In Silico Tools and Phosphoproteomic Software Exclusives


 and
and
Abstract
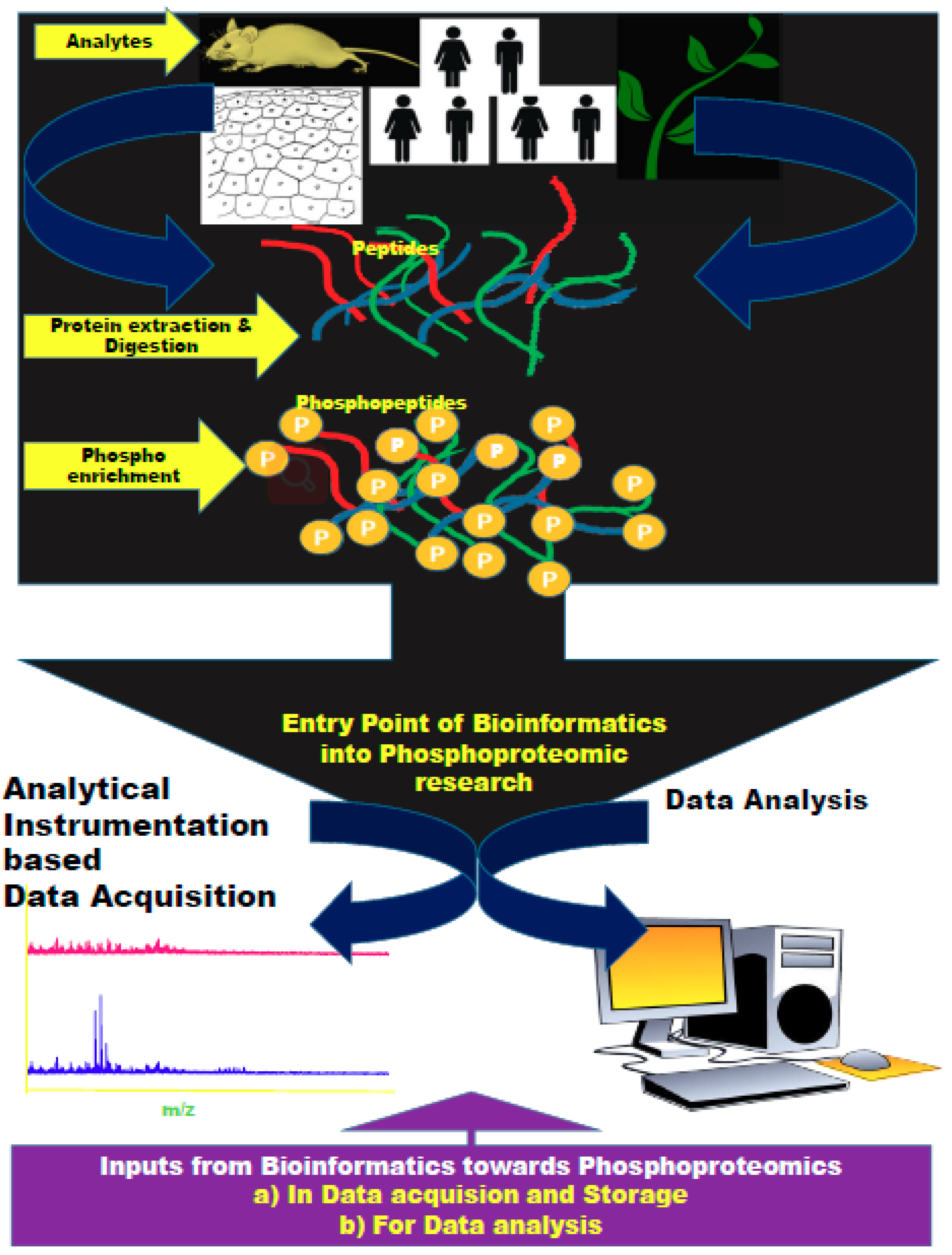
:1. Introduction
2. Biocomputational Tools for Proteomics—A Snapshot
3. Biocomputational Tools for Phosphoproteomics
3.1. Tools for Analysis of Phosphopeptide Data/Spectra
3.2. Tools for Phosphorylation Site Assignment
3.3. Tools for Prediction of Phosphorylation Sites
3.4. Tools for Detection of Phosphosites and Kinase Activity from Phosphopeptide Data
4. Future Direction—Implementation of Biocomputation Integrated Phosphoproteomics
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rao, V.S.; Das, S.K.; Rao, V.J.; Gedela, S. Recent developments in life sciences research: Role of Bioinformatics. Afr. J. Biotechnol. 2008, 7, 495–503. [Google Scholar]
- Ulloa, A.R.; Rodríguez, R. Bioinformatic tools for proteomic data analysis: An overview. Biotecnol. Apl. 2008, 25, 312–319. [Google Scholar]
- Nakagami, H.; Sugiyama, N.; Mochida, K.; Daudi, A.; Yoshida, Y.; Toyoda, T.; Tomita, M.; Ishihama, Y.; Shirasu, K. Large-scale comparative phosphoproteomics identifies conserved phosphorylation sites in plants. Plant Physiol. 2010, 153, 1161–1174. [Google Scholar] [CrossRef]
- Nilsson, T.; Mann, M.; Aebersold, R.; Yates, J.R., 3rd; Bairoch, A.; Bergeron, J.J. Mass spectrometry in high-throughput proteomics: Ready for the big time. Nat. Methods 2010, 7, 681–685. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, K.; Li, Q.; Nice, E.C.; Zhang, H.; Huang, C. Clinical proteomics-driven precision medicine for targeted cancer therapy: Current overview and future perspectives. Expert Rev. Proteom. 2016, 13, 367–381. [Google Scholar] [CrossRef] [PubMed]
- Guerin, M.; Gonçalves, A.; Toiron, Y.; Baudelet, E.; Audebert, S.; Boyer, J.B.; Borg, J.P.; Camoin, L. How may targeted proteomics complement genomic data in breast cancer? Expert Rev. Proteom. 2017, 14, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P. Proteomics retrenches. Nat. Biotechnol. 2010, 28, 665–670. [Google Scholar] [CrossRef]
- Searle, B.C. Scaffold: A bioinformatic tool for validating MS/MS-based proteomic studies. Proteomics 2010, 10, 1265–1269. [Google Scholar] [CrossRef] [PubMed]
- Varjosalo, M.; Sacco, R.; Stukalov, A.; van Drogen, A.; Planyavsky, M.; Hauri, S.; Aebersold, R.; Bennett, K.L.; Colinge, J.; Gstaiger, M.; et al. Interlaboratory reproducibility of large-scale human protein-complex analysis by standardized AP-MS. Nat. Methods 2013, 10, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Mann, M. Comparative analysis to guide quality improvements in proteomics. Nat. Methods 2009, 6, 717–719. [Google Scholar] [CrossRef] [PubMed]
- Stead, D.A.; Paton, N.W.; Missier, P.; Embury, S.M.; Hedeler, C.; Jin, B.; Brown, A.J.; Preece, A. Information quality in proteomics. Brief. Bioinform. 2008, 9, 174–188. [Google Scholar] [CrossRef] [PubMed]
- Tabb, D.L. Quality assessment for clinical proteomics. Clin. Biochem. 2013, 46, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Wang, X. Statistical assessment of QC metrics on raw LC-MS/MS data. Proteomics. In Proteomics; Comai, L., Katz, J.E., Mallick, P., Eds.; Springer: New York, NY, USA, 2017; pp. 325–337. [Google Scholar]
- Whiteaker, J.R.; Halusa, G.N.; Hoofnagle, A.N.; Sharma, V.; MacLean, B.; Yan, P.; Wrobel, J.A.; Kennedy, J.; Mani, D.R.; Zimmerman, L.J.; et al. Using the CPTAC Assay Portal to identify and implement highly characterized targeted proteomics assays. Methods MolBiol. 2016, 1410, 223–236. [Google Scholar]
- Sharma, K.; D’Souza, R.C.J.; Tyanova, S.; Schaab, C.; Wiśniewski, J.R.; Cox, J.; Mann, M. Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep. 2014, 8, 1583–1594. [Google Scholar] [CrossRef] [PubMed]
- Casado, P.; Hijazi, M.; Britton, D.; Cutillas, P.R. Impact of phosphoproteomics in the translation of kinase-targeted therapies. Proteomics 2017, 17, 1600235. [Google Scholar] [CrossRef] [PubMed]
- Cutillas, P.R. Role of phosphoproteomics in the development of personalized cancer therapies. Proteom. Clin. Appl. 2015, 9, 383–395. [Google Scholar] [CrossRef]
- Yang, J.-Y.; Yoshihara, K.; Tanaka, K.; Hatae, M.; Masuzaki, H.; Itamochi, H.; Cancer Genome Atlas (TCGA) Research Network; Takano, M.; Ushijima, K.; Tanyi, J.L. Predicting time to ovarian carcinoma recurrence using protein markers. J. Clin. Investig. 2013, 123, 3740–3750. [Google Scholar] [CrossRef]
- Parker, R.; Vella, L.J.; Xavier, D.; Amirkhani, A.; Parker, J.; Cebon, J.; Molloy, M.P. Phosphoproteomic analysis of cell-based resistance to BRAF inhibitor therapy in melanoma. Front. Oncol. 2015, 5, 95. [Google Scholar] [CrossRef]
- Wei, W.; Shin, Y.S.; Xue, M.; Matsutani, T.; Masui, K.; Yang, H.; Ikegami, S.; Gu, Y.; Herrmann, K.; Johnson, D.; et al. Single-cell phosphoproteomics resolves adaptive signaling dynamics and informs targeted combination therapy in glioblastoma. Cancer Cell 2016, 29, 563–573. [Google Scholar] [CrossRef]
- Locard-Paulet, M.; Lim, L.; Veluscek, G.; McMahon, K.; Sinclair, J.; van Weverwijk, A.; Worboys, J.D.; Yuan, Y.; Isacke, C.M.; Jørgensen, C. Phosphoproteomic analysis of interacting tumor and endothelial cells identifies regulatory mechanisms of transendothelial migration. Sci. Signal. 2016, 9, ra15. [Google Scholar] [CrossRef]
- Casado, P.; Alcolea, M.P.; Iorio, F.; Rodríguez-Prados, J.C.; Vanhaesebroeck, B.; Saez-Rodriguez, J.; Joel, S.; Cutillas, P.R. Phosphoproteomics data classify hematological cancer cell lines according to tumor type and sensitivity to kinase inhibitors. Genome Biol. 2013, 14, R37. [Google Scholar] [CrossRef] [PubMed]
- Casado, P.; Rodriguez-Prados, J.-C.; Cosulich, S.C.; Guichard, S.; Vanhaesebroeck, B.; Joel, S.; Cutillas, P.R. Kinasesubstrate enrichment analysis provides insights into the heterogeneity of signaling pathway activation in leukemia cells. Sci. Signal 2013, 6, rs6. [Google Scholar] [CrossRef] [PubMed]
- Eduati, F.; Doldan-Martelli, V.; Klinger, B.; Cokelaer, T.; Sieber, A.; Kogera, F.; Dorel, M.; Garnett, M.J.; Blüthgen, N.; Saez-Rodriguez, J. Drug resistance mechanisms in colorectal cancer dissected with cell type–specific dynamic logic models. Cancer Res. 2017, 77, 3364–3375. [Google Scholar] [CrossRef] [PubMed]
- Thiele, H.; Jörg, G.; Peter, H.; Gerhard, K.; Martin, B. Managing Proteomics Data: From Generation and Data Warehousing to Central Data Repository. J. Proteom. Bioinform. 2008, 1, 485–507. [Google Scholar] [CrossRef]
- Subramanian, R.; Muthurajan, R.; Ayyanar, M. Comparative Modeling and Analysis of 3-D Structure of EMV2, aLate Embryogenesis Abundant Protein of Vigna Radiata (Wilczek). J. Proteom. Bioinform. 2008, 1, 401–407. [Google Scholar]
- Wenger, C.D.; Phanstiel, D.H.; Lee, M.V.; Bailey, D.J.; Coon, J.J. COMPASS: A suite of pre- and post-search proteomics software tools for OMSSA. Proteomics 2011, 11, 1064–1074. [Google Scholar] [CrossRef]
- Kolker, E.; Higdon, R.; Welch, D.; Bauman, A.; Stewart, E.; Haynes, W.; Broomall, W.; Kolker, N. SPIRE: Systematic protein investigative research environment. J. Proteom. 2011, 75, 122–126. [Google Scholar] [CrossRef]
- Ma, Z.Q.; Chambers, M.C.; Ham, A.J.; Cheek, K.L.; Whitwell, C.W.; Aerni, H.R.; Schilling, B.; Miller, A.W.; Caprioli, R.M.; Tabb, D.L. ScanRanker: Quality assessment of tandem mass spectra via sequence tagging. J. Proteom. Res. 2011, 10, 2896–2904. [Google Scholar] [CrossRef]
- Courcelles, M.; Lemieux, S.; Voisin, L.; Meloche, S.; Thibault, P. ProteoConnections: A bioinformatics platform to facilitate proteome and phosphoproteome analyses. Proteomics 2011, 11, 2654–2671. [Google Scholar] [CrossRef]
- Haw, R.; Hermjakob, H.; D’Eustachio, P.; Stein, L. Reactome Pathway Analysis to Enrich Biological Discovery in Proteomics Datasets. Proteomics 2011, 11, 3598–3613. [Google Scholar] [CrossRef]
- Medina-Aunon, J.A.; Martinez-Bartolome, S.; Lopez-Garcia, M.A.; Salazar, E.; Navajas, R.; Jones, A.; Paradela, A.; Albar, J. The Proteored MIAPE Web Toolkit: A User-Friendly Framework to Connect and Share Proteomics Standards. Mol. Cell Proteom. 2011, 10, 8334. [Google Scholar] [CrossRef]
- Ponomarenko, E.A.; IlgisonisA, E.V.; Lisitsa, A.V. Knowledge-based technologies in proteomics. Bioorg. Khim. 2001, 37, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Sandro, A.; Gunnar, W.K.; Knut, R. Antilope–A Lagrangian Relaxation Approach to the de novo Peptide Sequencing Problem. IEEE/ACM Trans Comput. Biol. Bioinform. 2012, 9, 385–394. [Google Scholar]
- Vetrivel, U.; Sankar, P.; Nagarajan, N.K.; Subramanian, G. Peptidomimetics Based Inhibitor Design for HIV–1 gp120 Attachment Protein. J. Proteom. Bioinform. 2009, 2, 481–484. [Google Scholar] [CrossRef]
- Kikuta, K.; Tsunehiro, Y.; Yoshida, A.; Tochigi, N.; Hirohahsi, S.; Kawai, A.; Kondo, T. Proteome Expression Database of Ewing sarcoma: A segment of the Genome Medicine Database of Japan Proteomics. J. Proteom. Bioinform. 2009, 2, 500–504. [Google Scholar] [CrossRef]
- Sandra, O.; Henning, H. Standardising Proteomics Data–the work of the HUPO Proteomics Standards Initiative. J. Proteom. Bioinform. 2008, 1, 3–5. [Google Scholar]
- Neha, G.; Sachin, P.; Anil, P.; Anil, K. Primer Designing for Dreb1A, A Cold Induced Gene. J. Proteom. Bioinform. 2008, 1, 28–35. [Google Scholar]
- Allam, A.R.; Kiran, K.R.; Hanuman, T. Bioinformatic Analysis of Alzheimer’s Disease Using Functional Protein Sequences. J. Proteom. Bioinform. 2008, 1, 036–042. [Google Scholar] [CrossRef]
- Kush, A.; Raghava, G.P.S. AC2DGel: Analysis and Comparison of 2D Gels. J. Proteom. Bioinform. 2008, 1, 43–46. [Google Scholar] [CrossRef] [Green Version]
- Seenivasagan, R.; Kasimani, R.; Marimuthu, P.; Kalidoss, R.; Shanmughavel, P. Comparative Modeling of Viral Protein R (Vpr) From Human Immunodeficiency Virus 1 (Hiv 1). J. Proteom. Bioinform. 2008, 1, 73–76. [Google Scholar]
- Gnad, F.; Oroshi, M.; Birney, E.; Mann, M. MAPU 2.0: High-accuracy proteomes mapped to genomes. Nucleic Acids Res. 2009, 37, D902–D906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunil, K.; Priya, R.D.; Prakash, C.S. Prediction of 3-Dimensional Structure of Cathepsin L Protein of Rattus Norvegicus. J. Proteom. Bioinform. 2008, 1, 307–314. [Google Scholar]
- Paul, K.; Nathan, L.C.; Daniel, C.; Clarissa, D.; Patrice, H.; Joanna, H.; Eustache, P.; Marc, R.; Olivier, M.; Howard, M.C.; et al. Global Proteomics: Pharmacodynamic Decision Making via Geometric Interpretations of Proteomic Analyses. J. Proteom. Bioinform. 2008, 1, 315–328. [Google Scholar]
- ExPASy. SIB Bioinformatics Resource Portal-Proteomics Tools.html.
- Nanda, T.; Tripathy, K.; Ashwin, P. Integration of Bioinformatics Tools for Proteomics Research. J. Comput. Sci. Syst. Biol. 2001, S13. [Google Scholar] [CrossRef]
- Beausoleil, S.A.; Villén, J.; Gerber, S.A.; Rush, J.; Gygi, S.P. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 2006, 24, 1285–1292. [Google Scholar] [CrossRef]
- Olsen, J.V.; Blagoev, B.; Gnad, F.; Macek, B.; Kumar, C.; Mortensen, P.; Mann, M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 2006, 127, 635–648. [Google Scholar] [CrossRef] [Green Version]
- Taus, T.; Köcher, T.; Pichler, P.; Paschke, C.; Schmidt, A.; Henrich, C.; Mechtler, K. Universal and confident phosphorylation site localization using phosphoRS. J. Proteome Res. 2001, 10, 5354–5362. [Google Scholar] [CrossRef] [PubMed]
- Bodenmiller, B.; Campbell, D.; Gerrits, B.; Lam, H.; Jovanovic, M.; Picotti, P.; Schlapbach, R.; Aebersold, R. PhosphoPep—A database of protein phosphorylation sites in model organisms. Nat. Biotechnol. 2008, 26, 1339–1340. [Google Scholar] [CrossRef] [Green Version]
- Hummel, J.; Niemann, M.; Wienkoop, S.; Schulze, W.; Steinhauser, D.; Selbig, J.; Walther, D.; Weckwerth, W. ProMEX: A mass spectral reference database for proteins and protein phosphorylation sites. BMC Bioinform. 2007, 8, 216. [Google Scholar] [CrossRef] [Green Version]
- Suni, V.; Imanishi, S.Y.; Maiolica, A.; Aebersold, R.; Corthals, G.L. Confident site localization using a simulated phosphopeptide spectral library. J. Proteome Res. 2015, 14, 2348–2359. [Google Scholar] [CrossRef]
- Kessner, D.; Chambers, M.; Burke, R.; Agus, D.; Mallick, P. ProteoWizard: Open source software for rapid proteomics tools development. Bioinformatics 2008, 24, 2534–2536. [Google Scholar] [CrossRef] [PubMed]
- Keller, A.; Eng, J.; Zhang, N.; Li, X.J.; Aebersold, R. A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Mol. Syst. Biol. 2005, 1, 2005.0017. [Google Scholar] [CrossRef] [PubMed]
- Craig, R.; Beavis, R.C. TANDEM: Matching proteins with tandem mass spectra. Bioinformatics 2004, 20, 1466–1467. [Google Scholar] [CrossRef] [PubMed]
- Florian, G.; Shubin, R.; Juergen, C.; Jesper, V.O.; Boris, M.; Mario, O.; Matthias, M. PHOSIDA (Phosphorylation Site Database): Management, Structural and Evolutionary Investigation, and Prediction of Phosphosites. Genome Biol. 2007, 8. [Google Scholar] [CrossRef] [Green Version]
- Söderholm, S.; Hintsanen, P.; Öhman, T.; Aittokallio, T.; Nyman, T.A. PhosFox: A bioinformatics tool for peptide-level processing of LC-MS/MS-based phosphoproteomic data. Proteome Sci. 2014, 12, 36. [Google Scholar] [CrossRef] [Green Version]
- Välikangas, T.; Suomi, T.; Elo, L.L. A systematic evaluation of normalization methods in quantitative label-free proteomics. Brief. Bioinform. 2016, 19, 1–11. [Google Scholar] [CrossRef]
- Kauko, O.; Teemu, D.L.; Mikael, J.; Petteri, H.; Veronika, S.; Pekka, H.; Garry, C.; Tero, A.; Jukka, W.; Kauko, O. Label-free quantitative phosphoproteomics with novel pairwise abundance normalization reveals synergistic RAS and CIP2A signaling. Sci. Rep. 2015, 5, 13099. [Google Scholar] [CrossRef]
- Olsen, J.V.; Mann, M. Status of large-scale analysis of posttranslational modifications by mass spectrometry. Mol. Cell. Proteom. 2013, 12, 3444–3452. [Google Scholar] [CrossRef] [Green Version]
- Saraei, S.; Suomi, T.; Kauko, O.; Elo, L.L.; Stegle, O. Phosphonormalizer: An R package for normalization of MS-based label-free phosphoproteomics. Bioinformatics 2018, 34, 693–694. [Google Scholar] [CrossRef] [Green Version]
- Tikira, T.; Juergen, C. The MaxQuant computational platform for mass spectrometry-based shotgun proteomicsStefka Tyanova. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyanova, S.; Temu, T.; Carlson, A.; Sinitcyn, P.; Mann, M.; Cox, J. Visualization of LC-MS/MS proteomics data in MaxQuant. Proteomics 2015, 15, 1453–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pino, L.K.; Searle, B.C.; Bollinger, J.G.; Nunn, B.; MacLean, B.; MacCoss, M.J. The Skyline ecosystem: Informatics for quantitative mass spectrometry proteomics. Mass Spectrom. Rev. 2017, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Ruttenberg, B.E.; Pisitkun, T.; Knepper, M.A.; Hoffert, J.D. PhosphoScore: An open-source phosphorylation site assignment tool for MSn data. J. Proteome Res. 2008, 7, 3054–3059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffert, J.D.; Wang, G.; Pisitkun, T.; Shen, R.F.; Knepper, M.A. An automated platform for analysis of phosphoproteomic datasets: Application to kidney collecting duct phosphoproteins. J. Proteome. Res. 2007, 9, 3501–3508. [Google Scholar] [CrossRef] [Green Version]
- Fleuren, E.D.; Zhang, L.; Wu, J.; Daly, R.J. The kinome ‘at large’ in cancer. Nat. Rev. Cancer 2016, 16, 83–98. [Google Scholar] [CrossRef]
- Creixell, P.; Erwin, M.; Craig, D.S.; James, L.; Chad, J.M.; Hua, J.L.; Lara, P.; Thomas, R.C.; Nevena, Z.; Antonio, P.; et al. Kinome-wide decoding of network-attacking mutations rewiring cancer signaling. Cell 2015, 163, 202–217. [Google Scholar] [CrossRef] [Green Version]
- Kobe, B.; Kampmann, T.; Forwood, J.K.; Listwan, P.; Brinkworth, R.I. Substrate specificity of protein kinases and computational prediction of substrates. Biochim. Biophys. Acta 2005, 1754, 200–209. [Google Scholar] [CrossRef]
- Hjerrild, M.; Gammeltoft, S. Phosphoproteomics toolbox: Computational biology, protein chemistry and mass spectrometry. FEBS Lett. 2006, 580, 4764–4770. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.L.; Blom, N. Kinase-specific prediction of protein phosphorylation sites. Methods Mol. Biol. 2009, 527, 299–310. [Google Scholar]
- Blom, N.; Gammeltoft, S.; Brunak, S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 1999, 294, 1351–1362. [Google Scholar] [CrossRef] [PubMed]
- Hjerrild, M.; Stensballe, A.; Rasmussen, T.E.; Kofoed, C.B.; Blom, N.; Sicheritz-Ponten, T.; Larsen, M.R.; Brunak, S.; Jensen, O.N.; Gammeltoft, S. Identification of phosphorylation sites in protein kinase a substrates using artificial neural networks and mass spectrometry. J. Proteome Res. 2004, 3, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Gao, X.; Cao, J.; Liu, Z.; Jin, C.; Wen, L.; Yao, X.; Ren, J. A summary of computational resources for protein phosphorylation. Curr. Protein Pept. Sci. 2010, 11, 485–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trost, B.; Kusalik, A. Computational prediction of eukaryotic phosphorylation sites. Bioinformatics 2011, 27, 2927–2935. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Zeng, S.; Xu, C.; Qiu, W.; Liang, Y.; Joshi, T.; Xu, D. MusiteDeep: A deep-learning framework for general and kinase-specific phosphorylation site prediction. Bioinformatics 2017, 33, 3909–3916. [Google Scholar] [CrossRef] [Green Version]
- Luo, F.; Wang, M.; Liu, Y.; Zhao, X.; Li, A. DeepPhos: Prediction of protein phosphorylation sites with deep learning. Bioinformatics 2019, 35, 2766–2773. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Wang, H.; Wang, J.; Leier, A.; Marquez-Lago, T.; Yang, B.; Zhang, Z.; Akutsu, T.; Webb, G.; Daly, R.J. PhosphoPredict: A bioinformatics tool for prediction of human kinase-specific phosphorylation substrates and sites by integrating heterogeneous feature selection. Sci. Rep. 2017, 7, 6862. [Google Scholar] [CrossRef] [Green Version]
- Newman, R.H.; Zhang, J.; Zhu, H. Toward a systems-level view of dynamic phosphorylation networks. Front. Genet. 2014, 5, 263. [Google Scholar] [CrossRef] [Green Version]
- Glickman, J.F. Assay Development for Protein Kinase Enzymes; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2012. [Google Scholar]
- Cutillas, P.R.; Khwaja, A.; Graupera, M.; Pearce, W.; Gharbi, S.; Waterfield, M.; Vanhaesebroeck, B. Ultrasensitive and absolute quantification of the phosphoinositide 3-kinase/Akt signal transduction pathway by mass spectrometry. Proc. Natl. Acad. Sci. USA 2006, 103, 8959–8964. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Anjum, R.; Kubota, K.; Rush, J.; Villen, J.; Gygi, S.P. A site-specific, multiplexed kinase activity assay using stable-isotope dilution and high-resolution mass spectrometry. Proc. Natl. Acad. Sci. USA 2009, 106, 11606–11611. [Google Scholar] [CrossRef] [Green Version]
- Qi, L.; Liu, Z.; Wang, J.; Cui, Y.; Guo, Y.; Zhou, T.; Zhou, Z.; Guo, X.; Xue, Y.; Sha, J. Systematic analysis of the phosphoproteome and kinase-substrate networks in the mouse testis. Mol. Cell. Proteom. 2014, 13, 3626–3638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, P.; Zheng, X.; Jayaswal, V.; Hu, G.; Yang, J.Y.H.; Jothi, R. Knowledge Based Analysis for Detecting Key Signaling Events from Time-Series Phosphoproteomics Data. PLoS Comput. Biol. 2015, 11, e1004403. [Google Scholar] [CrossRef] [PubMed]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinkel, H.; Chica, C.; Via, A.; Gould, C.M.; Jensen, L.J.; Gibson, T.J.; Diella, F. Phospho.ELM: A database of phosphorylation sites–update 2011. Nucleic Acids Res. 2011, 39, D261–D267. [Google Scholar] [CrossRef] [Green Version]
- Perfetto, L.; Briganti, L.; Calderone, A.; Cerquone Perpetuini, A.; Iannuccelli, M.; Langone, F.; Licata, L.; Marinkovic, M.; Mattioni, A.; Pavlidou, T.; et al. SIGNOR: A database of causal relationships between biological entities. Nucleic Acids Res. 2016, 44, D548–D554. [Google Scholar] [CrossRef]
- Gnad, F.; Gunawardena, J.; Mann, M. PHOSIDA 2011: The posttranslational modification database. Nucleic Acids Res. 2011, 39, D253–D260. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Rho, H.S.; Newman, R.H.; Zhang, J.; Zhu, H.; Qian, J. PhosphoNetworks: A database for human phosphorylation networks. Bioinformatics 2014, 30, 141–142. [Google Scholar] [CrossRef]
- Sadowski, I.; Breitkreutz, B.J.; Stark, C.; Su, T.C.; Dahabieh, M.; Raithatha, S.; Bernhard, W.; Oughtred, R.; Dolinski, K.; Barreto, K.; et al. The PhosphoGRID Saccharomyces cerevisiae protein phosphorylation site database: Version 2.0 update. Database (Oxford) 2013, 2013, bat026. [Google Scholar] [CrossRef] [Green Version]
- Duan, G.; Li, X.; Köhn, M. The human DEPhOsphorylation database DEPOD: A 2015 update. Nucleic Acids Res. 2015, 43, D531–D535. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Zha, X.; Tan, Y.; Hornbeck, P.V.; Mastrangelo, A.J.; Alessi, D.R.; Polakiewicz, R.D.; Comb, M.J. Phosphoprotein analysis using antibodies broadly reactive against phosphorylated motifs. J. Biol. Chem. 2002, 277, 39379–39387. [Google Scholar] [CrossRef] [Green Version]
- Lemeer, S.; Heck, A.J. The phosphoproteomics data explosion. Curr. Opin. Chem. Biol. 2009, 13, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Obenauer, J.C.; Cantley, L.C.; Yaffe, M.B. Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003, 31, 3635–3641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Turk, B.E. Analysis of serine-threonine kinase specificity using arrayed positional scanning peptide libraries. Curr. Protoc. Mol. Biol. 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidhu, S.S.; Koide, S. Phage display for engineering and analyzing protein interaction interfaces. Curr. Opin. Struct. Biol. 2007, 17, 481–487. [Google Scholar] [CrossRef]
- Miller, M.L.; Jensen, L.J.; Diella, F.; Jørgensen, C.; Tinti, M.; Li, L.; Hsiung, M.; Parker, S.A.; Bordeaux, J.; Sicheritz-Ponten, T.; et al. Linear motif atlas for phosphorylation-dependent signaling. Sci. Signal. 2008, 1, ra2. [Google Scholar] [CrossRef]
- Linding, R.; Jensen, L.J.; Pasculescu, A.; Olhovsky, M.; Colwill, K.; Bork, P.; Yaffe, M.B.; Pawson, T. NetworKIN: A resource for exploring cellular phosphorylation networks. Nucleic Acids Res. 2008, 36, D695–D699. [Google Scholar] [CrossRef] [Green Version]
- Horn, H.; Schoof, E.M.; Kim, J.; Robin, X.; Miller, M.L.; Diella, F.; Palma, A.; Cesareni, G.; Jensen, L.J.; Linding, R. KinomeXplorer: An integrated platform for kinome biology studies. Nat. Methods 2014, 1, 603–604. [Google Scholar] [CrossRef]
- Song, C.; Ye, M.; Liu, Z.; Cheng, H.; Jiang, X.; Han, G.; Songyang, Z.; Tan, Y.; Wang, H.; Ren, J.; et al. Systematic analysis of protein phosphorylation networks from phosphoproteomic data. Mol. Cell. Proteom. 2012, 11, 1070–1083. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Wagih, O.; Sugiyama, N.; Ishihama, Y.; Beltrao, P. Uncovering Phosphorylation-Based Specificities through Functional Interaction Networks. Mol. Cell. Proteom. 2016, 15, 236–245. [Google Scholar] [CrossRef] [Green Version]
- Wirbel, J.; Cutillas, P.; Saez-Rodriguez, J. Phosphoproteomics-Based Profiling of Kinase Activities in Cancer Cells. Methods Mol. Biol. 2018, 1711, 103–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mischnik, M.; Sacco, F.; Cox, J.; Schneider, H.C.; Schäfer, M.; Hendlich, M.; Crowther, D.; Mann, M.; Klabunde, T. IKAP: A heuristic framework for inference of kinase activities from Phosphoproteomics data. Bioinformatics 2016, 32, 424–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, P.; Patrick, E.; Humphrey, S.J.; Ghazanfar, S.; James, D.E.; Jothi, R.; Yang, J.Y. KinasePA: Phosphoproteomics data annotation using hypothesis driven kinase perturbation analysis. Proteomics 2016, 16, 1868–1871. [Google Scholar] [CrossRef] [PubMed]
- Lachmann, A.; Ma’ayan, A. KEA: Kinase enrichment analysis. Bioinformatics 2009, 25, 684–686. [Google Scholar] [CrossRef] [Green Version]
- Wiredja, D.D.; Koyutürk, M.; Chance, M.R. The KSEA App: A web-based tool for kinase activity inference from quantitative phosphoproteomics. Bioinformatics 2017, 33, 3489–3491. [Google Scholar] [CrossRef]
- Martin, D.M.; Nett, I.R.; Vandermoere, F.; Barber, J.D.; Morrice, N.A.; Ferguson, M.A. Prophossi: Automating expert validation of phosphopeptide-spectrum matches from tandem mass spectrometry. Bioinformatics 2010, 26, 2153–2159. [Google Scholar] [CrossRef]
- Brinkworth, R.I.; Breinl, R.A.; Kobe, B. Structural basis and prediction of substrate specificity in protein serine/threonine kinases. Proc. Natl. Acad. Sci. USA 2003, 100, 74–79. [Google Scholar] [CrossRef] [Green Version]
- Iakoucheva, L.M.; Radivojac, P.; Brown, C.J.; O’Connor, T.R.; Sikes, J.G.; Obradovic, Z.; Dunker, A.K. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004, 32, 1037–1049. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Lee, J.; Oh, B.; Kimm, K.; Koh, I. Prediction of phosphorylation sites using SVMs. Bioinformatics 2004, 20, 3179–3184. [Google Scholar] [CrossRef] [Green Version]
- Koenig, M.; Grabe, N. Highly specific prediction of phosphorylation sites in proteins. Bioinformatics 2004, 20, 3620–3627. [Google Scholar] [CrossRef] [Green Version]
- Zhou, F.-F.; Xue, Y.; Chen, G.L.; Yao, X. GPS: A novel group-based phosphorylation predicting and scoring method. Biochem. Biophys. Res. Commun. 2004, 325, 1443–1448. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-D.; Lee, T.Y.; Tzeng, S.W.; Wu, L.C.; Horng, J.T.; Tsou, A.P.; Huang, K.T. Incorporating hidden Markov models for identifying protein kinase-specific phosphorylation sites. J. Comput. Chem. 2005, 26, 1032–1041. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Li, A.; Wang, L.; Feng, H.; Yao, X. PPSP: Prediction of PK-specific phosphorylation site with Bayesian decision theory. BMC Bioinform. 2006, 7, 163. [Google Scholar]
- Linding, R.; Jensen, L.J.; Ostheimer, G.J.; van Vugt, M.A.; Jørgensen, C.; Miron, I.M.; Diella, F.; Colwill, K.; Taylor, L.; Elder, K.; et al. Systematic discovery of in vivo phosphorylation networks. Cell 2007, 129, 1415–1426. [Google Scholar] [CrossRef] [Green Version]
- Wong, Y.-H.; Lee, T.Y.; Liang, H.K.; Huang, C.M.; Wang, T.Y.; Yang, Y.H.; Chu, C.H.; Huang, H.D.; Ko, M.T.; Hwang, J.K. KinasePhos 2.0: A web server for identifying protein kinase-specific phosphorylation sites based on sequences and coupling patterns. Nucleic Acids Res. 2007, 35, W588–W594. [Google Scholar] [CrossRef]
- Plewczyński, D.; Tkacz, A.; Wyrwicz, L.; Rychlewski, L. AutoMotif Server for prediction of phosphorylation sites in proteins using support vector machine: 2007 update. J. Mol. Model 2008, 14, 69–76. [Google Scholar] [CrossRef]
- Heazlewood, J.L.; Durek, P.; Hummel, J.; Selbig, J.; Weckwerth, W.; Walther, D.; Schulze, W.X. PhosPhAt: A database of phosphorylation sites in Arabidopsis thaliana and a plant-specific phosphorylation site predictor. Nucleic Acids Res. 2008, 36, D1015–D1021. [Google Scholar] [CrossRef]
- Li, T.; Li, F.; Zhang, X. Prediction of kinase-specific phosphorylation sites with sequence features by a log-odds ratio approach. Proteins 2008, 70, 404–414. [Google Scholar] [CrossRef]
- Wan, J.; Kang, S.; Tang, C.; Yan, J.; Ren, Y.; Liu, J.; Gao, X.; Banerjee, A.; Ellis, L.B.M.; Li, T. Meta-prediction of phosphorylation sites with weighted voting and restricted grid search parameter selection. Nucleic Acids Res. 2008, 36, e22. [Google Scholar] [CrossRef] [Green Version]
- Yoo, P.D.; Ho, Y.S.; Zhou, B.B.; Zomaya, A.Y. SiteSeek: Post-translational modification analysis using adaptive locality-effective kernel methods and new profiles. BMC Bioinform. 2008, 9, 272. [Google Scholar] [CrossRef] [Green Version]
- Saunders, N.F.W. Predikin and PredikinDB: A computational framework for the prediction of protein kinase peptide specificity and an associated database of phosphorylation sites. BMC Bioinform. 2008, 9, 245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, Y. GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Mol. Cell. Proteom. 2008, 7, 1598–1608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, T.H. Prediction of kinase-specific phosphorylation sites using conditional random fields. Bioinformatics 2008, 24, 2857–2864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durek, P.; Schudoma, C.; Weckwerth, W.; Selbig, J. Walther D. Detection and characterization of 3D-signature phosphorylation site motifs and their contribution towards improved phosphorylation site prediction in proteins. BMC Bioinform. 2009, 10, 117. [Google Scholar] [CrossRef] [Green Version]
- Biswas, A.K.; Noman, N.; Sikder, A.R. Machine learning approach to predict protein phosphorylation sites by incorporating evolutionary information. BMC Bioinform. 2010, 11, 273. [Google Scholar] [CrossRef] [Green Version]
- Sobolev, B. Functional classification of proteins based on projection of amino acid sequences: Application for prediction of protein kinase substrates. BMC Bioinform. 2010, 11, 313. [Google Scholar] [CrossRef] [Green Version]
- Jung, I. PostMod: Sequence based prediction of kinase-specific phosphorylation sites with indirect relationship. BMC Bioinform. 2010, 11, S10. [Google Scholar] [CrossRef] [Green Version]
- Xue, Y. GPS 2.1: Enhanced prediction of kinase-specific phosphorylation sites with an algorithm of motif length selection. Protein Eng. Des. Sel. 2011, 24, 255–260. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Xu, D. The Musite open-source framework for phosphorylation-site prediction. BMC Bioinform. 2010, 11, S9. [Google Scholar] [CrossRef] [Green Version]
- Aravind, S.; Pablo, T.; Vamsi, K.M.; Sayan, M.; Benjamin, L.E.; Michael, A.G.; Amanda, P.; Scott, L.P.; Todd, R.G.; Eric, S.L.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar]

{kind=link}
| Software Function/Application | Bioinformatics Tool | Specified Function | Website | Ref. |
|---|---|---|---|---|
| Analysis of phosphopeptide data/spectra | SimPhospho | Search, simulate phosphopeptide spectra and tandem mass spectra | https://sourceforge.net/projects/simphospho/ | [53,54,55] |
| PHOSIDA | Storage, management and recovery of phosphopeptide data, predicting putative phosphorylation sites, acetylation and other post-translational modification sites and analyses phosphorylation events of proteins of interest | http://www.phosida.com | [56,89] | |
| Prophossi | Automating expert validation of phosphopeptide–spectrum matches from tandem mass spectrometry | http://www.compbio.dundee.ac.uk/prophossi | [109] | |
| PhosFox | Peptide-level processing of phosphoproteomic data generated by Mascot, Sequest, and Paragon, qualitative and quantitative phosphoproteomics | https://bitbucket.org/phintsan/phosfox | [57] | |
| R package, Phospho normalizer | Normalization of phosphoproteomics data | https://bioconductor.org/packages/phosphonormalizer | [61] | |
| Correct phosphorylation site assignment | PhosphoScore | Phosphorylation site assignment | https://omictools.com/phosphoscore-tool | [66] |
| Ascore | Phosphorylation site assignment | http://ascore.med.harvard.edu/ascore.php | [67] | |
| Phosphorylation site prediction | NetPhos | Machine learning methods, artificial neural networks (ANNs) | cbs.dtu.dk/services/NetPhos | [73,74] |
| Scansite | Machine learning methods, position-specific scoring matrices (PSSMs) used | scansite.mit.edu | [95] | |
| Predikin 1.0 | Structural analysis (SA) used | predikin.biosci.uq.edu.au | [110] | |
| DISPHOS | Logistic regression (LA) used | www.dabi.temple.edu/disphos | [111] | |
| NetPhosK | ANN used | cbs.dtu.dk/services/NetPhos | [74] | |
| PredPhospho | Support vector machines (SVMs) used | (website no longer accessible) | [112] | |
| PHOSITE | PSSM | (website no longer accessible) | [113] | |
| GPS 1.0 | PSSM, Markov clustering (MC) used | gps.biocuckoo.org | [114] | |
| KinasePhos 1.0 | Hidden Markov Model (HMM) used | kinasephos.mbc.nctu.edu.tw | [115] | |
| PPSP | Bayesian probability (BP) based | ppsp.biocuckoo.org | [116] | |
| NetworKIN /KinomeXplorer | ANN, PSSM based | networkin.info | [100,101,117] | |
| KinasePhos 2.0 | SVM | kinasephos2.mbc.nctu.edu.tw | [118] | |
| AutoMotif | SVM | (website no longer accessible) | [119] | |
| PhosPhAt | SVM | phosphat.mpimp-golm.mpg.de | [120] | |
| PhoScan | PSSM | bioinfo.au.tsinghua.edu.cn/phoscan | [121] | |
| MetaPredPS | Meta-predictor (MP) | metapred.biolead.org/MetaPredPS | [122] | |
| SiteSeek | Non specified | (no web implementation available) | [123] | |
| Predikin 2.0 | HMM, SA | predikin.biosci.uq.edu.au | [124] | |
| GPS 2.0 | PSSM, genetic algorithm (GA) | gps.biocuckoo.org | [125] | |
| CRPhos | Conditional random fields (CRF) | www.ptools.ua.ac.be/CRPhos | [126] | |
| Phos3D | SVM | phos3d.mpimp-golm.mpg.de | [127] | |
| PPRED | PSSM, SVM | ashiskb.info/research/ppred | [128] | |
| PAAS | PSSM | (website no longer accessible) | [129] | |
| PostMod | PSSM | pbil.kaist.ac.kr/PostMod | [130] | |
| GPS 2.1 | PSSM, GA | gps.biocuckoo.org | [131] | |
| Musite | SVM | musite.sourceforge.net | [132] | |
| MusiteDeep | Predicting general and kinase-specific phosphorylation sites | https://github.com/duolinwang/MusiteDeep | [77] | |
| DeepPhos | Prediction of protein phosphorylation, kinase-specific prediction | https://github.com/USTCHIlab/DeepPhos | [78] | |
| PhosphoPredict | Prediction of kinase-specific substrates and associated phosphorylation sites | http://phosphopredict.erc.monash.edu/ | [79] | |
| Inference of kinase activity from phosphoproteomics data/detection of phosphosites | Kinase-Substrate Enrichment Analysis (KSEA) | Computational characterization of differential kinase activity from phosphoproteomics datasets | https://casecpb.shinyapps.io/ksea/ | [108] |
| CLUE (CLUster Evaluation) include IKAP, KinasePA, KAA (Kinase activity analysis) and KEA | Computational analysis of the detected phosphorylation sites (phosphosites) | https://omictools.com/clue-tool | [83,84,106,107,108] | |
| GSEA (Gene Set Enrichment Analysis) | Inference of kinase activity from phosphoproteomics data | http://software.broadinstitute.org/gsea/ | [133] | |
| PhosphoSitePlus | Database for expert-edited and curated interactions between kinases and individual phosphosites | https://www.phosphosite.org/homeAction.action | [86] | |
| Phospho.ELM | Computes a score for the conservation of a phosphosite | http://phospho.elm.eu.org | [87] | |
| Signor | Focuses on interactions with proteins involved in signal transduction | https://signor.uniroma2.it/ | [88] | |
| Netphorest | Classifies phosphorylation sites | http://www.netphorest.info/ | [98] | |
| PhosphoGRID | Related information for Saccharomyces cerevisiae | https://phosphogrid.org/ | [91] | |
| DEPhOsphorylation database DEPOD | Supports phosphatase–kinase substrate networks | http://www.koehn.embl.de/depod | [92] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paul, P.; Muthu, M.; Chilukuri, Y.; Haga, S.W.; Chun, S.; Oh, J.-W. In Silico Tools and Phosphoproteomic Software Exclusives. Processes 2019, 7, 869. https://doi.org/10.3390/pr7120869
Paul P, Muthu M, Chilukuri Y, Haga SW, Chun S, Oh J-W. In Silico Tools and Phosphoproteomic Software Exclusives. Processes. 2019; 7(12):869. https://doi.org/10.3390/pr7120869
Chicago/Turabian StylePaul, Piby, Manikandan Muthu, Yojitha Chilukuri, Steve W. Haga, Sechul Chun, and Jae-Wook Oh. 2019. "In Silico Tools and Phosphoproteomic Software Exclusives" Processes 7, no. 12: 869. https://doi.org/10.3390/pr7120869