

Research on the Influence of a Magnesium-Based Carbon Dioxide Battery System on CO2 Storage Performance

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. The Reaction Mechanism of the Mg-CO2 Battery
2.2. Preparation of CO2 Battery Materials
- (1)
- CeO2 carrier preparation
- (2)
- Pd/CeO2 catalyst preparation
- (3)
- Pd/CeO2 cathode coating
2.3. Characterization
3. Results and Discussion
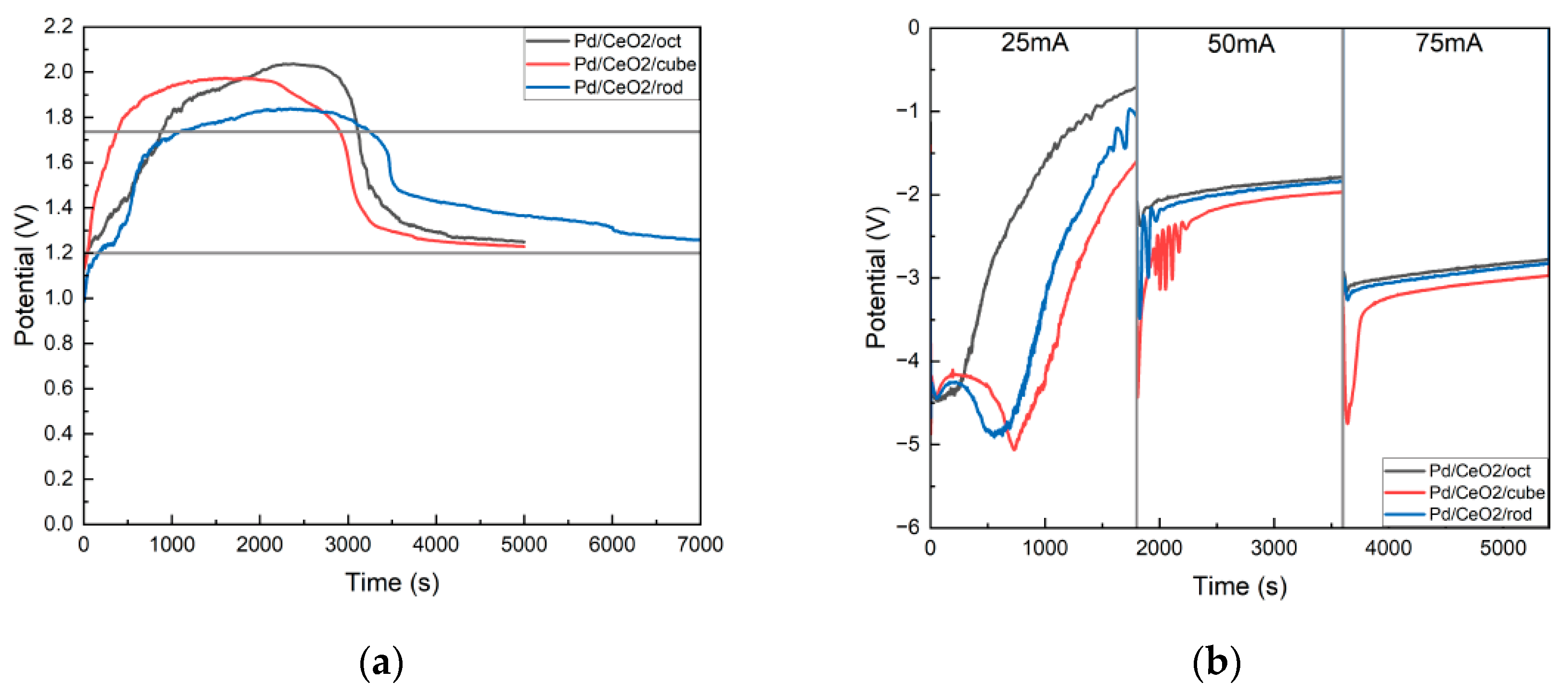
3.1. The Effect of the Catalytic Electrode on the Conversion Efficiency of Mg-CO2 Batteries with Molecules
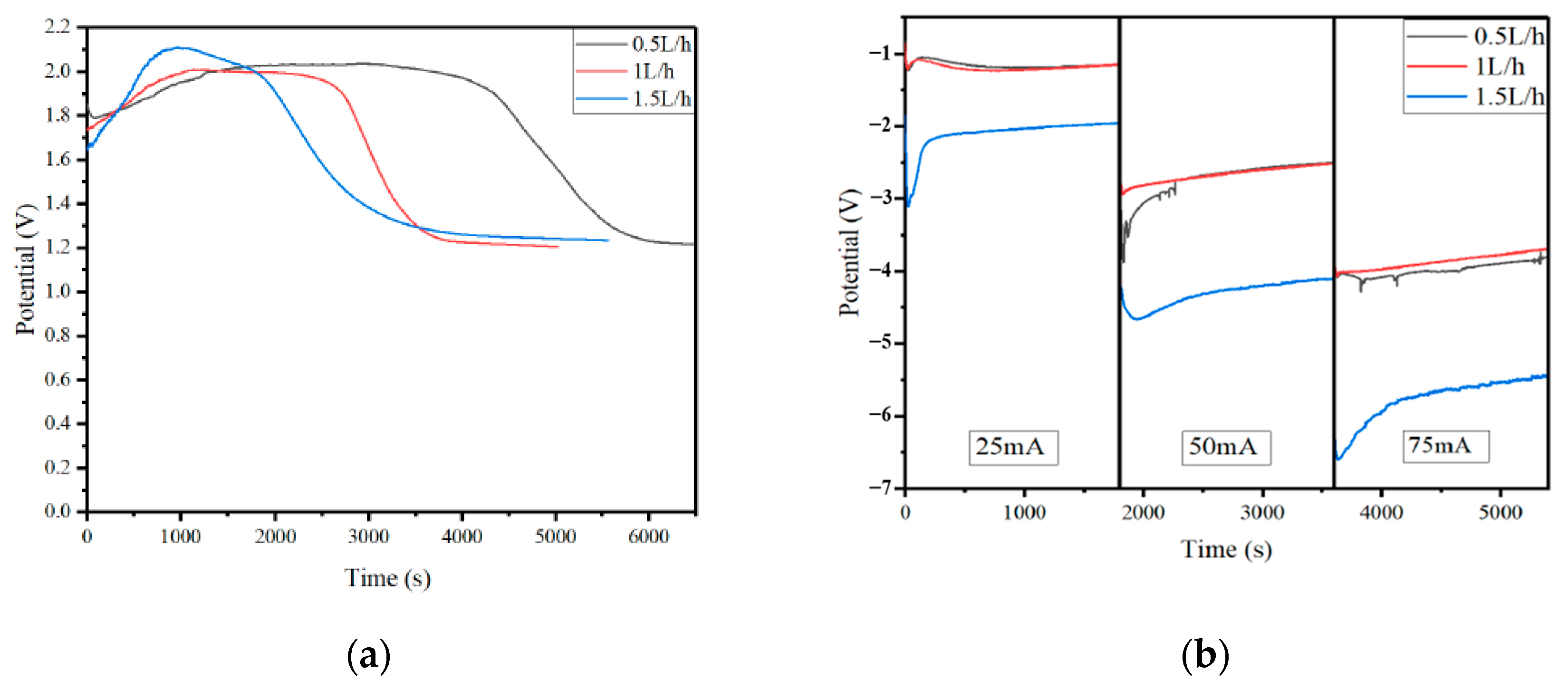
3.2. The Effect of the Electrolyte Concentration on the Conversion Efficiency of the CO2 Molecules
3.3. The Effect of CO2 Flow Change on the Conversion Efficiency of CO2 Molecules
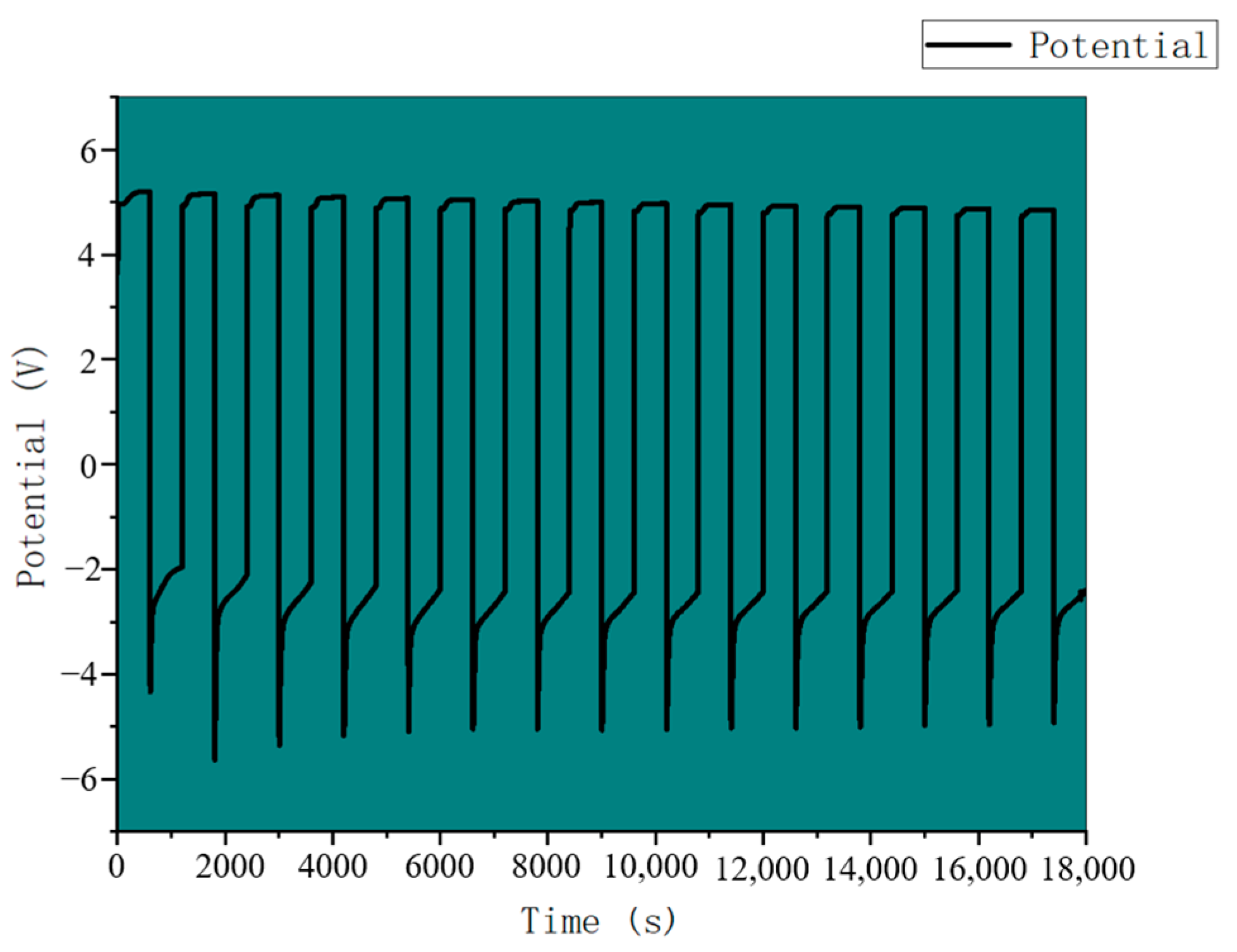
3.4. CO2 Battery System Performance Analysis
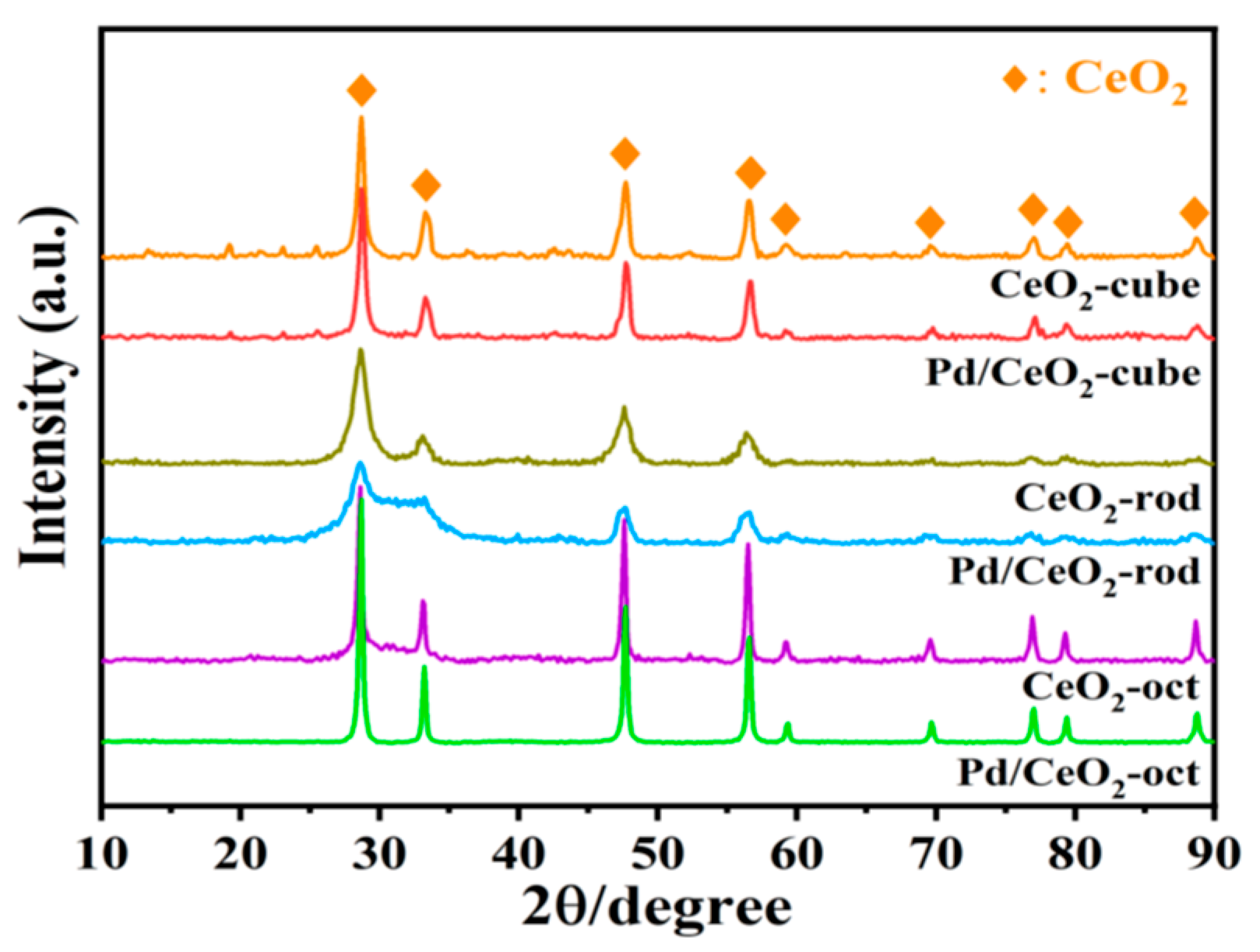
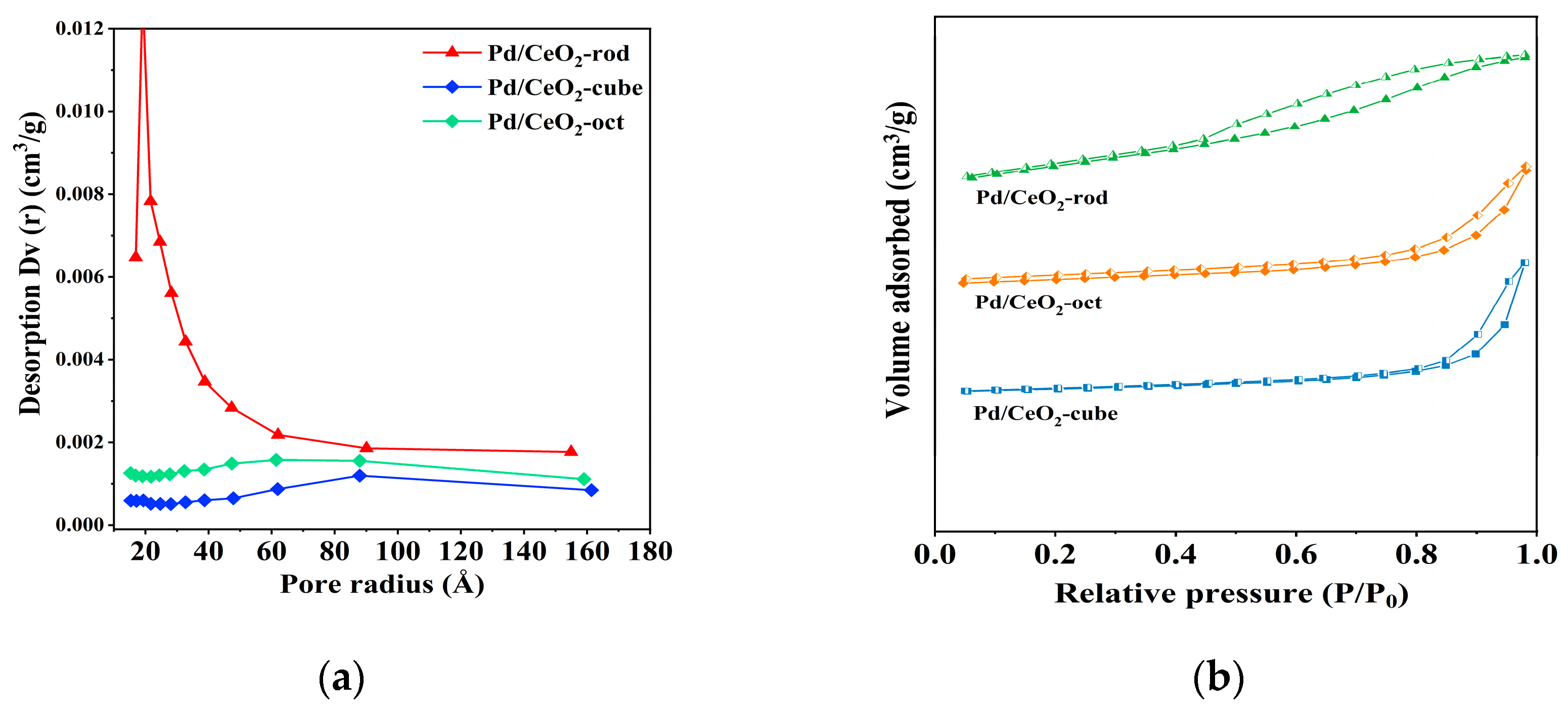
3.5. Characterization of Catalyst Structure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- BP. BP Statistical Review of World Energy 2022; BP p.l.c.: London, UK, 2022. [Google Scholar]
- Marbán, G.; Valdés-Solís, T. Towards the hydrogen economy? Int. J. Hydrogen Energy 2007, 32, 1625–1637. [Google Scholar] [CrossRef]
- Mao, Z.; Ma, A.; Zhang, Z. Towards carbon neutrality in shipping: Impact of European Union’s emissions trading system for shipping and China’s response. Ocean. Coast. Manag. 2024, 249, 107006. [Google Scholar] [CrossRef]
- IPCC. Climate Change 2021—The Physical Science Basis. Chem. Int. 2021, 43, 22–23. [Google Scholar] [CrossRef]
- Xie, Z.; Zhang, X.; Zhang, Z. Metal–CO2 batteries on the road: CO2 from contamination gas to energy source. Adv. Mater. 2017, 29, 1605891. [Google Scholar] [CrossRef]
- Folger, P. Carbon Capture and Sequestration (CCS) in the United States; Congressional Research Service: Washington, DA, USA, 2017. [Google Scholar]
- Czernichowski-Lauriol, I.; Berenblyum, R.; Bigi, S.; Car, M.; Liebscher, A.; Persoglia, S.; Poulsen, N.; Stead, R.; Vercelli, S.; Vincent, C.J.; et al. CO2GeoNet Perspective on CO2 Capture and Storage: A Vital Technology for Completing the Climate Change Mitigation Portfolio. Energy Procedia 2017, 114, 7480–7491. [Google Scholar] [CrossRef]
- Hua, W.; Sha, Y.; Zhang, X.; Cao, H. Research Progress of Carbon Capture and Storage (CCS) Technology Based on the Shipping Industry. Ocean. Eng. 2023, 281, 114929. [Google Scholar] [CrossRef]
- Abanades, J.C.; Rubin, E.S.; Mazzotti, M. On the climate change mitigation potential of CO2 conversion to fuels. Energy Environ. Sci. 2017, 10, 2491–2499. [Google Scholar] [CrossRef]
- Vega, F.; Baena-Moreno, F.M.; Fernández, L.M.G.; Portillo, E.; Navarrete, B.; Zhang, Z. Current Status of CO2 Chemical Absorption Research Applied to CCS: Towards Full Deployment at Industrial Scale. Appl. Energy 2020, 260, 114313. [Google Scholar]
- Xie, J.; Wang, X.; Lv, J.; Huang, Y.; Wu, M.; Wang, Y.; Yao, J.Q. Frontispiece: Reversible Aqueous Zinc–CO2 Batteries Based on CO2–HCOOH Interconversion. Angew. Chem. 2018, 57, 16996–17001. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Kim, J.; Joo, S.; Bu, Y.; Liu, M.; Cho, J.; Kim, G. Efficient CO2 Utilization via a Hybrid Na-CO2 System Based on CO2 Dissolution. iScience 2018, 9, 278–285. [Google Scholar] [CrossRef]
- Kim, J.; Seong, A.; Yang, Y.; Joo, S.; Kim, C.; Jeon, D.H.; Dai, L.; Kim, G. Indirect surpassing CO2 utilization in membrane-free CO2 battery. Nano Energy 2021, 82, 105741. [Google Scholar] [CrossRef]
- Takechi, K.; Shiga, T.; Asaoka, T. A Li–O2/CO2 battery. Chem. Commun. 2011, 47, 3463–3465. [Google Scholar] [CrossRef] [PubMed]
- Subbaraman, R.; Tripkovic, D.; Strmcnik, D.; Chang, K.C.; Uchimura, M.; Paulikas, A.P.; Stamenkovic, V.; Markovic, N.M. Enhancing Hydrogen Evolution Activity in Water Splitting by Tailoring Li+-Ni(OH)2-Pt Interfaces. Science 2011, 334, 1256–1260. [Google Scholar] [CrossRef]
- Morales-Guio, C.G.; Stern, L.-A.; Hu, X. Nanostructured hydrotreating catalysts for electrochemical hydrogen evolution. Chem. Soc. Rev. 2014, 43, 6555–6569. [Google Scholar] [CrossRef] [PubMed]
- Green, T.; Britz, D. Kinetics of the deuterium and hydrogen evolution reactions at palladium in alkaline solution. J. Electroanal. Chem. 1996, 412, 59–66. [Google Scholar] [CrossRef]
- Andonoglou, P.P.; Jannakoudakis, A.D. Palladium deposition on activated carbon fibre supports and electrocatalytic activity of the modified electrodes towards the hydrogen evolution reaction. Electrochim. Acta 1997, 42, 1905–1913. [Google Scholar] [CrossRef]
- Li, J.; Zhou, P.; Li, F.; Ren, R.; Liu, Y.; Niu, J.; Ma, J.; Zhang, X.; Tian, M.; Jin, J.; et al. Ni@Pd/PEI–rGO stack structures with controllable Pd shell thickness as advanced electrodes for efficient hydrogen evolution. J. Mater. Chem. A 2015, 3, 11261–11268. [Google Scholar] [CrossRef]
- Kukunuri, S.; Austeria, P.M.; Sampath, S. Electrically conducting palladium selenide (Pd4Se, Pd17Se15, Pd7Se4) phases: Synthesis and activity towards hydrogen evolution reaction. Chem. Commun. 2016, 52, 206–209. [Google Scholar] [CrossRef]
- Cargnello, M.; Doan-Nguyen, V.V.T.; Gordon, T.R.; Diaz, R.E.; Stach, E.A.; Gorte, R.J.; Fornasiero, P.; Murray, C.B. Control of Metal Nanocrystal Size Reveals Metal-Support Interface Role for Ceria Catalysts. Science 2013, 341, 771–773. [Google Scholar] [CrossRef]
- Lewera, A.; Timperman, L.; Roguska, A. Metal–support interactions between nanosized Pt and metal oxides (WO3 and TiO2) studied using X-ray photoelectron spectroscopy. J. Phys. Chem. C 2011, 115, 20153–20159. [Google Scholar] [CrossRef]
- Li, Z.Y.; Liu, Z.; Liang, J.C. Facile synthesis of Pd–Mn3O4/C as high-efficient electrocatalyst for oxygen evolution reaction. J. Materials Chem. A 2014, 2, 18236–18240. [Google Scholar] [CrossRef]
- Pradhan, D.; Su, Z.; Sindhwani, S.; Honek, J.F.; Leung, K.T. Electrochemical growth of ZnO nanobelt-like structures at 0 C: Synthesis, characterization, and in-situ glucose oxidase embedment. J. Phys. Chem. C 2011, 115, 18149–18156. [Google Scholar] [CrossRef]
- Titkov, A.I.; Salanov, A.N.; Koscheev, S.V. Mechanisms of Pd(1 1 0) surface reconstruction and oxidation: XPS, LEED and TDS study. Surf. Sci. 2006, 600, 4119–4125. [Google Scholar] [CrossRef]
- Spiel, C.; Blaha, P.; Suchorski, Y. CeO2/Pt(111) interface studied using first-principles density functional theory calculations. Phys. Rev. B 2011, 84, 045412. [Google Scholar] [CrossRef]
- Peng, C.; Xue, L.; Zhao, Z.; Guo, L.; Zhang, C.; Wang, A.; Mao, J.; Dou, S.X.; Guo, Z. Boosted Mg−CO2 Batteries by Amide-Mediated CO2 Capture Chemistry and Mg2+-Conducting Solid-Electrolyte Interphases. Angew. Chemie 2023, 136, e202313264. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, A.; Guo, L.; Yi, J.; Luo, J. A Moisture-Assisted Rechargeable Mg-CO2 Battery. Angew. Chem. 2022, 61, e202200181. [Google Scholar] [CrossRef]
- Wei, S.; Shi, H.; Li, X.; Hu, X.; Xu, C.; Wang, X. A Green and Efficient Method for Preparing Graphene Using CO2@Mg In-Situ Reaction and Its Application in High-Performance Lithium-Ion Batteries. J. Alloys Compd. 2022, 902, 163700. [Google Scholar] [CrossRef]
- Miller, H.A.; Lavacchi, A.; Vizza, F. A Pd/C-CeO2 Anode Catalyst for High-Performance Platinum-Free Anion Exchange Membrane Fuel Cells. Angew. Chemie 2016, 55, 6004–6007. [Google Scholar] [CrossRef]
- Miller, H.A.; Vizza, F.; Marelli, M.; Zadick, A.; Dubau, L.; Chatenet, M.; Geiger, S.; Cherevko, S.; Doan, H.; Pavlicek, R.K.; et al. Highly active nanostructured palladium-ceria electrocatalysts for the hydrogen oxidation reaction in alkaline medium. Nano Energy 2017, 33, 293–305. [Google Scholar] [CrossRef]
- Wright, S.E. Comparison of the Theoretical Performance Potential of Fuel Cells and Heat Engines. Renew. Energy 2004, 29, 179–195. [Google Scholar] [CrossRef]
- Cabello, M.; Medina, A.; Alcántara, R.; Nacimiento, F.; Pérez-Vicente, C.; Tirado, J.L. A Theoretical and Experimental Study of Hexagonal Molybdenum Trioxide as Dual-Ion Electrode for Rechargeable Magnesium Battery. J. Alloys Compd. 2020, 831, 154795. [Google Scholar] [CrossRef]
- Yamada, Y.; Wang, J.; Ko, S.; Watanabe, E.; Yamada, A. Advances and Issues in Developing Salt-Concentrated Battery Electrolytes. Nat. Energy 2019, 4, 269–280. [Google Scholar] [CrossRef]
- Guo, X.; Li, Y.; Chen, N.; Wang, X.; Xie, Q. Construction of Sustainable Release Antimicrobial Microspheres Loaded with Potassium Diformate. J. Inorg. Mater. 2021, 36, 181–187. [Google Scholar] [CrossRef]
- Duan, J.; Chen, S.; Zhao, C. Ultrathin Metal-Organic Framework Array for Efficient Electrocatalytic Water Splitting. Nat. Commun. 2017, 8, 15341. [Google Scholar] [CrossRef]
- Ikäläinen, S.; Lantto, P.; Vaara, J. Fully Relativistic Calculations of Faraday and Nuclear Spin-Induced Optical Rotation in Xenon. J. Chem. Theory Comput. 2011, 8, 91–98. [Google Scholar] [CrossRef]
- Ciuparu, D.; Lyubovsky, M.R.; Altman, E.; Pfefferle, L.D.; Datye, A. CATALYTIC COMBUSTION OF METHANE OVER PALLADIUM-BASED CATALYSTS. Catal. Rev. 2002, 44, 593–649. [Google Scholar] [CrossRef]
- Xu, X.; Shuai, K.; Xu, B. Review on Copper and Palladium Based Catalysts for Methanol Steam Reforming to Produce Hydrogen. Catalysts 2017, 7, 183. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, R.J.; Mo, A.C.; Chen, Z.Q.; Wu, H.K. Synthesis and Characterization of Yttrium/Hydroxyapatite Nanoparticles. Key Eng. Mater. 2007, 330–332, 295–298. [Google Scholar] [CrossRef]
- Yan, L.-G.; Yang, K.; Shan, R.-R.; Yan, T.; Wei, J.; Yu, S.-J.; Yu, H.-Q.; Du, B. Kinetic, Isotherm and Thermodynamic Investigations of Phosphate Adsorption onto Core–Shell Fe3O4@LDHs Composites with Easy Magnetic Separation Assistance. J. Colloid Interface Sci. 2015, 448, 508–516. [Google Scholar] [CrossRef]
- Chalgin, A.; Song, C.; Tao, P.; Shang, W.; Deng, T.; Wu, J. Effect of Supporting Materials on the Electrocatalytic Activity, Stability and Selectivity of Noble Metal-Based Catalysts for Oxygen Reduction and Hydrogen Evolution Reactions. Prog. Nat. Sci. 2020, 30, 289–297. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Item | Chemistry | Specifications | Manufacturer |
|---|---|---|---|---|
| 1 | Palladium nitrate | Pd(NO3)2·2H2O | 17.27 wt% | Guyan chem., Kunming, China |
| 2 | Cerium(III) nitrate | Ce(NO3)3·6H2O | AR | Macklin, Shanghai, China |
| 3 | Potassium hydroxide | KOH | 95% | Macklin, Shanghai, China |
| 4 | Sodium hydroxide | NaOH | 95% | Macklin, Shanghai, China |
| 5 | Carbon dioxide | CO2 | 99.999% | Peric, Handan, China |
| 6 | Nafion perfluorinated resin solution | - | 5 wt% | Macklin, Shanghai, China |
| 7 | Ethanol | CH3CH2OH | 99.5% | Macklin, Shanghai, China |
| 8 | Porous carbon nanotube | C | 97.5% | Macklin, Shanghai, China |
| 9 | VulcanXC-72 Carbon Black | C | 99.5% | CABOT, Boston, MA, USA |
| 10 | Magnesium flake | Mg | - | Huabei Metal, Wuxi, China |
| 11 | Nickel Foam Sheet | Ni | - | Suzhou Metal, Suzhou, China |
| 12 | Polyvinylpyrrolidone | PVP | AR | Macklin, Shanghai, China |
| 13 | Trisodium phosphate | Na3PO4 | 96% | Macklin, Shanghai, China |
| NaOH Concentration (mol/L) | CO2 Flow Rate (L/h) | Complete Reaction Generation NaHCO3 Time (min) |
|---|---|---|
| 0.5 | 1 | 107.52 |
| 0.1 | 21.50 | |
| 0.05 | 10.75 |
| CO2 Traffic (L/h) | NaOH Potency (mol/L) | Complete Reaction Time to Generate NaHCO3 (min) |
|---|---|---|
| 0.5 | 0.05 | 21.5 |
| 1 | 10.75 | |
| 1.5 | 7.17 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, H.; Wei, M.; Wang, B.; Wang, L.; Chen, Q.; Su, C.; Feng, Y.; Wang, X.; Li, K. Research on the Influence of a Magnesium-Based Carbon Dioxide Battery System on CO2 Storage Performance. Processes 2024, 12, 1896. https://doi.org/10.3390/pr12091896
Yang H, Wei M, Wang B, Wang L, Chen Q, Su C, Feng Y, Wang X, Li K. Research on the Influence of a Magnesium-Based Carbon Dioxide Battery System on CO2 Storage Performance. Processes. 2024; 12(9):1896. https://doi.org/10.3390/pr12091896
Chicago/Turabian StyleYang, Haoran, Mian Wei, Baodong Wang, Leqi Wang, Qiuyan Chen, Chang Su, Yongcheng Feng, Xing Wang, and Ke Li. 2024. "Research on the Influence of a Magnesium-Based Carbon Dioxide Battery System on CO2 Storage Performance" Processes 12, no. 9: 1896. https://doi.org/10.3390/pr12091896
APA StyleYang, H., Wei, M., Wang, B., Wang, L., Chen, Q., Su, C., Feng, Y., Wang, X., & Li, K. (2024). Research on the Influence of a Magnesium-Based Carbon Dioxide Battery System on CO2 Storage Performance. Processes, 12(9), 1896. https://doi.org/10.3390/pr12091896