesis of Nirmatrelvir: Development of an Efficient, Scalable Process to Generate the Western Fragment. Org. Process Res. Dev. 2023, 27, 2240–2249. [Google Scholar] [CrossRef]
- Algera, R.F.; Allais, C.; Becirovic, H.; Brown, A.R.; Chen, B.; Clarke, H.J.; Concannon, P.E.; Du, Y.; Georgian, W.; Lee, J.W.; et al. Synthesis of Nirmatrelvir: Development of Magnesium Sulfate-Mediated Aminolysis for the Manufacture of the Eastern Fragment. Org. Process Res. Dev. 2023, 27, 2271–2279. [Google Scholar] [CrossRef]
- Algera, R.F.; Allais, C.; Baldwin, A.F.; Busch, T.; Colombo, F.; Colombo, M.; Depretz, C.; Dumond, Y.R.; Faria Quintero, A.R.; Heredia, M.; et al. Synthesis of Nirmatrelvir: Development of a Scalable Cobalt-Catalyzed Cyclopropanation for Manufacture of the Bicyclic [3.1.0] Proline-Building Block. Org. Process Res. Dev. 2023, 27, 2260–2270. [Google Scholar] [CrossRef]
- Werth, J.; Uyeda, C. Cobalt-Catalyzed Reductive Dimethylcyclopropanation of 1,3-Dienes. Angew. Chem. Int. Ed. Engl. 2018, 57, 13902–13906. [Google Scholar] [CrossRef]
- Porta, R.; Benaglia, M.; Puglisi, A. Flow Chemistry: Recent Developments in the Synthesis of Pharmaceutical Products. Org. Process Res. Dev. 2016, 20, 2–25. [Google Scholar] [CrossRef]
- Preschel, H.D.; Otte, R.T.; Zhuo, Y.; Ruscoe, R.E.; Burke, A.J.; Kellerhals, R.; Horst, B.; Hennig, S.; Janssen, E.; Green, A.P.; et al. Multicomponent Synthesis of the SARS-CoV-2 Main Protease Inhibitor Nirmatrelvir. J. Org. Chem. 2023, 88, 12565–12571. [Google Scholar] [CrossRef] [PubMed]
- Kincaid, J.R.A.; Caravez, J.C.; Iyer, K.S.; Kavthe, R.D.; Fleck, N.; Aue, D.H.; Lipshutz, B.H. A sustainable synthesis of the SARS-CoV-2 M(pro) inhibitor nirmatrelvir, the active ingredient in Paxlovid. Commun. Chem. 2022, 5, 156. [Google Scholar] [CrossRef]
- Okabe, H.; Naraoka, A.; Isogawa, T.; Oishi, S.; Naka, H. Acceptor-Controlled Transfer Dehydration of Amides to Nitriles. Org. Lett. 2019, 21, 4767–4770. [Google Scholar] [CrossRef]
- Wood, A.B.; Kincaid, J.R.A.; Lipshutz, B.H. Dehydration of primary amides to nitriles in water. Late-stage functionalization and 1-pot multistep chemoenzymatic processes under micellar catalysis conditions. Green Chem. 2022, 24, 2853–2858. [Google Scholar] [CrossRef]
- Caravez, J.C.; Iyer, K.S.; Kavthe, R.D.; Kincaid, J.R.A.; Lipshutz, B.H. A 1-Pot Synthesis of the SARS-CoV-2 Mpro Inhibitor Nirmatrelvir, the Key Ingredient in Paxlovid. Org. Lett. 2022, 24, 9049–9053. [Google Scholar] [CrossRef] [PubMed]
- Shanahan, C.S.; Kadam, A.L.; Chiranjeevi, B.; Nunes, A.A.; Jayaraman, A.; Ahmad, S.; Aleshire, S.L.; Donsbach, K.O.; Gupton, B.F.; Nuckols, M.C. Efforts to develop a cost-effective and scalable synthetic process for nirmatrelvir. ChemRxiv 2022. [Google Scholar] [CrossRef]
- Algera, R.F.; Baldwin, A.F.; Bowles, P.; Clarke, H.J.; Connor, C.G.; Cordi, E.M.; Do, N.M.; Nicholson, L.D.; Georgian, W.; Happe, A.; et al. Synthesis of Nirmatrelvir: Design and Optimization of an Efficient Telescoped Amidation–Dehydration Sequence. Org. Process Res. Dev. 2023, 27, 2250–2259. [Google Scholar] [CrossRef]
- Hu, Y.N.; Lewandowski, E.M.; Tan, H.; Zhang, X.; Morgan, R.T.; Zhang, X.; Jacobs, L.M.C.; Butler, S.G.; Gongora, M.V.; Choy, J.; et al. Naturally Occurring Mutations of SARS-CoV-2 Main Protease Confer Drug Resistance to Nirmatrelvir. ACS Cent. Sci. 2023, 9, 1658–1669. [Google Scholar] [CrossRef] [PubMed]
- Dayan Elshan, N.G.R.; Wolff, K.C.; Riva, L.; Woods, A.K.; Grabovyi, G.; Wilson, K.; Pedroarena, J.; Ghorai, S.; Nazarian, A.; Weiss, F.; et al. Discovery of CMX990: A Potent SARS-CoV-2 3CL Protease Inhibitor Bearing a Novel Warhead. J. Med. Chem. 2024, 67, 2369–2378. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Yadav, M.; Iddum, S.; Ghazi, S.; Lendy, E.K.; Jayashankar, U.; Beechboard, S.N.; Takamatsu, Y.; Hattori, S.I.; Aamano, M.; et al. Exploration of P1 and P4 modifications of nirmatrelvir: Design, synthesis, biological evaluation, and X-ray structural studies of SARS-CoV-2 Mpro inhibitors. Eur. J. Med. Chem. 2024, 267, 116132. [Google Scholar] [CrossRef] [PubMed]
- Brewitz, L.; Dumjahn, L.; Zhao, Y.; Owen, C.D.; Laidlaw, S.M.; Malla, T.R.; Nguyen, D.; Lukacik, P.; Salah, E.; Crawshaw, A.D.; et al. Alkyne Derivatives of SARS-CoV-2 Main Protease Inhibitors Including Nirmatrelvir Inhibit by Reacting Covalently with the Nucleophilic Cysteine. J. Med. Chem. 2023, 66, 2663–2680. [Google Scholar] [CrossRef] [PubMed]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Advantages | Disadvantages |
|---|---|---|
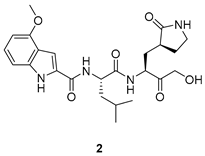 | High inhibitory potency Good antiviral activity | Low oral bioavailability |
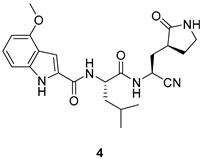 | Lower number of H-bond donors Nitrile warhead enhances oral absorption in rats | Antiviral and inhibitory activities lower than 2 |
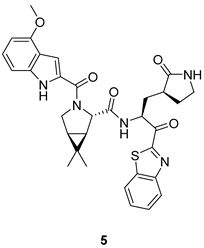 | Benzothiazole-7-yl ketone warhead increases permeability Replacement of leucine with modified proline allows a better fit into the S2 subsite | Antiviral and inhibitory activities lower than 2 and 4 |
 | Replacement of indole with methanesulfonamide allows a better fit into the S3 subsite Antiviral and inhibitory activities higher than 4 and 5 | Antiviral and inhibitory activities lower than 2 Oral absorption in rats and monkeys lower than 7 |
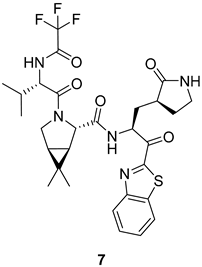 | Replacement of indole with trifluoroacetamide allows a better fit into the S3 subsite Antiviral and inhibitory activities higher than 4 and 5 Oral absorption in rats and monkeys higher than 6 | Antiviral and inhibitory activities lower than 2 |
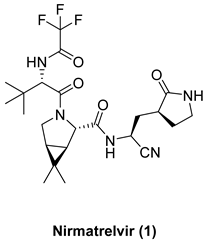 | Combination of nitrile warhead, modified proline at P2 position, and trifluoroacetamide at P3 position provides antiviral and inhibitory activities higher than 2 Oral bioavailability higher than all precursors |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galli, M.; Migliano, F.; Fasano, V.; Silvani, A.; Passarella, D.; Citarella, A. Nirmatrelvir: From Discovery to Modern and Alternative Synthetic Approaches. Processes 2024, 12, 1242. https://doi.org/10.3390/pr12061242
Galli M, Migliano F, Fasano V, Silvani A, Passarella D, Citarella A. Nirmatrelvir: From Discovery to Modern and Alternative Synthetic Approaches. Processes. 2024; 12(6):1242. https://doi.org/10.3390/pr12061242
Chicago/Turabian StyleGalli, Michela, Francesco Migliano, Valerio Fasano, Alessandra Silvani, Daniele Passarella, and Andrea Citarella. 2024. "Nirmatrelvir: From Discovery to Modern and Alternative Synthetic Approaches" Processes 12, no. 6: 1242. https://doi.org/10.3390/pr12061242