Figure 1.
Snapshot of a section of the extracted data from the NCBI Pathogen Detection Isolates Browser for the US.
Figure 2.
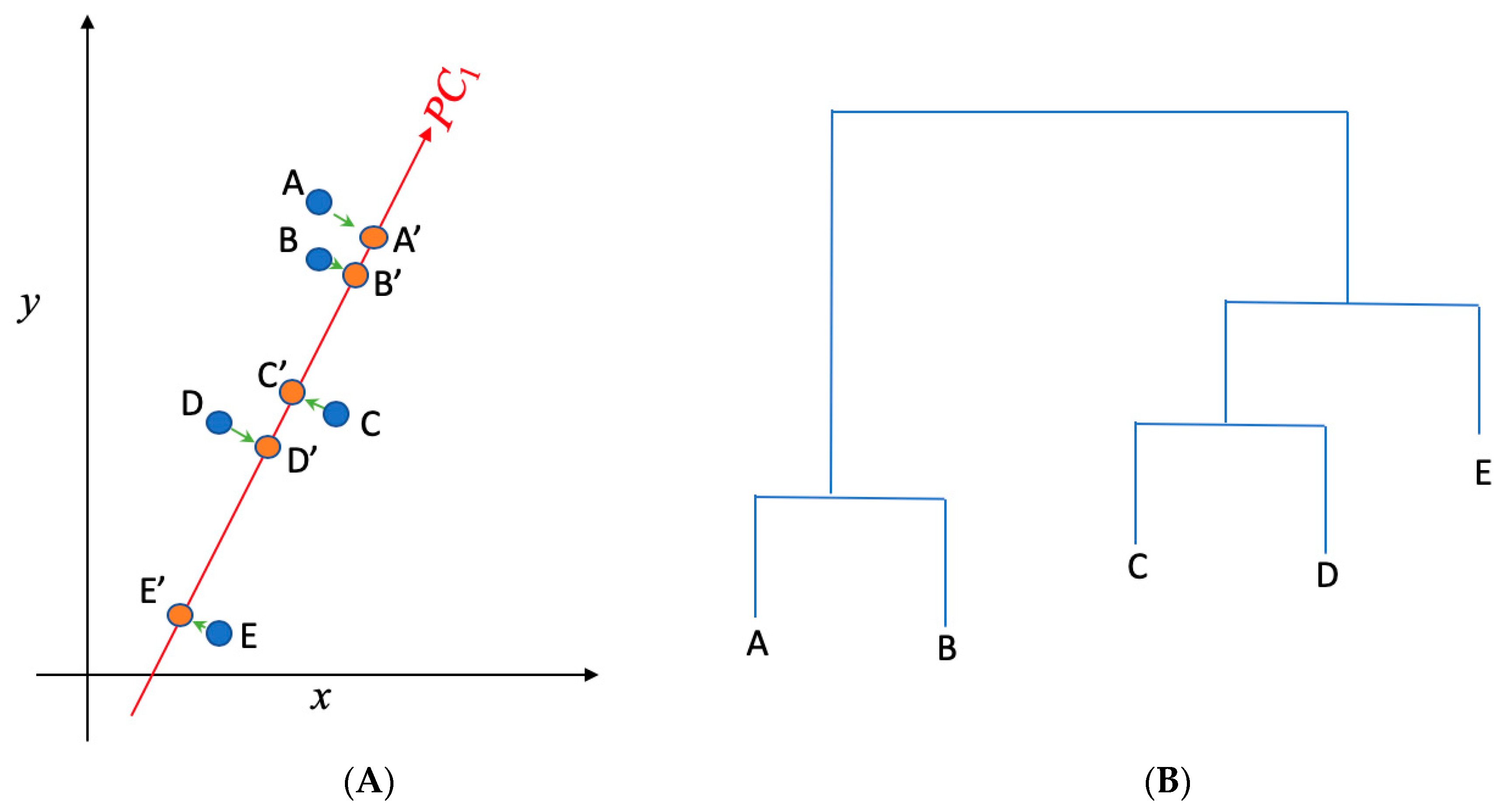
(A) Principal component analysis is used to present the two dimensional (x,y) gene data into one dimensional space (PC1); (B) hierarchical clustering is implemented to cluster genes in (A) into separate groups. A, B, C, D, and E present the data in the x~y space in (A), while A′, B′, C′, D′, and E′ present the projections of those data points in the PC1 space. While A, B, and C are not distinguishable in the x direction (as they have the similar projections onto x), they can be separated in the PC1 direction. PC1 represents the direction that returns the largest variance in the data projections onto it. Steps shown in (A) and (B) were used to separate and identify important AMR genes and clinical pathogens from the data.
Figure 2.
(A) Principal component analysis is used to present the two dimensional (x,y) gene data into one dimensional space (PC1); (B) hierarchical clustering is implemented to cluster genes in (A) into separate groups. A, B, C, D, and E present the data in the x~y space in (A), while A′, B′, C′, D′, and E′ present the projections of those data points in the PC1 space. While A, B, and C are not distinguishable in the x direction (as they have the similar projections onto x), they can be separated in the PC1 direction. PC1 represents the direction that returns the largest variance in the data projections onto it. Steps shown in (A) and (B) were used to separate and identify important AMR genes and clinical pathogens from the data.
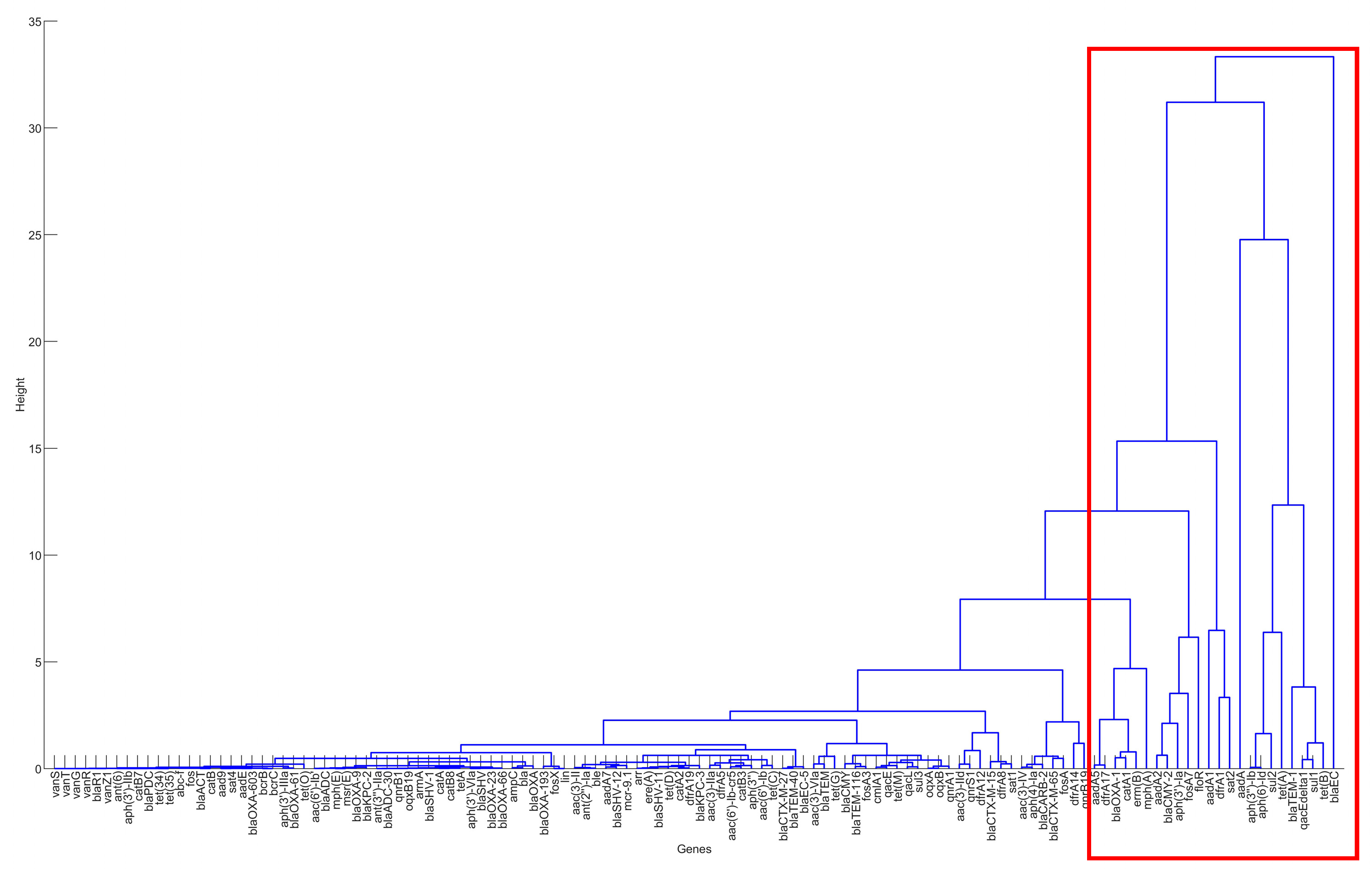
Figure 3.
The result of hierarchical clustering for the genes in the clinical samples for the United States, 2010–2019.
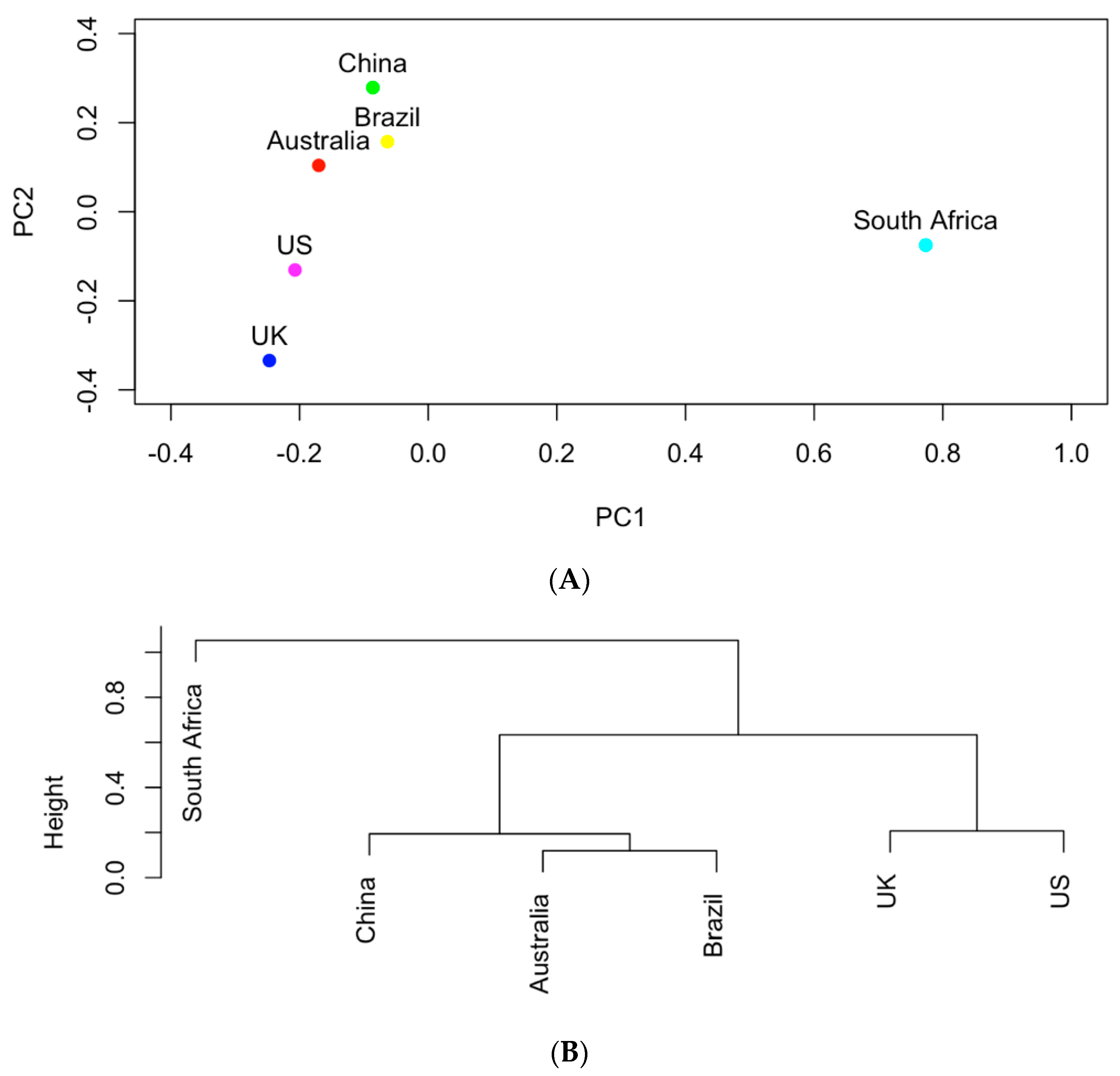
Figure 4.
(A) The projection of the six countries onto the PC1~PC2 space based on AMR genes; (B) hierarchical clustering analysis of the similarities of the six countries in carrying AMR genes.
Figure 5.
(
A) PCA analysis of countries based on the occurrence data of clinical pathogens listed in
Table 2; (
B) hierarchical clusters of countries based on the occurrence data of clinical pathogens listed in
Table 2.
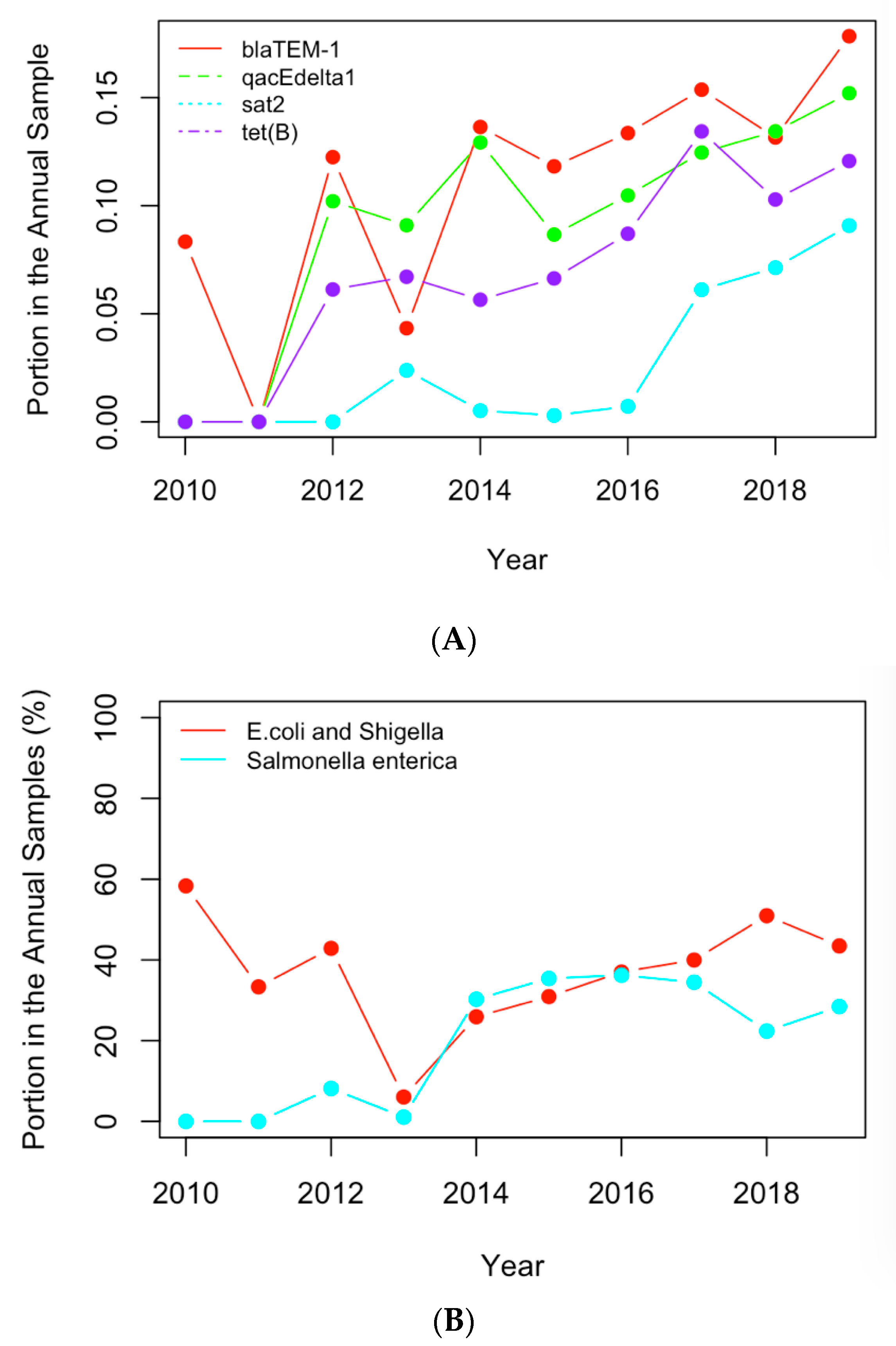
Figure 6.
(A) The historical profiles of the AMR genes with a generally increasing trend in their occurrence in the clinical samples collected in the US; (B) the historical profiles of the clinical pathogens with a generally increasing trend in their occurrence in the clinical samples collected in the US.
Figure 6.
(A) The historical profiles of the AMR genes with a generally increasing trend in their occurrence in the clinical samples collected in the US; (B) the historical profiles of the clinical pathogens with a generally increasing trend in their occurrence in the clinical samples collected in the US.
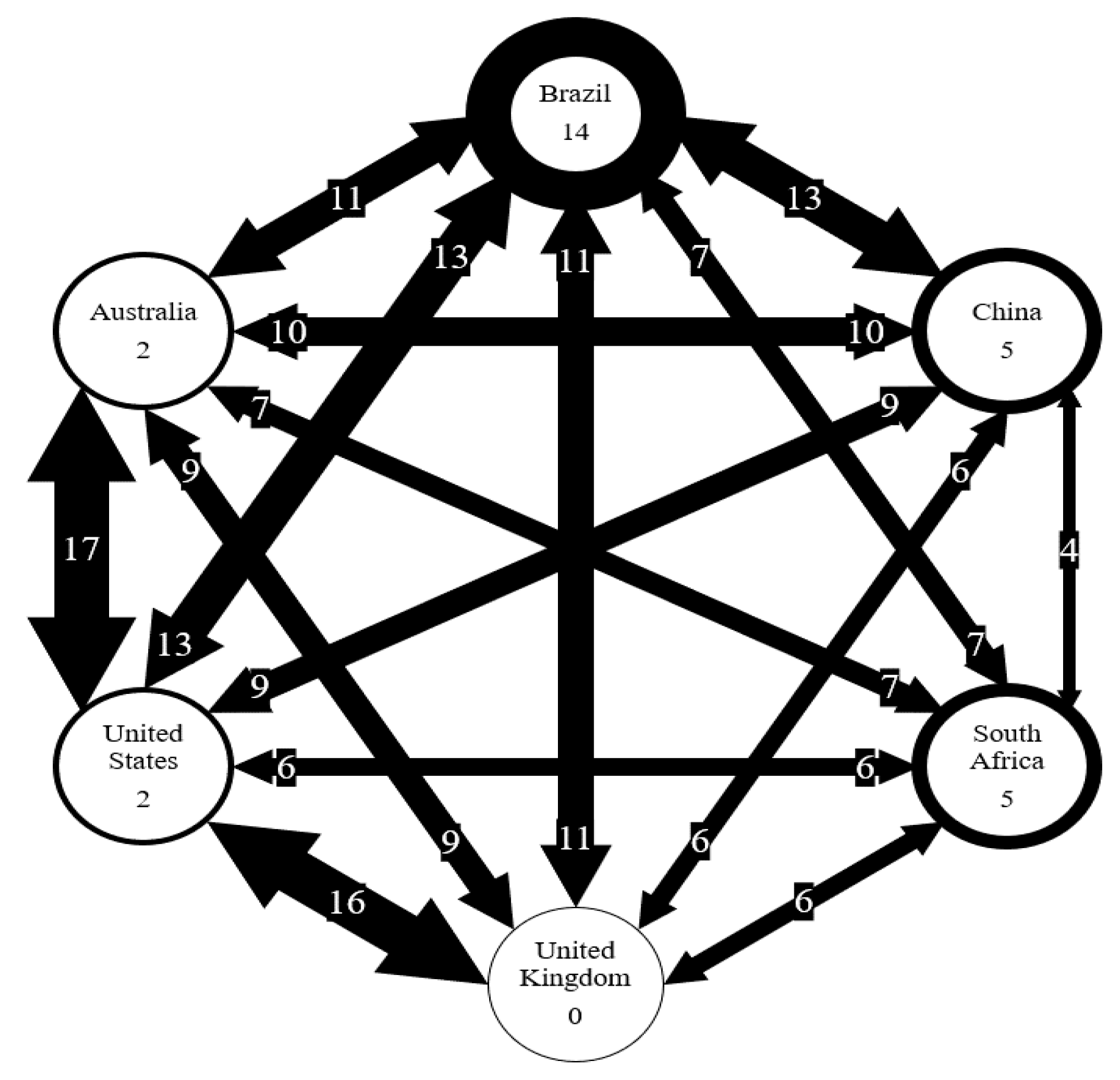
Figure 7.
Number of AMR genes shared between six countries. Number on arrows represent the number of genes shared. Thickness of arrow correlated to the number of genes shared. Number in circles represents the number of genes unique to each country.
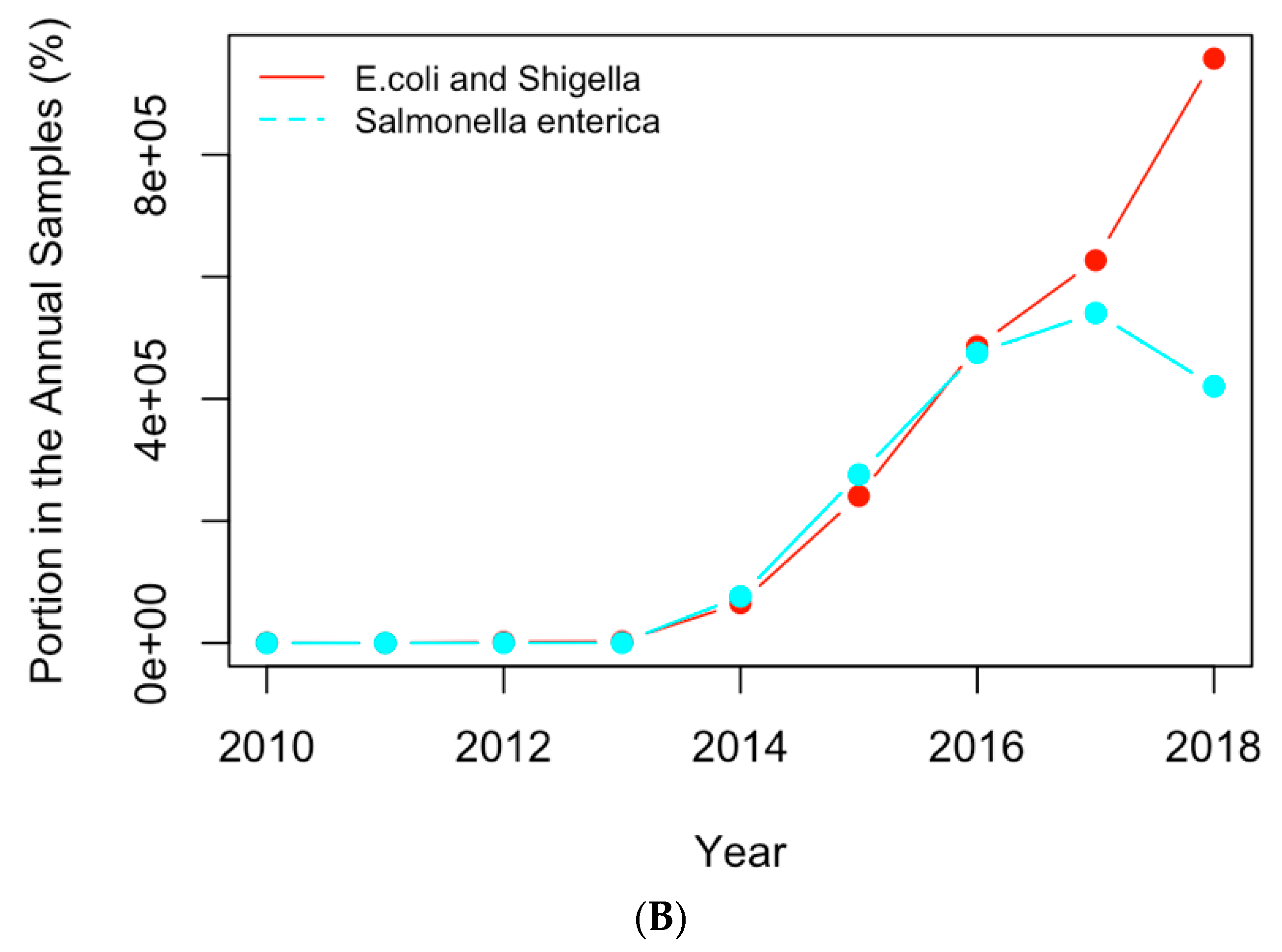
Figure 8.
Replotting
Figure 6 with the actual numbers of samples containing the AMR genes (
A) and clinical pathogens (
B) in USA.
Table 1.
The genes mostly involved in antimicrobial resistance in the six countries. Red text indicates the genes shared by all six countries, while blue denotes genes common to five countries.
| Australia | Brazil | China | South Africa | UK | US |
|---|
| aac(3)-I | aac(3)-IIa | aadA2 | aac(2′)-Ic | aadA | aadA |
| aadA1 | aac(6′)-Ib | ant(3″)-IIa | aph(3″)-Ib | aadA1 | aadA1 |
| aadA5 | aadA2 | aph(3″)-Ib | aph(6)-Id | aph(3″)-Ib | aadA2 |
| ant(3″)-IIa | aadA7 | aph(3′)-Ia | blaA | aph(6)-Id | aadA5 |
| aph(3″)-Ib | aph(3″)-Ib | aph(6)-Id | blaCTX-M-15 | blaEC | aph(3″)-Ib |
| aph(3′)-Ia | aph(3′)-IIb | armA | blaTEM-1 | blaOXA-1 | aph(3′)-Ia |
| aph(6)-Id | aph(3′)-Ia | blaEC | erm(37) | blaTEM-1 | aph(6)-Id |
| blaADC-30 | aph(6)-Id | blaKPC-2 | fosX | catA1 | blaCMY-2 |
| blaEC | blaCTX-M-15 | blaOXA-23 | lin | dfrA1 | blaEC |
| blaOXA-23 | blaEC | blaOXA-66 | qacEdelta1 | mph(A) | blaOXA-1 |
| blaOXA-66 | blaKPC-2 | blaSHV-11 | sul1 | qacEdelta1 | blaTEM-1 |
| blaTEM-1 | blaOXA | blaTEM-1 | sul2 | sat2 | catA1 |
| dfrA1 | blaOXA-494 | dfrA12 | | sul1 | dfrA1 |
| dfrA17 | blaOXA-56 | floR | | sul2 | dfrA17 |
| erm(B) | blaPDC-5 | fosA | | tet(A) | erm(B) |
| mph(A) | blaSHV-11 | mcr-1.1 | | tet(B) | floR |
| qacEdelta1 | blaSPM-1 | mph(A) | | | fosA7 |
| sat2 | blaTEM-1 | mph(E) | | | mph(A) |
| sul1 | catA1 | msr(E) | | | qacEdelta1 |
| sul2 | catB7 | oqxA | | | sat2 |
| tet(A) | cmx | oqxB | | | sul1 |
| tet(B) | crpP | qacEdelta1 | | | sul2 |
| | dfrA12 | | | | tet(A) |
| | fosA | | | | tet(B) |
| | mph(A) | | | | |
| | oqxA | | | | |
| | oqxB | | | | |
| | qacEdelta1 | | | | |
| | rmtD1 | | | | |
| | sul1 | | | | |
| | sul2 | | | | |
| | tet(A) | | | | |
Table 2.
The important pathogens for the six countries. Pathogens were identified through hierarchical clustering.
| Australia | Brazil | China | South Africa | UK | US |
|---|
| Acinetobacter baumannii | Acinetobacter baumannii | Acinetobacter baumannii | E. coli and Shigella | E. coli and Shigella | Acinetobacter baumannii |
| E. coli and Shigella | E. coli and Shigella | E. coli and Shigella | Enterobacter | Enterobacter | E. coli and Shigella |
| Klebsiella pneumoniae | Klebsiella pneumoniae | Enterobacter | Klebsiella pneumoniae | Klebsiella pneumoniae | Enterobacter |
| Mycobacterium tuberculosis | Pseudomonas aeruginosa | Klebsiella pneumoniae | Listeria monocytogenes | Salmonella enterica | Klebsiella pneumoniae |
| Salmonella enterica | Salmonella enterica | Salmonella enterica | Mycobacterium Tuberculosis | | Salmonella enterica |
| | | Vibrio cholerae | | | |
Table 3.
The important genes from
Table 1 with a generally increasing occurrence.
| China | South Africa | Brazil | Australia | UK | US |
|---|
| aadA2 | fosX | aadA7 | aac(3)-I | aadA1 | blaTEM-1 |
| blaEC | lim | aph(3″)-Ib | blaADC-30 | aph(3″)-Ib | qacEdelta1 |
| blaKPC-2 | | aph(3′)-IIb | blaOXA-66 | aph(6)-Id | sat2 |
| blaSHV-11 | | aph(3′)-Ia | aadA1 | blaTEM-1 | |
| blaTEM-1 | | aph(6)-Id | aph(3″)-Ib | dfrA1 | |
| dfrA12 | | blaCTX-M-15 | aph(6)-Id | mph(A) | |
| floR | | blaOXA-49 | sul2 | qacEdelta1 | |
| fosA | | blaPDC-5 | aadA5 | sat2 | |
| oqxA | | blaSHV-11 | dfrA17 | sul1 | |
| oqxB | | blaSPM-1 | dfrA1 | sul2 | |
| qacEdelta1 | | catB7 | erm(B) | tet(A) | |
| | | crpP | sat2 | tet(B) | |
| | | dfrA1 | | | |
| | | fosA | | | |
| | | mph(A) | | | |
| | | oqxB | | | |
| | | rmtD1 | | | |
| | | sul1 | | | |
Table 4.
The important pathogens from
Table 2 with an increasing occurrence.
| Australia | Brazil | China | South Africa | UK | US |
|---|
| Acinetobacter baumannii | Klebsiella pneumoniae | E. coli and Shigella | Listeria monocytogenes | None | E. coli and Shigella |
| E. coli and Shigella | Pseudomonas aeruginosa | Klebsiella pneumoniae | Mycobacterium Tuberculosis | None | Salmonella enterica |
Table 5.
Genes unique to each country.
| Australia | Brazil | China | South Africa | United Kingdom | United States |
|---|
| aac(3)-I | aac(3)-IIa | armA | aac(2′)-Ic | | blaCMY-2 |
| blaADC-30 | aac(6′)-Ib | blaSHV-11 | blaA | | fosA7 |
| | aadA7 | mcr-1.1 | erm37 | | |
| | aph(3′)-IIb | mph(E) | fosX | | |
| | blaCTX-M-15 | msr(E) | lin | | |
| | blaOXA | | | | |
| | blaOXA-494 | | | | |
| | blaOXA-56 | | | | |
| | blaPDC-5 | | | | |
| | blaSPM-1 | | | | |
| | catB7 | | | | |
| | cmx | | | | |
| | crpP | | | | |
| | rmtD1 | | | | |
Table 6.
Genes carried by different countries sorted by frequency. Only genes with a frequency of 3 or higher are shown.
| | Australia | Brazil | China | South Africa | UK | US | Total |
|---|
| aph(3″)-Ib | 1 | 1 | 1 | 1 | 1 | 1 | 6 |
| aph(6)-Id | 1 | 1 | 1 | 1 | 1 | 1 | 6 |
| blaTEM-1 | 1 | 1 | 1 | 1 | 1 | 1 | 6 |
| qacEdelta1 | 1 | 1 | 1 | 1 | 1 | 1 | 6 |
| blaEC | 1 | 1 | 1 | 0 | 1 | 1 | 5 |
| mph(A) | 1 | 1 | 1 | 0 | 1 | 1 | 5 |
| sul1 | 1 | 1 | 0 | 1 | 1 | 1 | 5 |
| sul2 | 1 | 1 | 0 | 1 | 1 | 1 | 5 |
| aph(3′)-Ia | 1 | 1 | 1 | 0 | 0 | 1 | 4 |
| tet(A) | 1 | 1 | 0 | 0 | 1 | 1 | 4 |
| aadA1 | 1 | 0 | 0 | 0 | 1 | 1 | 3 |
| aadA2 | 0 | 1 | 1 | 0 | 0 | 1 | 3 |
| catA1 | 0 | 1 | 0 | 0 | 1 | 1 | 3 |
| dfrA1 | 1 | 0 | 0 | 0 | 1 | 1 | 3 |
| sat2 | 1 | 0 | 0 | 0 | 1 | 1 | 3 |
| tet(B) | 1 | 0 | 0 | 0 | 1 | 1 | 3 |
Table 7.
Important pathogens carrying AMR genes from different countries sorted by frequency (Red: shared by all six countries; Orange - shared by five countries; Green: shared by four countries; and Blue: unique to one country.
| | Australia | Brazil | China | South Africa | UK | US | Total |
|---|
| E. coli and Shigella | 1 | 1 | 1 | 1 | 1 | 1 | 6 |
| Klebsiella pneumoniae | 1 | 1 | 1 | 1 | 1 | 1 | 6 |
| Salmonella enterica | 1 | 1 | 1 | 0 | 1 | 1 | 5 |
| Acinetobacter baumannii | 1 | 1 | 1 | 0 | 0 | 1 | 4 |
| Enterobacter | 0 | 0 | 1 | 1 | 1 | 1 | 4 |
| Listeria monocytogenes | 0 | 0 | 0 | 1 | 0 | 0 | 1 |
| Mycobacterium tuberculosis | 1 | 0 | 0 | 0 | 0 | 0 | 1 |
| Mycobacterium Tuberculosis | 0 | 0 | 0 | 1 | 0 | 0 | 1 |
| Pseudomonas aeruginosa | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| Vibrio cholerae | 0 | 0 | 1 | 0 | 0 | 0 | 1 |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}