Abstract
This study evaluates radioiodinated anastrozole ([125I]anastrozole) and epirubicin ([125I]epirubicin) for HER2-targeted cancer therapy, utilizing radiopharmaceutical therapy (RPT) for personalized treatment of HER2-positive cancers. Through molecular docking and dynamics simulations (200 ns), it investigates these compounds’ binding affinities and mechanisms to the HER2 receptor compared to lapatinib, a known HER2 inhibitor. Molecular docking studies identified [125I]epirubicin with the highest ΔGbind (−10.92 kcal/mol) compared to lapatinib (−10.65 kcal/mol) and [125I]anastrozole (−9.65 kcal/mol). However, these differences were not statistically significant. Further molecular dynamics (MD) simulations are required to better understand the implications of these findings on the therapeutic potential of the compounds. MD simulations affirmed a stable interaction with the HER2 receptor, indicated by an average RMSD of 4.51 Å for [125I]epirubicin. RMSF analysis pointed to significant flexibility at key receptor regions, enhancing the inhibitory action against HER2. The [125I]epirubicin complex maintained an average of four H-bonds, indicating strong and stable interactions. The average Rg values for [125I]anastrozole and [125I]epirubicin complexes suggest a modest increase in structural flexibility without compromising protein compactness, reflecting their potential to induce necessary conformational changes in the HER2 receptor function. These analyses reveal enhanced flexibility and specific receptor region interactions, suggesting adaptability in binding, which could augment the inhibitory action against HER2. MM-PBSA calculations indicate the potential of these radioiodinated compounds as HER2 inhibitors. Notably, [125I]epirubicin exhibited a free binding energy of −65.81 ± 0.12 kJ/mol, which is comparable to lapatinib at −64.05 ± 0.11 kJ/mol and more favorable than [125I]anastrozole at −57.18 ± 0.12 kJ/mol. The results suggest electrostatic interactions as a major contributor to the binding affinity. The computational analysis underscores that [125I]anastrozole and [125I]epirubicin may have a promising role as HER2 inhibitors, especially [125I]epirubicin due to its high binding affinity and dynamic receptor interactions. These findings, supported by molecular docking scores and MM-PBSA binding energies, advocate for their potential superior inhibitory capability against the HER2 receptor. To validate these computational predictions and evaluate the therapeutic potential of these compounds for HER2-targeted cancer therapy, it is essential to conduct empirical validation through both in vitro and in vivo studies.
1. Introduction
Radiopharmaceutical therapy (RPT) is characterized by the administration of radioactive atoms to targets associated with tumors [1]. This emerging therapeutic technique offers significant benefits over traditional cancer treatments [1]. Distinct from external beam radiotherapy, RPT administers radiation internally, either systemically or within specific regions, similar to the methods of chemotherapy or targeted biological treatments [2]. This method enables the accurate targeting of cytotoxic radiation specifically to cancer cells or their surrounding microenvironment [2]. This is often achieved through carriers that specifically attach to natural targets or are selectively taken up by processes unique to tumor growth, facilitating a precise attack on cancer [3]. In contrast to biological therapies, RPT’s effectiveness does not heavily rely on the comprehensive understanding of cellular signaling pathways or the identification of molecules that disrupt the critical pathways driving cancer [4]. It is important to note that the failure rate for clinical trials of targeted biological cancer treatments is remarkably high at 97% [5], a statistic that underscores the challenges in choosing the correct pathways to target in clinical trials [6].
Exploring further into RPT with an emphasis on targeting the human epidermal growth factor 2 (HER2) receptor, the employment of radiolabeled compounds has been crucial in the evolution of precise cancer treatment strategies [7]. Specifically, the utilization of zirconium−89 (89Zr) with antibodies such as Trastuzumab and Pertuzumab for PET imaging marks a significant advancement, offering a window into the biology of HER2-positive tumors and their response to treatments [8]. These compounds serve as a bridge between diagnostic assessments and therapeutic interventions, empowering healthcare professionals to customize patient care with unprecedented accuracy [7].
The introduction of additional PET imaging agents like copper−64 (64Cu) and gallium−68 (68Ga) [9], along with SPECT tracers including indium−111 (111In) and technetium−99 m (99mTc) [10], has expanded the toolbox available for diagnosing cancers. Moreover, the advent of smaller, precisely targeted entities such as nanobodies, affibodies, and minibodies marks a noteworthy advancement [11]. These smaller molecules facilitate more accurate tumor targeting and quicker elimination from the body, enhancing image quality while minimizing risks to the patient [11]. For example, the affibody molecule ABY−025, known for its minimal uptake in non-targeted organs like the liver, offers clearer imaging results, aiding in the evaluation of treatment efficacy [12]. Its successful labeling with both 68Ga for PET imaging and 111In for SPECT imaging demonstrates its versatility [13]. Additionally, the 99mTc-HYNIC-H6F peptide, known for its specific accumulation in HER2-positive tumors, stands out as a valuable tool for identifying HER2-rich cancer lesions [14].
Beyond imaging, these radiopharmaceuticals are instrumental in therapy, allowing for the direct targeting of cancer cells with radioactive substances. This is exemplified by the therapeutic use of lutetium−177 (177Lu) and yttrium−90 (90Y), which deliver lethal doses of radiation to tumor cells [15]. Such approaches represent a significant shift in cancer treatment philosophy, underlining RPT’s role in fostering personalized and more effective cancer management strategies. The merging of cutting-edge radiopharmaceuticals for both diagnosing and treating HER2-positive cancers signifies major progress in the fields of nuclear medicine and oncology [16]. These developments not only deepen our comprehension of cancer’s biological underpinnings but also set the stage for tailoring patient-specific treatment plans that aim to enhance survival rates and life quality for those afflicted by cancer [16].
Following the advancements in nuclear medicine and oncology, the exploration of [125I]anastrozole and [125I]epirubicin (Figure 1) introduces a new perspective in the targeting of HER2-positive cancers. These compounds, distinguished by their incorporation of radioactive iodine, have shown promising results in solid tumor imaging, highlighting their potential as innovative radiopharmaceutical agents [17]. Achieving high radiochemical yields and demonstrating significant in vitro stability, these agents exhibit favorable biological accumulation in tumor models, marking them as potential candidates for further clinical investigation in targeted cancer imaging [17].
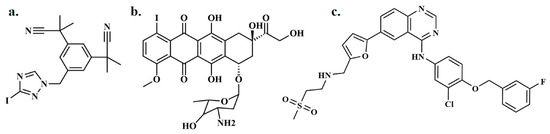
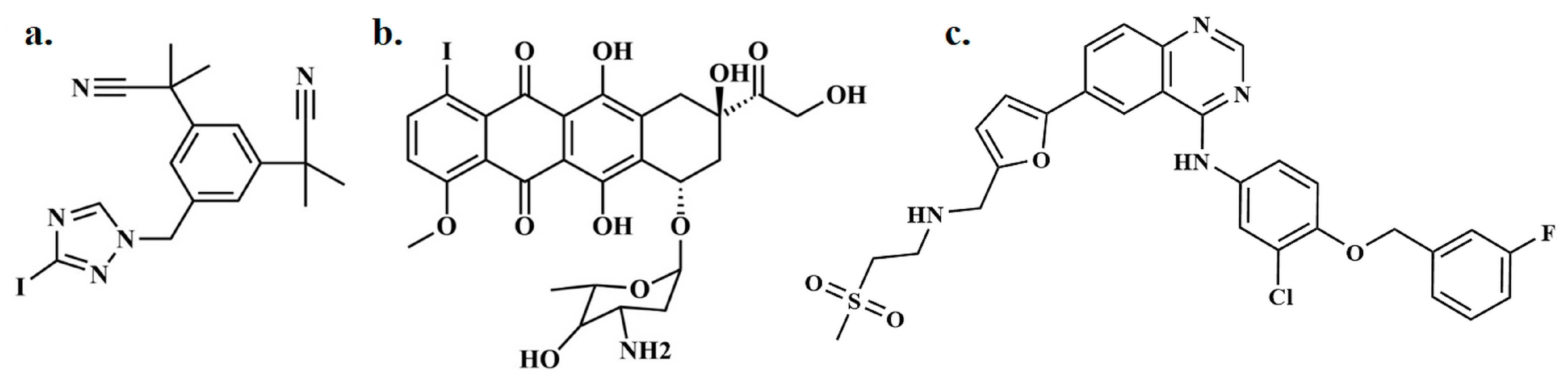
Figure 1.
Depiction of (a) radioiodinated anastrozole ([125I]anastrozole), (b) radioiodinated epirubicin ([125I]epirubicin), and (c) lapatinib.
This study progresses to examine the interactions between these radioiodinated compounds and the HER2 receptor, employing computational techniques to delineate their binding mechanisms. Through molecular docking and dynamics simulations, this research aims to uncover the affinity of [125I]anastrozole and [125I]epirubicin for the HER2 receptor, providing valuable insights into their potential efficacy as diagnostic and therapeutic tools. Lapatinib is a well-known HER2 inhibitor extensively studied and used in clinical settings for treating HER2-positive breast cancer [18]. It inhibits the tyrosine kinase activity associated with the HER2 and EGFR pathways, blocking the downstream signaling that promotes tumor growth and survival [19,20]. The binding constant (Ka) of lapatinib to human serum albumin (HAS) has been reported as 1.24 × 105 M−1 at 303 K, with a range of 1.49–1.01 × 105 M−1 obtained at different temperatures [21,22]. Given its established role and effectiveness, lapatinib serves as a valuable comparator in my study to benchmark the performance of [125I]anastrozole and [125I]epirubicin. This comparison not only helps in assessing the relative efficacy of the new compounds but also provides a deeper understanding of their potential clinical performance.
2. Results
2.1. Molecular Docking Simulation
This study includes molecular docking simulations for [125I]anastrozole, [125I]epirubicin, lapatinib, and the co-crystallized ligand (TAK-285) with the human HER2 receptor (PDB ID: 3RCD). The investigation of TAK-285 was included to validate the AutoDock software’s version 1.5.6 ability to accurately reproduce the known binding pose of this ligand within the HER2 receptor, ensuring reliability with the lowest possible root-mean-square deviation (RMSD) [23,24,25,26,27,28]. This validation step is crucial for confirming the accuracy of the docking parameters, which were then applied to the selected radioiodinated compounds [23,24,25,26,27,28]. The findings are summarized in detail in Table 1 and Figure 2.

Table 1.
Free binding energy (*ΔGbind) and molecular interactions for lapatinib, [125I]anastrozole, [125I]epirubicin, and the co-crystallized ligand TAK-285 within the HER2 receptor’s active site (PDB ID: 3RCD) were analyzed utilizing AutoDock 4.2.
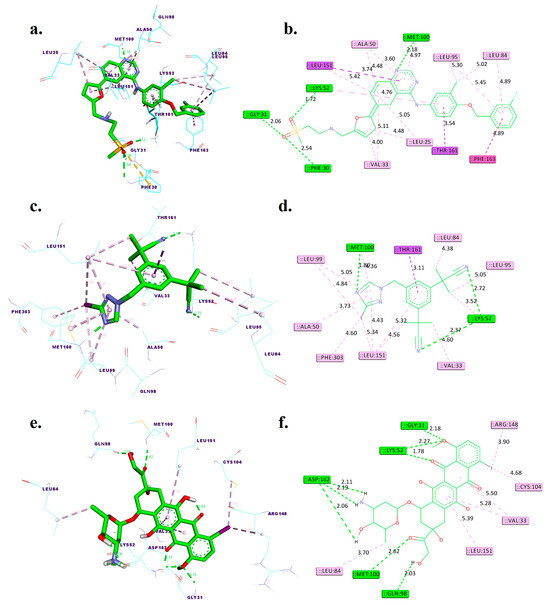
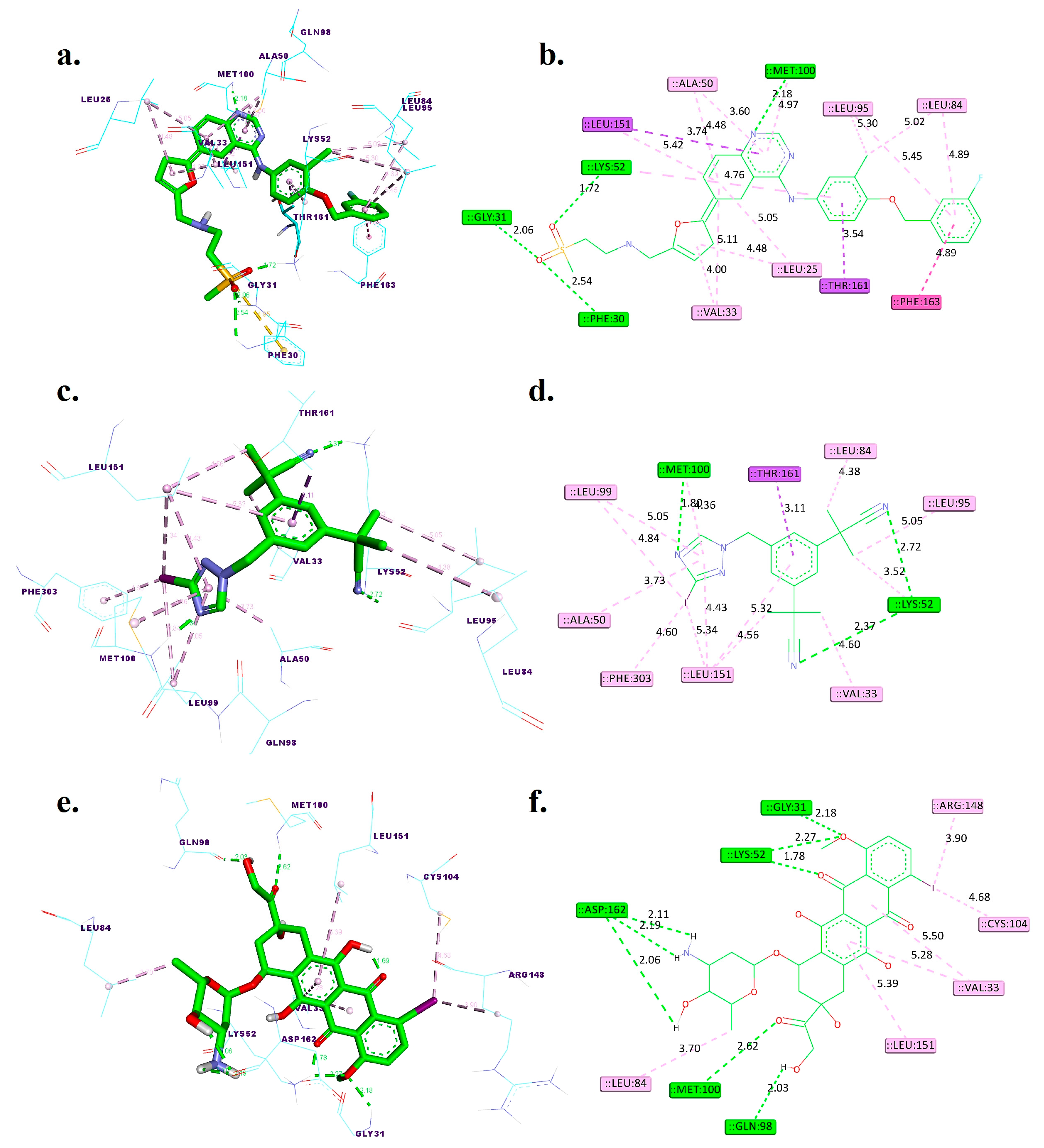
Figure 2.
Three-dimensional and two-dimensional interactions of lapatinib (a,b), [125I]anastrozole (c,d), and [125I]epirubicin (e,f) within the active binding site of the human HER2 receptor (PDB ID: 3RCD). Distances are given in angstroms (Å). Discovery Studio visualizer was used to generate these models.
2.2. Molecular Dynamic Simulation
A 200 ns molecular dynamics simulation was executed using the GROMACS 2016 software package, version 2016.3, to analyze the interaction mechanisms between [125I]anastrozole and [125I]epirubicin and the active site of the HER2 protein. This study compared the interaction profiles of the co-crystallized ligand, TAK-285, and the reference drug, lapatinib. The investigation included a detailed examination of several critical metrics, such as the root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), radius of gyration (Rg), and hydrogen bond interactions, along with MM-PBSA (Molecular Mechanics Poisson–Boltzmann Surface Area) calculations for the ligands’ interactions with the protein complex’s backbone atoms. The results from these in-depth analyses are presented in Figure 3, Figure 4, Figure 5 and Figure 6 and encapsulated within Table 2.
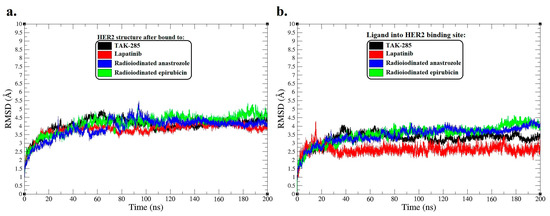
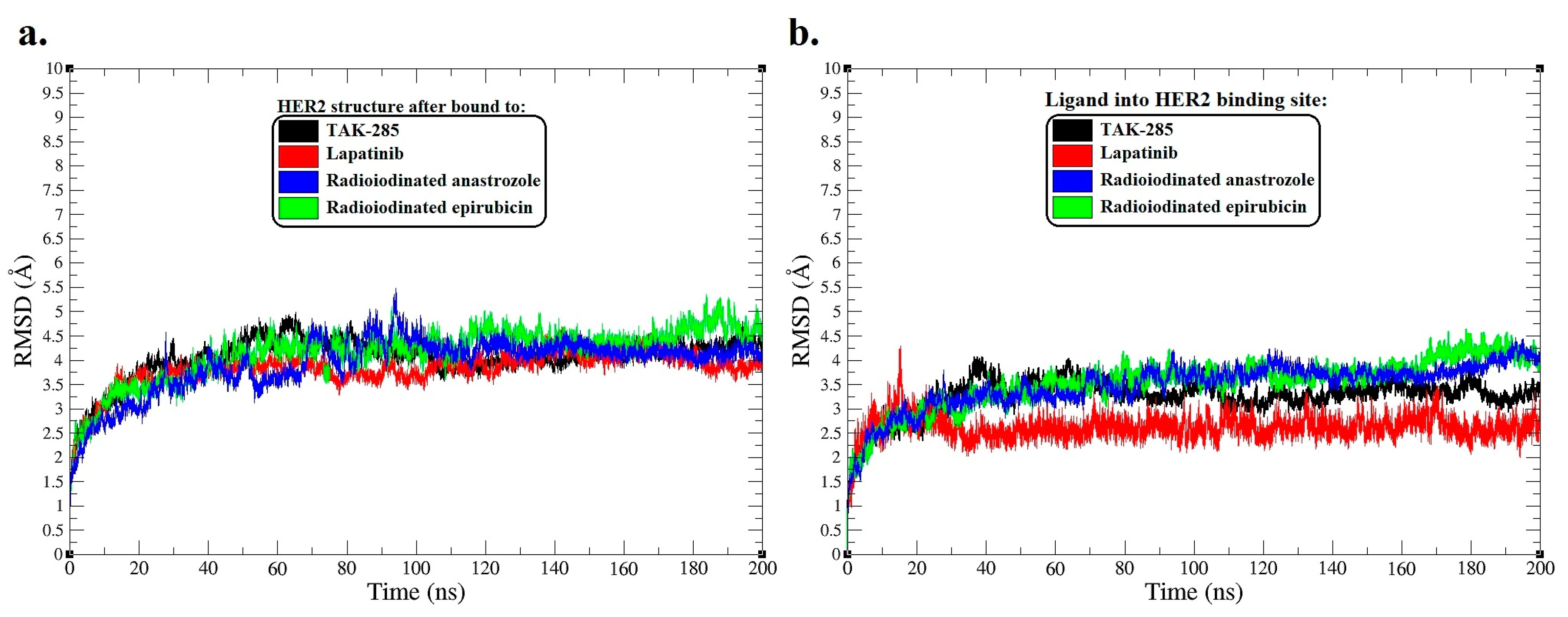
Figure 3.
Root-mean-square deviation (RMSD) analysis for molecular dynamics (MD) simulation trajectories over a span of 200 ns. (a) RMSD plots of the HER2 protein backbone, reflecting molecular variations after interaction with [125I]anastrozole, [125I]epirubicin, lapatinib, and the co-crystallized ligand TAK-285. (b) The RMSD plots also reveal structural modifications of [125I]anastrozole, [125I]epirubicin, lapatinib, and the co-crystallized ligand TAK-285 into the active binding site HER2.
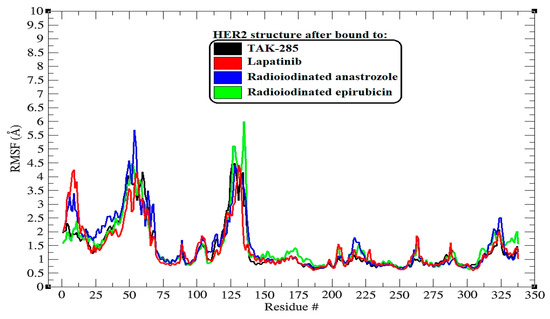
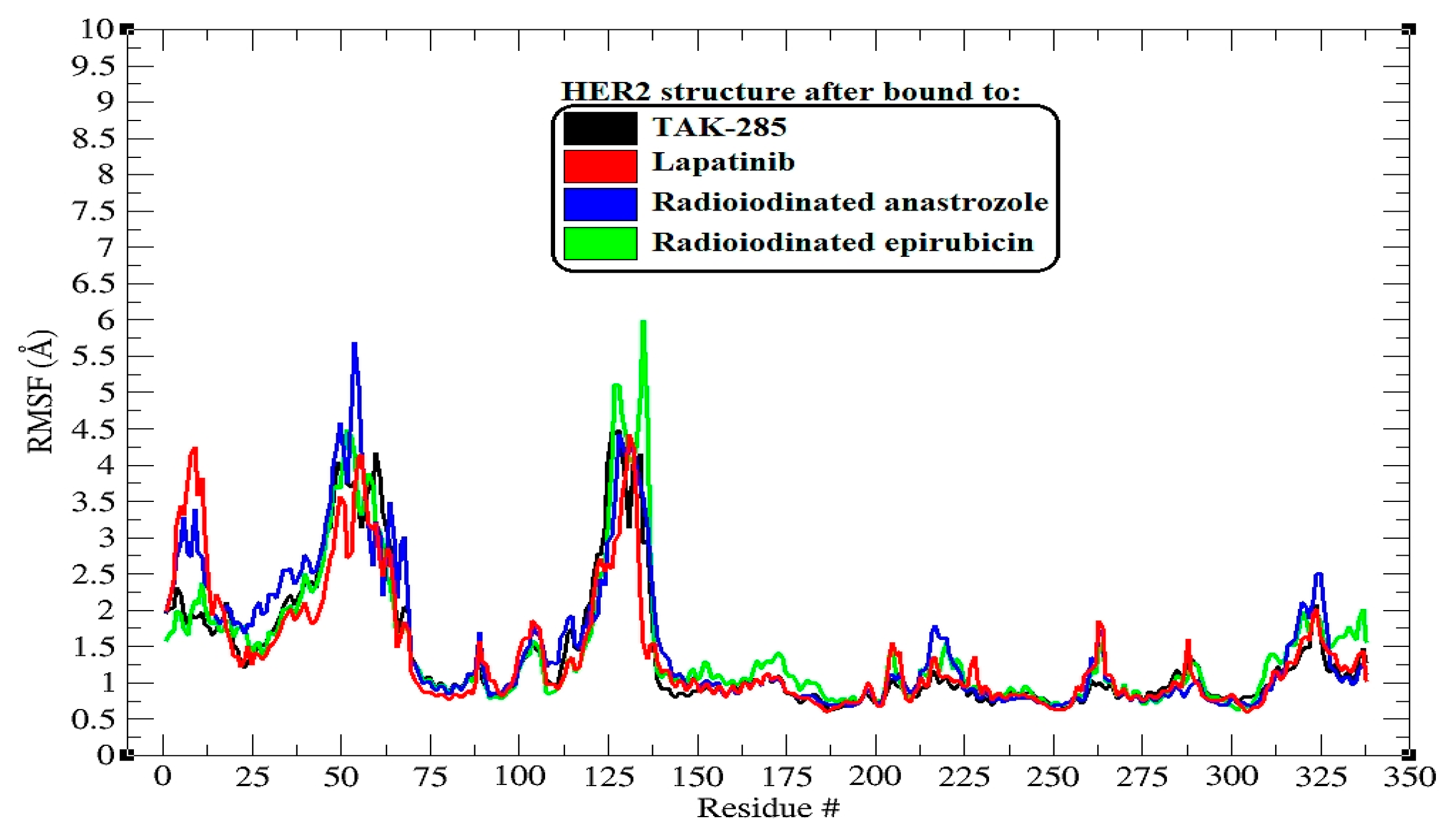
Figure 4.
RMSF (root-mean-square fluctuation) plots illustrating the behavior of backbone atoms in HER2 over a 200 ns MD simulation across all systems. The RMSF data show the fluctuations of individual protein residues as they interact with the ligands throughout the simulation.
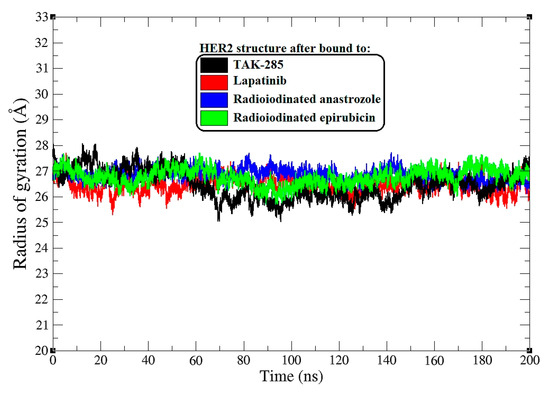
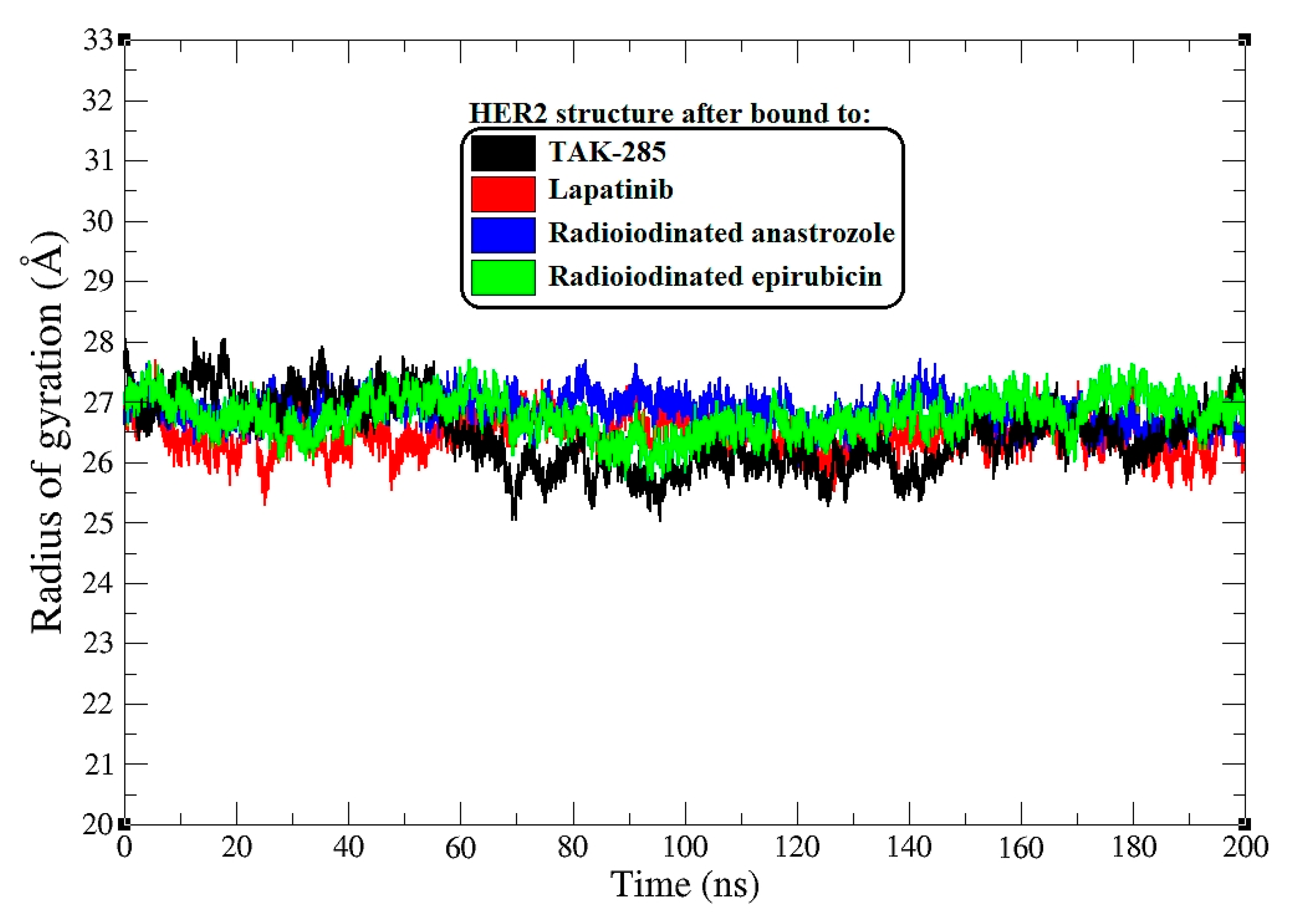
Figure 5.
Radius of gyration (Rg) plots for HER2 backbone atoms across all systems during a 200 ns MD simulation.
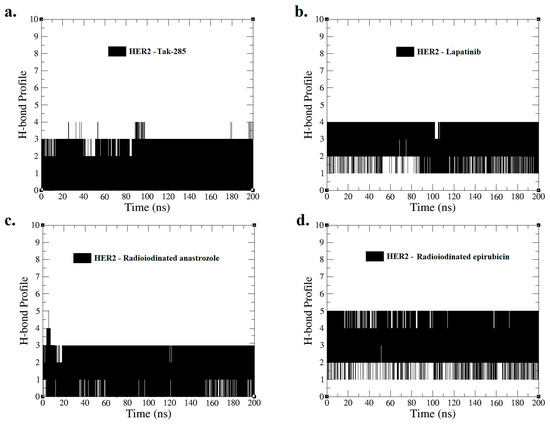
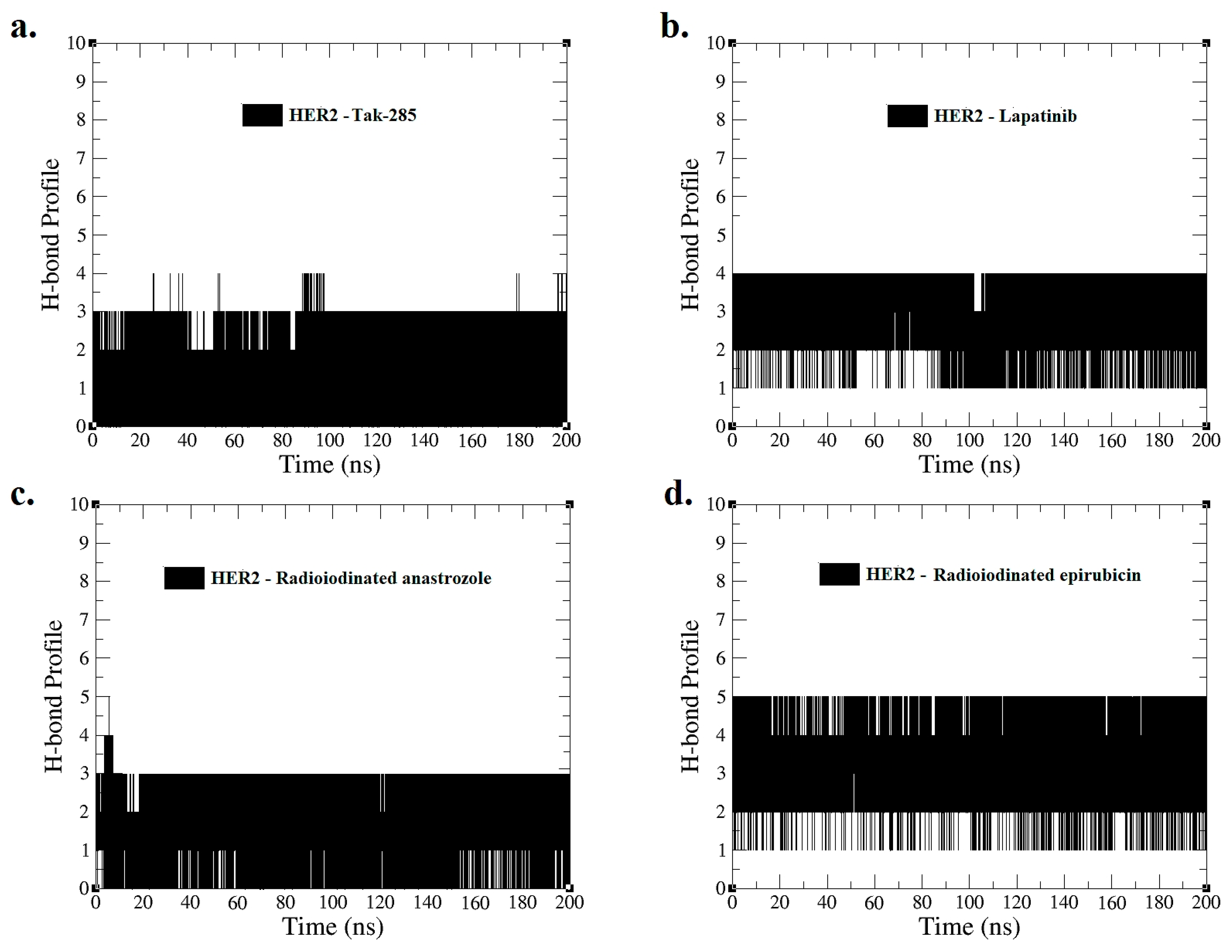
Figure 6.
Hydrogen bond profiles for interactions of (a) HER2–TAK-285, (b) HER2–lapatinib, (c) HER2–[125I]anastrozole, and (d) HER2–[125I]epirubicin were derived from MD simulations spanning 0–200 ns.
Table 2.
MM-PBSA binding energies (ΔGbind) of TAK-285, lapatinib, [125I]anastrozole, and [125I]epirubicin with the HER2 receptor during the final 20 ns of the MD simulation. Binding energies are reported in kJ/mol.
3. Discussion
3.1. Molecular Docking
Molecular docking is a vital computational method used to forecast the interaction between a protein and a small molecule, often resulting in the formation of a stable complex. This technique offers important insights into binding orientations and affinities [29]. It plays a pivotal role in the arena of drug discovery, enabling scientists to investigate the possible inhibitory impacts on specific target enzymes [30]. Redocking the co-crystallized ligand acts as a validation step for the docking parameters, confirming that the computational model can accurately replicate the experimentally observed position of the ligand within the enzyme’s active site [23].
Figure S1 provides a detailed visualization of the docking efficacy of TAK-285 within the active binding site of the human HER2 receptor. Panel (a) illustrates the superimposition of the co-crystallized ligand (in blue) against the re-docked ligand (in orange), highlighting the precision with which the re-docking process was able to replicate the original ligand orientation within the binding pocket. The root-mean-square deviation (RMSD) of 1.42 Å serves as a quantitative metric for this comparison, suggesting a close conformational similarity between the two poses.
Panel (b) and (c) of Figure S1 dissect the interaction profiles of the co-crystallized and re-docked ligands, respectively. The diagrams delineate key interactions, including hydrogen bonds, pi-sigma, pi-pi T-shaped, alkyl, and pi-alkyl interactions, that contribute to the stabilization of the ligand within the enzymatic cleft. Notably, the hydrogen bond between the ligand and SER82 is maintained at a distance of 1.77 Å in the co-crystallized ligand and a slightly longer yet acceptable distance of 1.82 Å in the re-docked ligand, indicating a minor deviation in binding interaction post re-docking.
The preservation of critical interactions, such as the pi-pi T-shaped interaction with MET100 and the alkyl interaction with LEU95, demonstrates the re-docking’s ability to emulate the original binding mode. The interaction distances in the re-docked ligand, ranging from 3.66 Å to 5.97 Å, are within acceptable limits when compared to the co-crystallized ligand, affirming the reliability of the software used for the docking process.
Given the RMSD value of 1.42 Å, the re-docking results are within the generally accepted range for high-quality docking simulations, as established by the literature standards [31,32]. This degree of precision suggests that the docking software can be confidently applied to similar molecular investigations, such as the study of [125I]anastrozole and [125I]epirubicin, and the reference drug (lapatinib) against the HER2 receptor.
Figure 2 and Table 1 illuminate the intricate interactions of lapatinib, [125I]anastrozole, and [125I]epirubicin within the HER2 receptor binding site. This detailed analysis examines each molecule’s binding affinity and interaction profile with a focus on hydrogen bonds and hydrophobic interactions.
Lapatinib showcases a robust binding affinity with a ΔGbind of −10.65 kcal/mol. It forms hydrogen bonds at distances of 2.54 Å with PHE30, 2.06 Å with GLY31, 1.72 Å with LYS52, and 2.18 Å with MET100, indicating strong electrostatic interactions that stabilize the ligand–receptor complex. Additionally, it engages in pi-sigma interactions with LEU151 and THR161. Hydrophobic interactions are noted with LEU25, VAL33, ALA50, LEU84, LEU95, LEU151, and PHE163, which are crucial for the nonpolar binding complementarity.
[125I]Anastrozole, with a ΔGbind of −9.65 kcal/mol, establishes hydrogen bonds with LYS52 at 2.37 Å and 2.72 Å and a notably strong bond with MET100 at 1.80 Å. It also forms a hydrophobic interaction with THR161. The hydrophobic interactions are extensive, involving VAL33, ALA50, LYS52, LEU84, LEU95, LEU99, LEU151, and PHE303, which altogether create an extensive network of nonpolar interactions to stabilize the ligand within the binding site.
[125I]Epirubicin presents the most favorable ΔGbind of −10.92 kcal/mol. It forms a network of hydrogen bonds with GLY31 at 2.18 Å, LYS52 at 1.78 Å and 2.27 Å, GLN98 at 2.03 Å, MET100 at 2.62 Å, and ASP162 at 2.06 Å, 2.11 Å, and 2.19 Å. These distances suggest tight and potentially highly specific binding. It does not form pi-sigma interactions but is involved in hydrophobic interactions with VAL33, LEU84, CYS104, ARG148, and LEU151.
Comparatively, [125I]anastrozole and [125I]epirubicin establish additional hydrogen bonds when compared to the co-crystallized ligand TAK-285, potentially indicating stronger or additional points of receptor engagement. The lack of pi-sigma interactions for [125I]epirubicin, unlike TAK-285, might suggest a different mode of binding, potentially leading to altered pharmacokinetic or pharmacodynamic properties. Additionally, the comparative analysis of these binding pockets reveals that [125I]epirubicin, with its multiple hydrogen bonds and highest ΔGbind, exhibits a binding mode that could potentially lead to more robust and stable interactions. In contrast, [125I]anastrozole, while forming significant hydrogen bonds and hydrophobic interactions, might offer a slightly different interaction profile that could affect its pharmacokinetic properties. These distinctions underscore the importance of evaluating both the number and nature of interactions to fully understand the therapeutic potential of these compounds.
The molecular interaction data propose that [125I]anastrozole and [125I]epirubicin are promising candidates as HER2 receptor inhibitors. Their interaction profiles demonstrate the necessary characteristics for potent and specific receptor–ligand interactions, with the potential to disrupt HER2 signaling in a manner akin to or surpassing that of TAK-285. However, while the binding energies and static interaction profiles are insightful, they represent a snapshot of a dynamic process. To comprehensively understand the behavior and stability of these molecules at the molecular level, molecular dynamics simulations are imperative. Such simulations will provide temporal insights into the flexibility, conformational changes, and resilience of the binding interactions under physiological conditions, which are critical parameters for the successful development of therapeutic agents.
3.2. Molecular Dynamics (MD) Simulation
In the exploration of HER2′s structural dynamics upon ligand binding, RMSD analyses were conducted over a 200 ns molecular dynamics simulation to assess the stability of and conformational changes in the protein–ligand complexes.
The RMSD trajectories for the HER2 protein backbone (Figure 3a) indicate that the complex with the co-crystallized ligand TAK-285 maintained an average RMSD of around 4.2 Å, signifying a stable interaction throughout the simulation. This serves as a stability benchmark for comparing the investigational ligands. The lapatinib-bound HER2 complex demonstrated a similar average RMSD of 3.95 Å, suggesting a comparable degree of conformational stability, which dovetails with its clinical efficacy as an HER2 inhibitor.
Diverging from this trend, the [125I]anastrozole and [125I]epirubicin complexes presented distinct dynamics. The [125I]anastrozole–HER2 complex exhibited an average RMSD value of 4.35 Å, signifying a stable yet slightly more dynamic interaction with the enzyme compared to TAK-285. Conversely, the [125I]epirubicin–HER2 complex displayed a higher average RMSD of 4.51 Å, indicative of a conformation that is notably more flexible, which may have implications for its binding affinity and therapeutic profile.
Correspondingly, the RMSD values of the ligands within the HER2 binding site (Figure 3b) convey further details about their conformational behavior upon binding. TAK-285 exhibited an average RMSD of 3.21 Å, reflecting a consistent and stable ligand conformation throughout the simulation. Lapatinib maintained a robust conformation with an average RMSD of 2.62 Å, underscoring its conformational fidelity within the active site. The radioiodinated compounds, though showing higher average RMSDs of 3.41 Å for anastrozole and 3.49 Å for [125I]epirubicin, remained within a range suggestive of stable binding. However, the episodic peaks observed in their RMSD trajectories may hint at transient conformational shifts that could play a role in their mechanism of action.
The RMSD data collectively highlight the varying stability profiles of the ligand–HER2 complexes. TAK-285 and lapatinib exhibited greater conformational stability within the active binding site of HER2, while the radioiodinated compounds displayed a nuanced balance between stability and conformational flexibility.
The root-mean-square fluctuation (RMSF) analysis extends our understanding of the dynamic interplay between the HER2 protein and the bound ligands, highlighting the flexibility of specific amino acid residues upon ligand binding [33]. The RMSF profiles, as illustrated in Figure 4, are critical for pinpointing regions within the protein that exhibit significant mobility, which may be crucial for the function and ligand interaction of HER2.
Upon binding to TAK-285, the HER2 protein exhibited pronounced flexibility at residues around the 125th position, with the highest peak reaching nearly 4.5 Å. This suggests that this region, possibly a loop or hinge domain, maintains a high degree of dynamic motion that may be intrinsic to the protein’s function or conformational changes upon ligand binding.
The lapatinib–HER2 complex displayed a similar pattern, with notable peaks at residue numbers in the vicinity of 125 and 135, reaching up to approximately 4.4 Å. These peaks may represent regions that are crucial for the structural accommodation of lapatinib, reflecting its mode of interaction with the protein.
In the complexes with [125I]anastrozole and [125I]epirubicin, there were fluctuations notably at residues near (47 to 67 and 125 to 135), with the highest peaks exceeding 5.7 Å and 6.1 Å, respectively. These heightened fluctuations could indicate areas of increased flexibility, which may be influenced by the binding of these radioiodinated compounds, potentially revealing allosteric effects or changes in the protein dynamics that could impact the efficacy of ligand binding.
Despite these localized increases in flexibility, it is apparent that the overall RMSF profiles did not deviate dramatically from the inherent flexibility pattern of the HER2 protein. This observation underscores the ligands’ ability to preserve the structural integrity of HER2 while potentially modulating its dynamic behavior at specific sites.
In a further analysis, I assessed the radius of gyration (Rg) as a measure of protein compactness upon ligand binding [34]. Figure 5 displays the Rg plots for the HER2 backbone atoms throughout the 0–200 ns MD simulation.
The average Rg values provide a quantitative measure of the overall shape and compactness of the protein–ligand complexes. TAK-285-bound HER2 maintained an average Rg of 26.6 Å, suggesting a tightly packed protein structure. Similarly, the lapatinib–HER2 complex showed an almost equivalent average Rg of 26.5 Å, indicating that lapatinib binding preserves the compactness of the HER2 protein.
The [125I]anastrozole–HER2 complex exhibited a slightly higher average Rg of 27.1 Å, pointing to a subtle increase in the protein’s radius of gyration, which may reflect a less compact protein structure. [125I]Epirubicin-bound HER2 showed an average Rg of 26.9 Å, which is closer to the compactness observed in the TAK-285 and lapatinib complexes yet still indicates a slight expansion in the protein structure.
Throughout the simulation, the Rg values fluctuated within a range, with the TAK-285 and lapatinib complexes showing tighter Rg distributions, reflecting less variation in the protein’s compactness over time. In contrast, the radioiodinated compounds exhibited a broader range of Rg values, suggesting a greater degree of structural flexibility. This could potentially correlate with an ability to adapt to different conformational states within the HER2 binding site.
While the Rg provides insight into the global protein structure, the variations within the Rg trajectories also hint at the dynamic nature of these complexes [35]. For instance, peaks and troughs in the Rg plots may correspond to transient expansions and contractions of the protein structure, which could have functional implications in terms of protein–ligand interactions [36].
The radius of gyration analysis complements the RMSD and RMSF findings, painting a picture of the HER2 complexes that are generally compact and maintain structural integrity upon ligand binding. However, the subtle differences in the average Rg values and the range of fluctuations observed for the radioiodinated compounds suggest nuanced differences in how these ligands may influence the protein’s conformational landscape.
The integrity and functionality of protein–ligand interactions are not solely defined by the compactness of the protein structure but also by the specificity and strength of individual interactions, such as hydrogen bonds [37]. As we transition from the analysis of the radius of gyration, which provided us with insights into the overall protein compactness, we now focus on the hydrogen bond (H-bond) profile, which offers a more detailed view of the dynamic interactions at the molecular level.
Figure 6 presents the H-bond profile obtained from the MD simulation for a duration of 0–200 ns, illustrating the interactions of HER2 with (a) TAK-285, (b) lapatinib, (c) [125I]anastrozole, and (d) [125I]epirubicin. The average number of hydrogen bonds between the ligands and the HER2 receptor during the simulation provides a quantitative measure of interaction strength and stability.
The HER2–TAK-285 and HER2–[125I]anastrozole complexes each sustained an average of two H-bonds, suggesting a consistent yet comparatively moderate interaction potential. On the other hand, the HER2–lapatinib complex formed an average of three H-bonds, indicating a potentially stronger and more stable interaction, which aligns with its established clinical profile as a potent HER2 inhibitor.
Notably, the HER2–[125I]epirubicin complex exhibited an average of four H-bonds, the highest among the complexes analyzed. This implies a particularly robust interaction, which may translate to a higher binding affinity and possibly enhanced inhibitory effects on the HER2 receptor.
The temporal H-bond profiles across the simulation period also reflect the dynamic nature of these molecular interactions. Fluctuations in H-bond counts suggest transient states in the binding process, which are essential for understanding the kinetics of ligand binding and release. Complementing these findings, MM-PBSA (Molecular Mechanics Poisson–Boltzmann Surface Area) calculations could offer valuable support by providing insights into the binding free energies of these complexes, reinforcing the significance of the H-bond interactions observed.
Table 2 shows the MM-PBSA binding energies for TAK-285, lapatinib, [125I]anastrozole, and [125I]epirubicin. These energies offer insights into the different energetic factors contributing to the overall binding affinity at the HER2 receptor’s active site.
The binding free energy (ΔGbind) for the HER2–TAK-285 complex was −54.52 ± 0.13 kJ/mol, establishing a reference point for comparing the other ligands. The electrostatic contribution was −27.42 ± 0.14 kJ/mol, while van der Waals interactions contributed −29.68 ± 0.17 kJ/mol, balanced by polar and non-polar solvation energies. The substantial negative values of the electrostatic interactions indicate their significant role in binding affinity.
Lapatinib, used as the reference drug, showed a more favorable binding free energy of −64.05 ± 0.11 kJ/mol. This indicates a stronger binding affinity compared to TAK-285, primarily due to the greater electrostatic interactions, measured at −36.36 ± 0.13 kJ/mol. The van der Waals interactions were slightly lower than those of TAK-285, yet the overall balance of forces resulted in a stronger interaction.
[125I]Anastrozole showed a ΔG binding of −57.18 ± 0.12 kJ/mol, with electrostatic interactions contributing −31.25 ± 0.14 kJ/mol to this energy. Though its binding energy was more favorable than that of TAK-285, it was less so compared to lapatinib. The energy components reveal that while the electrostatic interactions significantly contributed, they were less pronounced than those in the lapatinib complex.
The HER2–[125I]epirubicin complex demonstrated the most favorable binding energy of −65.81 ± 0.12 kJ/mol. The major contributor to this strong binding was the electrostatic component at −37.44 ± 0.17 kJ/mol, surpassing even that of lapatinib and suggesting a potent interaction with the receptor. The van der Waals component was comparable to the other ligands, and the balanced solvation energies support a strong overall binding affinity.
The MM-PBSA binding energy analysis reveals that although all the ligands show potential as HER2 inhibitors, [125I]epirubicin exhibited the highest binding affinity, with lapatinib closely following. The investigational ligands, particularly [125I]epirubicin, indicate potential as effective HER2 inhibitors due to their favorable binding free energies and substantial electrostatic interactions. Although these computational predictions are encouraging, binding affinity alone does not fully determine a compound’s potential as an inhibitor. It is crucial to validate these in silico results with in vitro and in vivo studies to confirm the therapeutic efficacy and safety of these compounds. These studies will verify the investigational ligands’ inhibitory effects on the HER2 receptor and assess their suitability for clinical development. Additionally, future work will include the application of quantum mechanics/molecular mechanics (QM/MM) methods to further investigate charge transferability between residues and ligands at the binding site, providing deeper insights into the binding interactions and enhancing our understanding of the system.
4. Conclusions
This investigation into [125I]anastrozole and [125I]epirubicin as potential radiopharmaceuticals targeting HER2-positive cancers has provided critical insights into their binding interactions, stability, and inhibitory capabilities against the HER2 receptor. This study uniquely combined molecular docking and dynamics simulations to evaluate these new compounds, comparing their efficacy with the established HER2 inhibitor lapatinib.
The molecular docking results revealed [125I]epirubicin as having the highest binding affinity (−10.92 kcal/mol), outperforming lapatinib (−10.65 kcal/mol) and indicating strong inhibitory potential. [125I]Anastrozole also showed promising affinity (−9.65 kcal/mol), suggesting its capability as an HER2 inhibitor. The molecular dynamics simulations supported these findings, demonstrating stable interactions with the HER2 receptor, highlighted by RMSD values of 4.51 Å for [125I]epirubicin and 4.35 Å for [125I]anastrozole, indicating a balanced mix of flexibility and stability crucial for effective binding. The RMSF analysis identified regions of increased flexibility, suggesting these compounds could induce beneficial conformational changes within the HER2 receptor. Notably, the average number of hydrogen bonds formed by [125I]epirubicin was the highest among the complexes, emphasizing its strong interaction with the receptor. The radius of gyration (Rg) values pointed to slight increases in structural flexibility without compromising the overall compactness of the protein–ligand complexes.
The MM-PBSA calculations underscored the superior binding energy of [125I]epirubicin (−65.81 kJ/mol), with significant contributions from electrostatic interactions, suggesting a potent interaction with HER2. [125I]Anastrozole also exhibited favorable binding energy (−57.18 kJ/mol), reinforcing its potential as a therapeutic agent. Overall, these findings advocate for the therapeutic viability of [125I]anastrozole and [125I]epirubicin as HER2 inhibitors, with [125I]epirubicin showing particularly compelling evidence of efficacy. These results, while promising, underscore the need for empirical validation through in vitro and in vivo studies to confirm the therapeutic potential and safety profile of these compounds in the clinical development of targeted cancer therapy.
5. Materials and Methods
5.1. Molecular Docking Preparation
In this research, the focal point was the HER2 receptor, analyzed in relation to its interaction with the potent inhibitor TAK-285, referenced in the literature [38]. The selection of HER2-positive cancer as the subject of this study stems from its significant presence in certain cases of breast cancer and the critical role that the receptor’s inhibition plays in regulating cancer cell growth and viability [39,40]. I obtained crystallographic details of the HER2 receptor complexed with TAK-285 from the Protein Data Bank (PDB) [41], accession number 3RCD, with the dataset retrieved on 10 January 2024. For comparative analysis, the co-crystalized inhibitor TAK-285 and lapatinib, a renowned orally administered medication for breast cancer and other solid malignancies, were employed as reference compounds.
To prepare the protein target for molecular docking, various pre-processing steps were conducted to ensure its suitability for analysis. This involved removing unnecessary water molecules and heteroatoms using the Biovia Discovery Studio Visualizer [42], resulting in a refined protein structure in PDB format. Missing amino acids in the enzyme’s structure were added using the YASARA web server [31,43,44]. The ionization states of titratable amino acids were calculated at a neutral pH of 7.4 using the H++ web server [45]. Polar hydrogen atoms and Kollman charges were then added, converting the structure to PDBQT format for further analysis using AutoDock Tools version 1.5.6 [46].
This study also focused on the preparation and optimization of ligands for molecular docking simulations to enhance the accuracy and reliability of the results. The ChemDraw JS web server [31] was used to create the [125I]anastrozole and [125I]epirubicin molecules, based on structures from previous studies [17], and these were saved in structural data file (SDF) format. The ligands were subjected to energy minimization using the universal force field and a conjugate gradient optimization algorithm, as described in reference [47]. This process, performed over a thousand iterations, was carried out using the Open Babel software version 2.3 [48], and the optimized structures were saved in PDB format. Gasteiger charges were assigned using AutoDock Tools version 1.5.6, preparing the structures for docking simulations in PDBQT format.
To elucidate the interactions between the ligands and the active site of HER2, molecular docking was executed employing AutoDock 4.2 [46]. This phase employed a Lamarckian genetic algorithm to optimize the ligand configurations within the binding site, permitting ligand flexibility while keeping the macromolecule stationary [49]. The grid box dimensions were set to 40 × 40 × 40 Å along the X, Y, and Z axes, with the central grid coordinates set at 12.48, 2.96, and 28.01. The docking procedure was conducted over 100 trials with a maximum of 25,000,000 evaluations per run, adhering to default parameters to ensure uniformity in the analyses.
5.2. Molecular Dynamics
This study focused on analyzing the interactions and stability of [125I]anastrozole and [125I]epirubicin within the active sites of the HER2 receptor using 200 ns MD simulations. The starting structure for these simulations was the human crystallographic form of the HER2 kinase domain complexed with the inhibitor TAK-285. Using the GROMACS 2016.3 software package and the Gromos96 54a7 force field, we conducted a comprehensive analysis of ligand–protein interactions [50]. These 200 ns simulations provided valuable insights into the binding dynamics and molecular stability within the receptor sites.
To begin the simulations, topology files were created for both the ligands and the protein. Ligand topology was prepared using the GROMACS ‘pdb2gmx’ function, while protein topology was generated via the PRODRG server, accessed on 23 January 2024 [http://davapc1.bioch.dundee.ac.uk/cgi-bin/prodrg]. The simulation systems were then solvated in a TIP3P water model and neutralized with counterions to ensure balanced conditions for interaction studies. Energy minimization was subsequently performed using the steepest descent method for 50,000 steps, each quantified at 0.01 energy units, to achieve optimal system stability.
System equilibration was performed in two stages: initially using the NVT ensemble for 100 picoseconds at 310 Kelvin with the v-rescale thermostat, followed by the NPT ensemble for an additional 100 picoseconds at 1.0 bar using the Berendsen pressure coupling method [33,51]. These steps ensured proper stabilization of the systems for the subsequent MD simulations. The simulations were then run for 200 ns at a constant temperature of 310 K and pressure of 1 bar. Short-range non-bonded interactions were managed with a 1.0 nm cut-off, while the Particle Mesh Ewald method handled long-range electrostatics [52]. The LINCS algorithm was employed to constrain bonds involving hydrogen atoms, maintaining structural integrity [53]. Utilizing a timestep of 2 fs and coordinate resets every 5000 steps (10 ps), this methodology ensured the precision and reliability of the simulation results [31,32].
I employed a variety of analytical techniques to assess the accuracy and stability of ligand–enzyme interactions. By analyzing MD trajectory data through RMSD, RMSF, radius of gyration (Rg), and hydrogen bond profiling, I demonstrated my dedication to obtaining precise and insightful conclusions regarding the molecular behavior being investigated.
5.3. Molecular Mechanics Poisson–Boltzmann Surface Area (MM-PBSA) Calculations
Within the scope of this investigation, the Molecular Mechanics Poisson–Boltzmann Surface Area (MM-PBSA) methodology was employed, a technique renowned for its precision in quantifying binding free energies derived from molecular dynamics (MD) simulations. My attention was particularly directed towards the production phase of these simulations to ascertain the binding free energies between the HER2 receptor and each of the following ligands: [125I]anastrozole, [125I]epirubicin, lapatinib, and TAK-285.
Throughout the duration of these simulations, snapshots were methodically taken at 100 ps intervals, specifically in the span from 180 to 200 ns. This process was facilitated by the utilization of the g_mmpbsa tool, a component of the Gromacs software suite, as per references [54,55]. The core objective of this approach was to calculate the binding free energy within the solvent environment for the ligand–protein complexes. This calculation was conducted by deducing the sum of the individual total energies of the protein (Gprotein) and the ligand (Gligand) in their separate solvent states from the total free energy of the unified protein–ligand complex (Gcomplex). The formula employed for this calculation is as follows [56]:
ΔGbind = [Gcomplex − Gprotein + Gligand]
In this analysis, the determination of free energy for each unique state—namely, the complex, protein, and ligand—relied on an aggregate of the average molecular mechanics potential energy in vacuum conditions (EMM) alongside the solvation free energy (Gsolvation), as delineated in the following equation [55]:
Gx = EMM + Gsolvation
This formula encapsulates the energetic assessment critical for understanding the thermodynamic properties of molecular interactions. The computation of a molecule’s potential energy in a vacuum is an intricate process that aggregates energies derived from bonded (Ebonded) and non-bonded (Enon-bonded) interactions. More precisely, Ebonded accounts for energy contributions emanating from bonds, angles, dihedrals, and improper interactions, forming a foundational component of the molecular mechanics potential. Conversely, Enon-bonded comprises the cumulative energy from electrostatic (Eelec) and van der Waals (Evdw) forces, as represented in the following equation [55]:
EMM = Ebonded + Enon_bonded = Ebonded + [Evdw + Eelec]
Furthermore, the solvation free energy is divided into two main categories: the non-polar solvation free energy (Gnon-polar) and the electrostatic solvation free energy (Gpolar), which are integrated as follows [55]:
Gsolvation = Gpolar + Gnon-polar
To accurately calculate the electrostatic solvation free energy (Gpolar), this study employed the Poisson–Boltzmann equation [57], a sophisticated approach that encapsulates the electrostatic interactions within a solvent. In parallel, the estimation of the non-polar solvation free energy (Gnon-polar) utilizes the solvent-accessible surface area (SASA) method, adhering to the following formula:
Gnon-polar = γSASA + b
In the conducted research, two specific coefficient pairs, γ and b, were utilized to estimate the solvation free energy, as specified in Equation (5). When performing simulations with energy units expressed as kcal/mol, I applied the coefficients γ= 0.0054 kcal/mol·Å2 and b = 0.916 kcal/mol. Alternatively, in the case of simulations utilizing energy units of kJ/mol, the coefficients were chosen as γ= 0.02267 kJ/mol·Å2 and b= 3.849 kJ/mol. The selection between these coefficient groups depended on the chosen unit system for the simulations. Notably, the assumption made in these calculations was that the change in the bonded energy ΔEbonded was nil [58]. The computation of the binding free energies is crucial, as it provides deep insights into the energetic interactions that regulate the binding processes between the protein and ligand molecules, thereby playing a key role in the discovery of potential therapeutic agents.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/pr12081659/s1, Figure S1. (a) Superimposition and (b) 2D interaction analysis of the co-crystallized ligand TAK-285 (depicted in blue for carbon (C), red for oxygen (O), navy for nitrogen (N), and cyan for fluorine (F)). (c) Re-docked ligand (depicted in orange for carbon (C), red for oxygen (O), navy for nitrogen (N), and cyan for fluorine (F)) in the crystal structure of the human HER2 receptor complexed with TAK-285 (PDB ID: 3RCD).
Funding
This research received no external funding.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding author/s.
Acknowledgments
The author wishes to express his gratitude to Imam Mohammad Ibn Saud Islamic University (IMSIU) for their unending support of this research.
Conflicts of Interest
The author declares no conflicts of interest.
References
- Sgouros, G.; Dewaraja, Y.K.; Escorcia, F.; Graves, S.A.; Hope, T.A.; Iravani, A.; Pandit-Taskar, N.; Saboury, B.; James, S.S.; Zanzonico, P.B. Tumor response to radiopharmaceutical therapies: The knowns and the unknowns. J. Nucl. Med. 2021, 62, 12S–22S. [Google Scholar] [PubMed]
- Salerno, K.E.; Roy, S.; Ribaudo, C.; Fisher, T.; Patel, R.B.; Mena, E.; Escorcia, F.E. A primer on radiopharmaceutical therapy. Int. J. Radiat. Oncol. Biol. Phys. 2023, 115, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Khan, F.; Mishra, R.K.; Khan, R. Precision cancer nanotherapy: Evolving role of multifunctional nanoparticles for cancer active targeting. J. Med. Chem. 2019, 62, 10475–10496. [Google Scholar] [CrossRef]
- Park, J.H.; de Lomana, A.L.G.; Marzese, D.M.; Juarez, T.; Feroze, A.; Hothi, P.; Cobbs, C.; Patel, A.P.; Kesari, S.; Huang, S. A systems approach to brain tumor treatment. Cancers 2021, 13, 3152. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.H.; Siah, K.W.; Lo, A.W. Estimation of clinical trial success rates and related parameters. Biostatistics 2019, 20, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Giuliano, C.J.; Palladino, A.; John, K.M.; Abramowicz, C.; Yuan, M.L.; Sausville, E.L.; Lukow, D.A.; Liu, L.; Chait, A.R. Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci. Transl. Med. 2019, 11, eaaw8412. [Google Scholar] [CrossRef] [PubMed]
- Miladinova, D. Molecular imaging of HER2 receptor: Targeting HER2 for imaging and therapy in nuclear medicine. Front. Mol. Biosci. 2023, 10, 1144817. [Google Scholar] [CrossRef] [PubMed]
- Melendez-Alafort, L.; Ferro-Flores, G.; De Nardo, L.; Ocampo-García, B.; Bolzati, C. Zirconium immune-complexes for PET molecular imaging: Current status and prospects. Coord. Chem. Rev. 2023, 479, 215005. [Google Scholar] [CrossRef]
- Morgan, K.A.; Rudd, S.E.; Noor, A.; Donnelly, P.S. Theranostic Nuclear Medicine with Gallium-68, Lutetium-177, Copper-64/67, Actinium-225, and Lead-212/203 Radionuclides: Focus Review. Chem. Rev. 2023, 123, 12004–12035. [Google Scholar] [CrossRef]
- Crișan, G.; Moldovean-Cioroianu, N.S.; Timaru, D.-G.; Andrieș, G.; Căinap, C.; Chiș, V. Radiopharmaceuticals for PET and SPECT imaging: A literature review over the last decade. Int. J. Mol. Sci. 2022, 23, 5023. [Google Scholar] [CrossRef]
- Herrero Álvarez, N.; Bauer, D.; Hernández-Gil, J.; Lewis, J.S. Recent advances in radiometals for combined imaging and therapy in cancer. ChemMedChem 2021, 16, 2909–2941. [Google Scholar] [CrossRef] [PubMed]
- Rinne, S.S.; Orlova, A.; Tolmachev, V. PET and SPECT imaging of the EGFR family (RTK class I) in oncology. Int. J. Mol. Sci. 2021, 22, 3663. [Google Scholar] [CrossRef]
- Gawne, P.J.; Man, F.; Blower, P.J.; TM de Rosales, R. Direct cell radiolabeling for in vivo cell tracking with PET and SPECT imaging. Chem. Rev. 2022, 122, 10266–10318. [Google Scholar] [CrossRef]
- Wu, Y.; Li, L.; Wang, Z.; Shi, J.; Hu, Z.; Gao, S.; Miao, W.; Ma, Q.; Dong, C.; Wang, F. Imaging and monitoring HER2 expression in breast cancer during trastuzumab therapy with a peptide probe 99m Tc-HYNIC-H10F. Eur. J. Nucl. Med. 2020, 47, 2613–2623. [Google Scholar] [CrossRef] [PubMed]
- Bober, B.; Saracyn, M.; Zaręba, K.; Lubas, A.; Mazurkiewicz, P.; Wilińska, E.; Kamiński, G. Early complications of radioisotope therapy with lutetium-177 and yttrium-90 in patients with neuroendocrine neoplasms—A preliminary study. J. Clin. Med. 2022, 11, 919. [Google Scholar] [CrossRef] [PubMed]
- Dhoundiyal, S.; Srivastava, S.; Kumar, S.; Singh, G.; Ashique, S.; Pal, R.; Mishra, N.; Taghizadeh-Hesary, F. Radiopharmaceuticals: Navigating the frontier of precision medicine and therapeutic innovation. Eur. J. Med. Res. 2024, 29, 26. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.; Sakr, T.; Khoweysa, O.; Motaleb, M.; Abd El-Bary, A.; El-Kolaly, M. Radioiodinated anastrozole and epirubicin as potential targeting radiopharmaceuticals for solid tumor imaging. J. Radioanal. Nucl. Chem. 2015, 303, 967–975. [Google Scholar] [CrossRef]
- Elwaie, T.A.; Abbas, S.E.; Aly, E.I.; George, R.F.; Ali, H.; Kraiouchkine, N.; Abdelwahed, K.S.; Fandy, T.E.; El Sayed, K.A.; Abd Elmageed, Z.Y. HER2 kinase-targeted breast cancer therapy: Design, synthesis, and in vitro and in vivo evaluation of novel lapatinib congeners as selective and potent HER2 inhibitors with favorable metabolic stability. J. Med. Chem. 2020, 63, 15906–15945. [Google Scholar] [CrossRef] [PubMed]
- Xuhong, J.-C.; Qi, X.-W.; Zhang, Y.; Jiang, J. Mechanism, safety and efficacy of three tyrosine kinase inhibitors lapatinib, neratinib and pyrotinib in HER2-positive breast cancer. Am. J. Cancer Res. 2019, 9, 2103. [Google Scholar]
- Shi, H.; Zhang, W.; Zhi, Q.; Jiang, M. Lapatinib resistance in HER2+ cancers: Latest findings and new concepts on molecular mechanisms. Tumor Biol. 2016, 37, 15411–15431. [Google Scholar] [CrossRef]
- Kabir, M.Z.; Mukarram, A.K.; Mohamad, S.B.; Alias, Z.; Tayyab, S. Characterization of the binding of an anticancer drug, lapatinib to human serum albumin. J. Photochem. Photobiol. B Biol. 2016, 160, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Lainez, G.; El Ouardi, M.; Moreno, A.; Lence, E.; González-Bello, C.; Miranda, M.A.; Andreu, I. Singlet oxygen and radical-mediated mechanisms in the oxidative cellular damage photosensitized by the protease inhibitor simeprevir. Free. Radic. Biol. Med. 2023, 194, 42–51. [Google Scholar] [CrossRef]
- Alhawarri, M.B.; Al-Thiabat, M.G.; Dubey, A.; Tufail, A.; Fouad, D.; Alrimawi, B.H.; Dayoob, M. ADME profiling, molecular docking, DFT, and MEP analysis reveal cissamaline, cissamanine, and cissamdine from Cissampelos capensis Lf as potential anti-Alzheimer’s agents. RSC Adv. 2024, 14, 9878–9891. [Google Scholar] [CrossRef] [PubMed]
- Alhawarri, M.B.; Olimat, S. Potential Serotonin 5-HT2A Receptor Agonist of Psychoactive Components of Silene undulata Aiton: LC-MS/MS, ADMET, and Molecular Docking Studies. Curr. Pharm. Biotechnol. 2024. [Google Scholar] [CrossRef] [PubMed]
- Yunos, N.M.; Al-Thiabat, M.G.; Sallehudin, N.J. Quassinoids from Eurycoma longifolia as Potential Dihydrofolate Reductase Inhibitors: A Computational Study. Curr. Pharm. Biotechnol. 2024, 25, 2154–2165. [Google Scholar] [CrossRef] [PubMed]
- Alhawarri, M.B.; Dianita, R.; Rawa, M.S.A.; Nogawa, T.; Wahab, H.A. Potential Anti-Cholinesterase Activity of Bioactive Compounds Extracted from Cassia grandis Lf and Cassia timoriensis DC. Plants 2023, 12, 344. [Google Scholar] [CrossRef]
- Larue, L.; Kenzhebayeva, B.; Al-Thiabat, M.G.; Jouan–Hureaux, V.; Mohd–Gazzali, A.; Wahab, H.A.; Boura, C.; Yeligbayeva, G.; Nakan, U.; Frochot, C. tLyp–1: A peptide suitable to target NRP–1 receptor. Bioorg. Chem. 2023, 130, 106200. [Google Scholar] [CrossRef]
- Amir Rawa, M.S.; Al-Thiabat, M.G.; Nogawa, T.; Futamura, Y.; Okano, A.; Wahab, H.A. Naturally Occurring 8ß, 13ß-kaur-15-en-17-al and Anti-Malarial Activity from Podocarpus polystachyus Leaves. Pharmaceuticals 2022, 15, 902. [Google Scholar] [CrossRef]
- De Ruyck, J.; Brysbaert, G.; Blossey, R.; Lensink, M.F. Molecular docking as a popular tool in drug design, an in silico travel. Adv. Appl. Bioinform. Chem. 2016, 9, 1–11. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Shalayel, M.H.F.; Al-Mazaideh, G.M.; Alanezi, A.A.; Almuqati, A.F.; Alotaibi, M. Diosgenin and Monohydroxy Spirostanol from Prunus amygdalus var amara Seeds as Potential Suppressors of EGFR and HER2 Tyrosine Kinases: A Computational Approach. Pharmaceuticals 2023, 16, 704. [Google Scholar] [CrossRef]
- Shalayel, M.H.F.; Al-Mazaideh, G.M.; Alanezi, A.A.; Almuqati, A.F.; Alotaibi, M. The Potential Anti-Cancerous Activity of Prunus amygdalus var. amara Extract. Processes 2023, 11, 1277. [Google Scholar] [CrossRef]
- Berendsen, H.J.; Postma, J.V.; Van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Lobanov, Y.; Bogatyreva, N.; Galzitskaya, O. Radius of gyration as an indicator of protein structure compactness. Mol. Biol. 2008, 42, 623–628. [Google Scholar] [CrossRef]
- Yan, C.; Dodd, T.; He, Y.; Tainer, J.A.; Tsutakawa, S.E.; Ivanov, I. Transcription preinitiation complex structure and dynamics provide insight into genetic diseases. Nat. Struct. Mol. Biol. 2019, 26, 397–406. [Google Scholar] [CrossRef]
- Chang, Y.-P.; Mahadeva, R.; Patschull, A.O.; Nobeli, I.; Ekeowa, U.I.; McKay, A.R.; Thalassinos, K.; Irving, J.A.; Haq, I.; Nyon, M.P. Targeting serpins in high-throughput and structure-based drug design. Methods Enzymol. 2011, 501, 139–175. [Google Scholar]
- Papaleo, E.; Saladino, G.; Lambrughi, M.; Lindorff-Larsen, K.; Gervasio, F.L.; Nussinov, R. The role of protein loops and linkers in conformational dynamics and allostery. Chem. Rev. 2016, 116, 6391–6423. [Google Scholar] [CrossRef]
- Ishikawa, T.; Seto, M.; Banno, H.; Kawakita, Y.; Oorui, M.; Taniguchi, T.; Ohta, Y.; Tamura, T.; Nakayama, A.; Miki, H. Design and synthesis of novel human epidermal growth factor receptor 2 (HER2)/epidermal growth factor receptor (EGFR) dual inhibitors bearing a pyrrolo [3, 2-d] pyrimidine scaffold. J. Med. Chem. 2011, 54, 8030–8050. [Google Scholar] [CrossRef]
- Lodi, M.; Voilquin, L.; Alpy, F.; Molière, S.; Reix, N.; Mathelin, C.; Chenard, M.-P.; Tomasetto, C.-L. STARD3: A New Biomarker in HER2-Positive Breast Cancer. Cancers 2023, 15, 362. [Google Scholar] [CrossRef]
- Zimmer, A.S.; Van Swearingen, A.E.; Anders, C.K. HER2-positive breast cancer brain metastasis: A new and exciting landscape. Cancer Rep. 2022, 5, e1274. [Google Scholar] [CrossRef]
- Westbrook, J.; Feng, Z.; Chen, L.; Yang, H.; Berman, H.M. The protein data bank and structural genomics. Nucleic Acids Res. 2003, 31, 489–491. [Google Scholar] [CrossRef] [PubMed]
- Biovia, D.S. Discovery Studio Visualizer; Biovia: San Diego, CA, USA, 2017; p. 936. [Google Scholar]
- Land, H.; Humble, M.S. YASARA: A tool to obtain structural guidance in biocatalytic investigations. Protein Eng. 2018, 1685, 43–67. [Google Scholar]
- Abdelbagi, M.E.; Al-Mazaideh, G.M.; Ahmed, A.E.; Al-Rimawi, F.; Ayyal Salman, H.; Almutairi, A.; Abuilaiwi, F.A.; Wedian, F. Exploring Securigera securidaca Seeds as a Source of Potential CDK1 Inhibitors: Identification of Hippeastrine and Naringenin as Promising Hit Candidates. Processes 2023, 11, 1478. [Google Scholar] [CrossRef]
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufriev, A. H++: A server for estimating p K as and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005, 33, W368–W371. [Google Scholar] [CrossRef] [PubMed]
- Norgan, A.P.; Coffman, P.K.; Kocher, J.-P.A.; Katzmann, D.J.; Sosa, C.P. Multilevel parallelization of AutoDock 4.2. J. Cheminform. 2011, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo-Botero, A.; Naserifar, S.; Goddard, W.A., III. General multiobjective force field optimization framework, with application to reactive force fields for silicon carbide. J. Chem. Theory Comput. 2014, 10, 1426–1439. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, J.; Rurainski, A.; Lenhof, H.P.; Neumann, D. A new Lamarckian genetic algorithm for flexible ligand-receptor docking. J. Comput. Chem. 2010, 31, 1911–1918. [Google Scholar] [CrossRef] [PubMed]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; Van Gunsteren, W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843–856. [Google Scholar] [CrossRef]
- Rühle, V. Pressure coupling/barostats. J. Club. 2008, 19, 1–5. [Google Scholar]
- Petersen, H.G. Accuracy and efficiency of the particle mesh Ewald method. J. Chem. Phys. 1995, 103, 3668–3679. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Consortium, O.S.D.D.; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Verma, S.; Grover, S.; Tyagi, C.; Goyal, S.; Jamal, S.; Singh, A.; Grover, A. Hydrophobic interactions are a key to MDM2 inhibition by polyphenols as revealed by molecular dynamics simulations and MM/PBSA free energy calculations. PLoS ONE 2016, 11, e0149014. [Google Scholar] [CrossRef]
- Woo, H.-J.; Roux, B. Calculation of absolute protein–ligand binding free energy from computer simulations. Proc. Natl. Acad. Sci. USA 2005, 102, 6825–6830. [Google Scholar] [CrossRef]
- Forli, S.; Olson, A.J. A force field with discrete displaceable waters and desolvation entropy for hydrated ligand docking. J. Med. Chem. 2012, 55, 623–638. [Google Scholar] [CrossRef]
- Homeyer, N.; Gohlke, H. Free energy calculations by the molecular mechanics Poisson− Boltzmann surface area method. Mol. Inform. 2012, 31, 114–122. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).