
Mathematical Models for Estimating Diffusion Coefficients in Concentrated Polymer Solutions from Experimental Data

 and
and
Abstract
1. Introduction
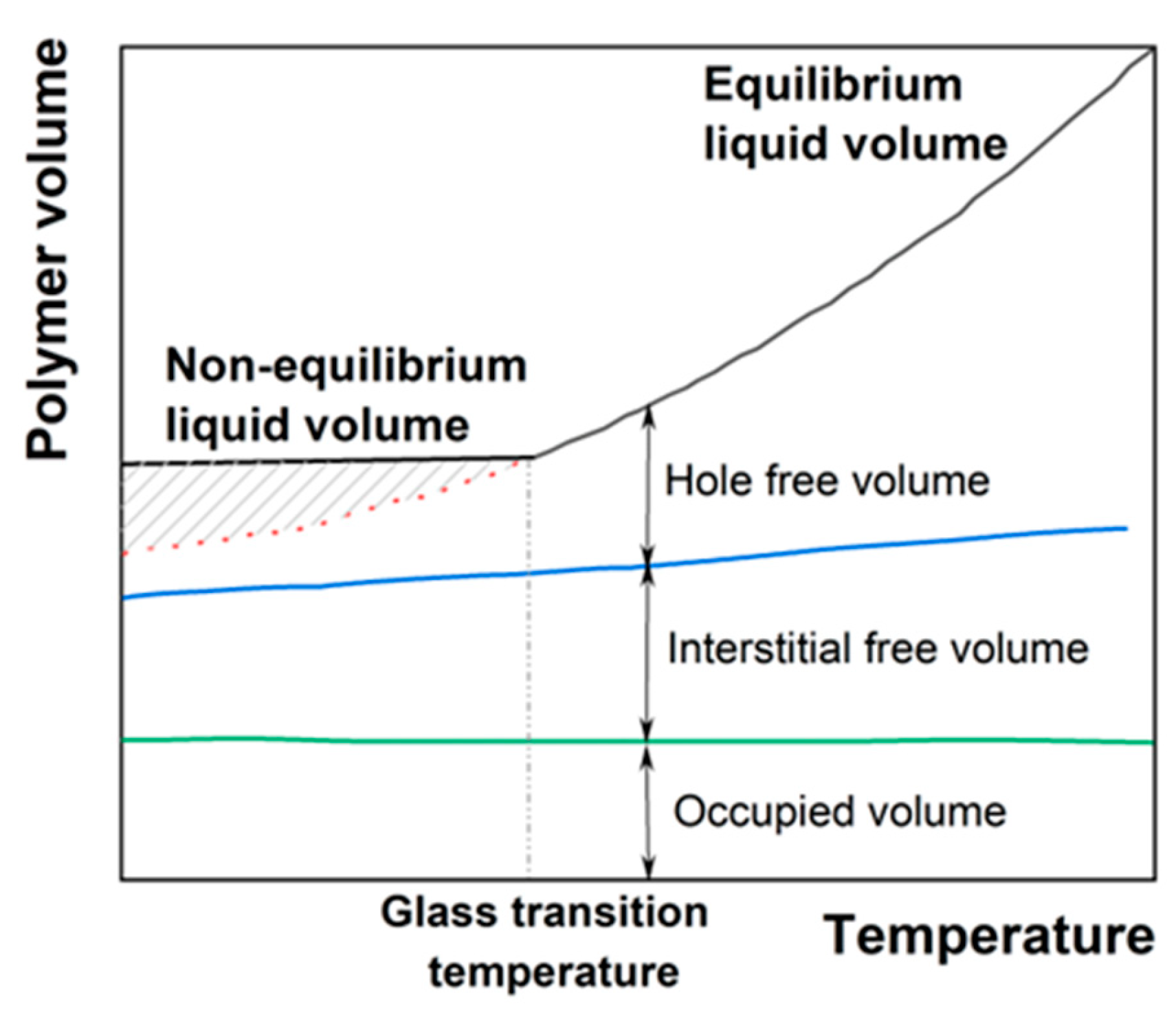
2. Diffusion Coefficients from Experimental Data
2.1. Methodology
2.2. Fick or Non-Fick Diffusion
3. Materials and Methods
4. Results and Discussion
4.1. Fick Diffusion (Case I)
4.1.1. Diffusion Coefficients from Drying Experiments CA-THF
4.1.2. Diffusion Coefficients from Drying Experiments PVA-H2O
4.2. Two-Stage Diffusion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Notations
| A | pre-exponential constant, a proportionality factor, m2 s−1. |
| b | a numerical factor of the order of unity. |
| B | a parameter depending only on the particle size, Equation (3). |
| D | is the diffusion coefficient of the molecule, m2 s−1 (Equations (4) and (5)). |
| D0i | is a pre-exponential factor of component I, m2 s−1. |
| D1 | is the solvent self-diffusion coefficient, m2 s−1. |
| fp | free volume due to the polymer. |
| fs | free volume due to the solvent. |
| fν | average free volume per molecule. |
| is the free volume of the solvent in the polymer solution. | |
| l | half of film thickness, m. |
| Meq | is the penetrate amount at equilibrium. |
| Mt | is the amount of penetrant diffused into polymer at time, t. |
| R | gas constant, J mol−1 K−1. |
| T | temperature, K. |
| ν* | the minimum required volume of the void. |
| is the average hole free volume per gram of mixture, m3 Kg−1. | |
| is the specific critical hole free volume required for a jump, m3 Kg−1. | |
| Greek symbols: | |
| γ | constant in Equation (1). |
| δ | thickness of film, membrane, m. |
| Θ | is a thermodynamic factor. |
| γ1p | is an overlap factor considering that free volume is available to more than one molecule. |
| ωi | is the weight fraction of component i. |
| ξ | is the ratio of the critical molar volume of solvent jumping units to the critical molar volume of jumping units of polymer. |
| χ1p | Florry Huggins interaction parameter, dimensionless. |
| ω1 | is the mass fraction of the solvent. |
| Φp | polymer volume fraction, dimensionless. |
| Φs | solvent volume fraction, dimensionless. |
References
- Sanders, D.F.; Smith, Z.P.; Guo, R.; Robeson, L.M.; McGrath, J.E.; Paul, D.R.; Freeman, B.D. Energy-efficient polymeric gas separation membranes for a sustainable future: A review. Polymer 2013, 54, 4729–4761. [Google Scholar] [CrossRef]
- Li, J.; Mooney, D.J. Designing hydrogels for controlled drug delivery. Nat. Rev. Mater. 2016, 1, 16071. [Google Scholar] [CrossRef] [PubMed]
- Poling-Skutvik, R.; Roberts, R.C.; Slim, A.H.; Narayanan, S.; Krishnamoorti, R.; Palmer, J.C.; Conrad, J.C. Structure dominates localization of tracers within aging nanoparticle glasses. J. Phys. Chem. Lett. 2019, 10, 1784–1789. [Google Scholar] [CrossRef]
- Bilchak, C.R.; Jhalaria, M.; Huang, Y.; Abbas, Z.; Midya, J.; Benedetti, F.M.; Parisi, D.; Egger, W.; Dickmann, M.; Minelli, M.; et al. Tuning selectivities in gas separation membranes based on polymer-grafted nanoparticles. ACS Nano 2020, 14, 17174–17183. [Google Scholar] [CrossRef]
- Vrentas, J.S.; Vrentas, C.M. Diffusion and Mass Transfer, 1st ed.; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar] [CrossRef]
- Barnett, J.W.; Kumar, S.K. Modeling gas transport in polymer-grafted nanoparticle membranes. Soft Matter 2019, 15, 424–432. [Google Scholar] [CrossRef]
- Blaiszik, B.J.; Kramer, S.; Olugebefola, S.; Moore, J.; Sottos, N.; White, S. Self-healing polymers and composites. Annu. Rev. Mater. Res. 2010, 40, 179–211. [Google Scholar] [CrossRef]
- Geise, G.M.; Paul, D.R.; Freeman, B.D. Fundamental water and salt transport properties of polymeric materials. Prog. Polym. Sci. 2014, 39, 1–42. [Google Scholar] [CrossRef]
- Ali, W.; Gebert, B.; Hennecke, T.; Graf, K.; Ulbricht, M.; Gutmann, J.S. Design of thermally responsive polymeric hydrogels for brackish water desalination: Effect of architecture on swelling, deswelling, and salt rejection. ACS Appl. Mater. Interfaces 2015, 7, 15696–15706. [Google Scholar] [CrossRef]
- Peppas, N.A. Hydrogels and drug delivery. Curr. Opin. Colloid Interface Sci. 1997, 2, 531–537. [Google Scholar] [CrossRef]
- Vrentas, J.S.; Duda, J.L. Diffusion in polymer—Solvent systems. I. Reexamination of the freevolume theory. J. Polym. Sci. B Polym. Phys. 1977, 15, 403–416. [Google Scholar] [CrossRef]
- Ramesh, N.; Davis, P.K.; Zielinski, J.M.; Danner, R.P.; Duda, J.L. Application of free-volume theory to self diffusion of solvents in polymers below the glass transition temperature: A review. J. Polym. Sci. B Polym. Phys. 2011, 49, 1629–1644. [Google Scholar] [CrossRef]
- Cohen, M.H.; Turnbull, D. Molecular Transport in Liquids and Glasses. J. Chem. Phys. 1959, 31, 1164–1169. [Google Scholar] [CrossRef]
- Duda, J.L. Molecular diffusion in polymeric systems. Pure Appl. Chem. 1985, 57, 1681–1690. [Google Scholar] [CrossRef]
- Fujita, H. Polymer Solutions, 1st ed.; Elsevier: Amsterdam, The Netherlands, 1990; ISBN 9780444596635. [Google Scholar]
- Yasuda, H.; Lamaze, C.E.; Ikenberry, L.D. Permeability of solutes through hydrated polymer membranes. Part I. Diffusion of sodium chloride. Makromol. Chem. 1968, 118, 19–35. [Google Scholar] [CrossRef]
- Vrentas, J.S.; Duda, J.L.; Ling, H.C. Self Diffusion in Polymer-Solvent-Solvent Systems. J. Polym. Sci. Polym. Phys. Ed. 1984, 22, 459–469. [Google Scholar] [CrossRef]
- Duda, J.L.; Zielinski, J.M. Free-volume theory. In Diffusion in Polymers; Neogi, P., Ed.; Marcel Dekker: New York, NY, USA, 1996; pp. 143–171. [Google Scholar]
- McNaught, A.D.; Wilkinson, A. IUPAC Compendium of Chemical Terminology, 2nd ed.; Blackwell Scientific Publications: Oxford, UK, 1997. [Google Scholar] [CrossRef]
- Albright, J.G.; Mills, R. A study of diffusion in the ternary system labelled urea-urea-water, at 25 °C by measurements of intradiffusion coefficients of urea. J. Phys. Chem. 1965, 69, 3120–3126. [Google Scholar] [CrossRef]
- Vrentas, J.S.; Vrentas, C.M. Evaluation of Free-Volume Theories for Solvent Self-Diffusion in Polymer-Solvent Systems. J. Polym. Sci. Part B Polym. Phys. 1993, 31, 69–76. [Google Scholar] [CrossRef]
- Vrentas, J.S.; Vrentas, C.M. A New Equation Relating Self-Diffusion and Mutual Diffusion Coefficients in Polymer-Solvent Systems. Macromolecules 1993, 26, 6129–6131. [Google Scholar] [CrossRef]
- Cukier, R.I. Diffusion of Brownian spheres in semidilute polymer solutions. Macromolecules 1984, 17, 252–255. [Google Scholar] [CrossRef]
- Crank, J. The Mathematics of Diffusion, 2nd ed.; Calderon Press: Oxford, UK, 1975; ISBN 9780198534112. [Google Scholar]
- Satterfleld, M.B.; Benziger, J.B. Non-Fickian water vapor sorption dynamics by nafion membranes. J. Phys. Chem. B 2008, 112, 3693–3704. [Google Scholar] [CrossRef]
- Mamaliga, I.; Schabel, W.; Kind, M. Measurements of sorption isotherms and diffusion coefficients by means of a magnetic suspension balance. Chem. Eng. Process. Process. Intensif. 2004, 43, 753–763. [Google Scholar] [CrossRef]
- Crank, J.; Park, G.S. Diffusion in Polymers, 1st ed.; Academic Press: London, UK; New York, NY, USA, 1968; ISBN 0121970507. [Google Scholar]
- Burnett, D.J.; Garcia, A.R.; Thielmann, F. Measuring moisture sorption and diffusion kinetics on proton exchange membranes using a gravimetric vapor sorption apparatus. J. Power Sources 2006, 160, 426–430. [Google Scholar] [CrossRef]
- Morris, D.R.; Sun, X. Water-sorption and transport properties of Nafion 117 H. J. Appl. Polym. Sci. 1993, 50, 1445–1452. [Google Scholar] [CrossRef]
- Rivin, D.; Kendrick, C.E.; Gibson, P.W.; Schneider, N.S. Solubility and transport behavior of water and alcohols in Nafion. Polymer 2001, 42, 623–635. [Google Scholar] [CrossRef]
- Yılmaz, L.; Tosun, İ.; Gürkan, T.; Gülçat, Ü. Anomalous diffusion of liquids in glassy polymers. Math. Model. 1983, 4, 535–543. [Google Scholar] [CrossRef]
- Arya, R.K.; Thapliyal, D.; Sharma, J.; Verros, G.D. Glassy Polymers—Diffusion, Sorption, Ageing and Applications. Coatings 2021, 11, 1049. [Google Scholar] [CrossRef]
- Sauer, B.B.; Walsh, D.J. Effect of solvent casting on reduced entanglement density in thin films studied by ellipsometry and neutron reflection. Macromolecules 1994, 27, 432–440. [Google Scholar] [CrossRef]
- Durning, C.J.; Hassan, M.M.; Tong, H.M.; Lee, K.W. A Study of Case II Transport by Laser Interferometry. Macromolecules 1995, 28, 4234–4248. [Google Scholar] [CrossRef]
- Lin, C.B.; Liu, K.S. Methanol-induced_opacity_in_poly_methyl_methacrylate. J. Polym. Sci. Part B Polym. Phys. 1991, 29, 1457–1466. [Google Scholar] [CrossRef]
- Mamaliga, I.; Negoescu, C. Some aspects of two stage diffusion in polymer films and membranes. EEMJ 2012, 11, 2091–2099. [Google Scholar] [CrossRef]
- Fischer, S.; Thümmler, K.; Volkert, B.; Hettrich, K.; Schmidt, I.; Fischer, K. Properties and Applications of Cellulose Acetate. In Macromolecular Symposia; Wiley: Hoboken, NJ, USA, 2008; Volume 262, pp. 89–96. [Google Scholar] [CrossRef]
- Mamaliga, I.; Schabel, W. Diffusion close to Glass Transition Temperature of Dichloromethane in Cellulose Triacetate Films. Cellul. Chem. Technol. 2008, 42, 345–351. [Google Scholar] [CrossRef]
- Fortu, I.O.; Negoescu, C.; Mamaliga, I. Effects of drying conditions on polyvinil alcohol-water and cellulose acetate-tetrahydrofuran films. Cellul. Chem. Technol. 2019, 53, 527–535. [Google Scholar] [CrossRef]
- Valente, A.J.M.; Polishchuk, A.Y.; Burrows, H.D.; Lobo, V.M.M. Permeation of water as a tool for characterizing the effect of solvent, film thickness and water solubility in cellulose acetate membranes. Eur. Polym. J. 2005, 41, 275–281. [Google Scholar] [CrossRef]
- Policastro, S.A.; Anderson, R.M.; Hangarter, C.M.; Arcari, A.; Iezzi, E.B. Experimental and Numerical Investigation into the Effect of Water Uptake on the Capacitance of an Organic Coating. Materials 2023, 16, 3623. [Google Scholar] [CrossRef] [PubMed]
- Negoescu, C. Studies on Drying Process of Polymeric Films and Membranes. Ph.D. Thesis, Technical University of Iasi, Iași, Romania, 2013. [Google Scholar]
- Berens, A.R.; Hopfenberg, H.B. Diffusion and relaxation in glassy polymer powders: 2. Separation of diffusion and relaxation parameters. Polymer 1978, 19, 489–496. [Google Scholar] [CrossRef]
- Neogi, P. Anomalous diffusion of vapours through solid polymers. Am. J. Chem. Eng. 1983, 29, 829–839. [Google Scholar] [CrossRef]
- Perry, K.L.; McDonald, P.J.; Clough, A.S. Case II diffusion in the PVC and acetone system. Magn. Reson. Imaging 1994, 12, 217–219. [Google Scholar] [CrossRef]
- Long, F.A.; Richman, D. Concentration Gradients for Difusion of Vapors in Glassy Polymers and their Relation to Time Dependent Diffusion Phenomena. J. Am. Chem. Soc. 1960, 82, 513–519. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Xm, kg kg−1 | Φ, - | D·1011, m2 s−1 | k·104, - |
|---|---|---|---|
| 0.27 | 0.48 | 8.0 | 4.5 |
| 0.36 | 0.51 | 6.5 | 6.0 |
| 0.50 | 0.72 | 4.5 | 7.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asoltanei, A.M.; Iacob-Tudose, E.T.; Secula, M.S.; Mamaliga, I. Mathematical Models for Estimating Diffusion Coefficients in Concentrated Polymer Solutions from Experimental Data. Processes 2024, 12, 1266. https://doi.org/10.3390/pr12061266
Asoltanei AM, Iacob-Tudose ET, Secula MS, Mamaliga I. Mathematical Models for Estimating Diffusion Coefficients in Concentrated Polymer Solutions from Experimental Data. Processes. 2024; 12(6):1266. https://doi.org/10.3390/pr12061266
Chicago/Turabian StyleAsoltanei, Adriana Mariana, Eugenia Teodora Iacob-Tudose, Marius Sebastian Secula, and Ioan Mamaliga. 2024. "Mathematical Models for Estimating Diffusion Coefficients in Concentrated Polymer Solutions from Experimental Data" Processes 12, no. 6: 1266. https://doi.org/10.3390/pr12061266
APA StyleAsoltanei, A. M., Iacob-Tudose, E. T., Secula, M. S., & Mamaliga, I. (2024). Mathematical Models for Estimating Diffusion Coefficients in Concentrated Polymer Solutions from Experimental Data. Processes, 12(6), 1266. https://doi.org/10.3390/pr12061266