Mapping Tyrosine Kinase Receptor Dimerization to Receptor Expression and Ligand Affinities


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
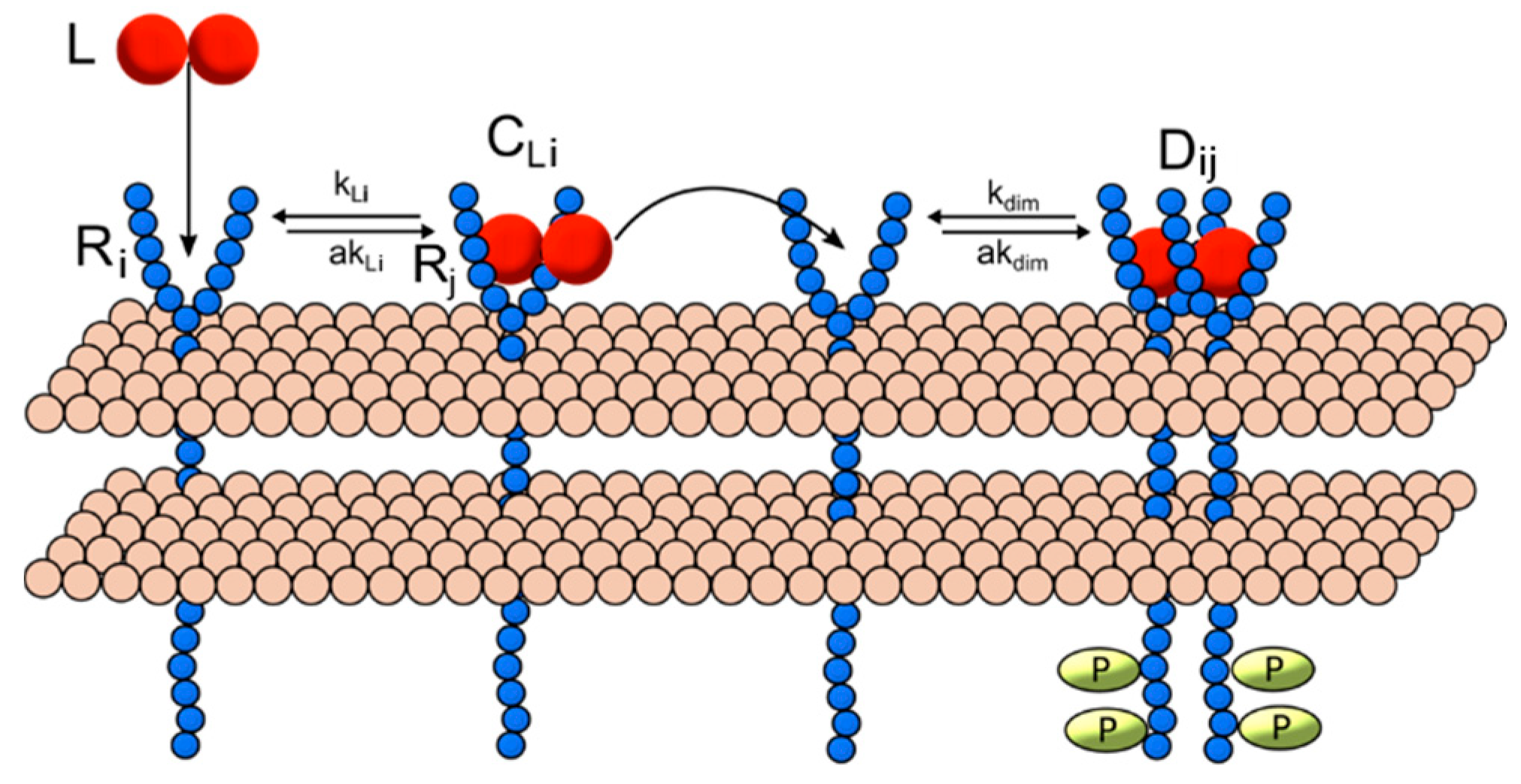
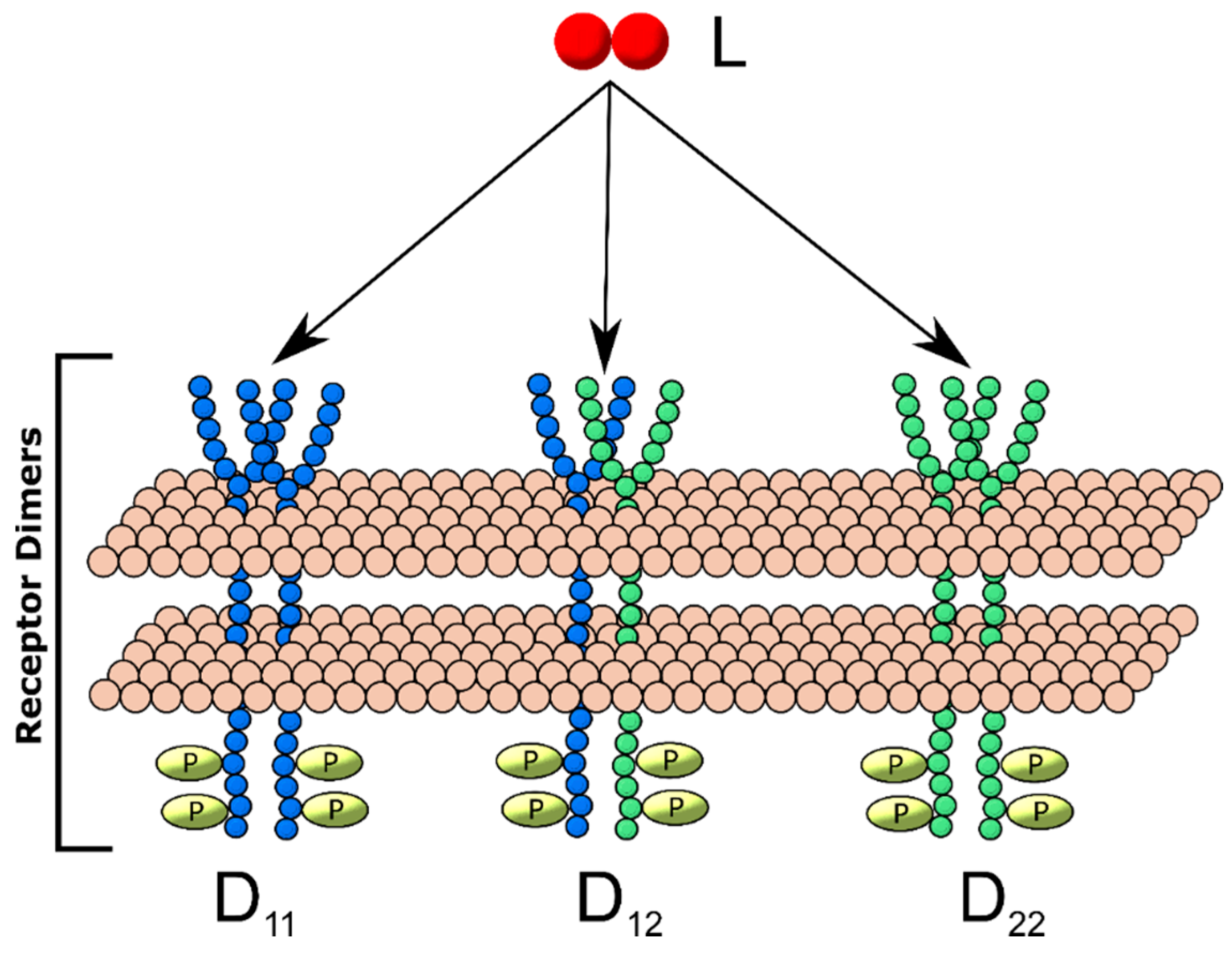
2.1. Ligand-Induced Dimerization Reaction Scheme
2.2. Model Parameters
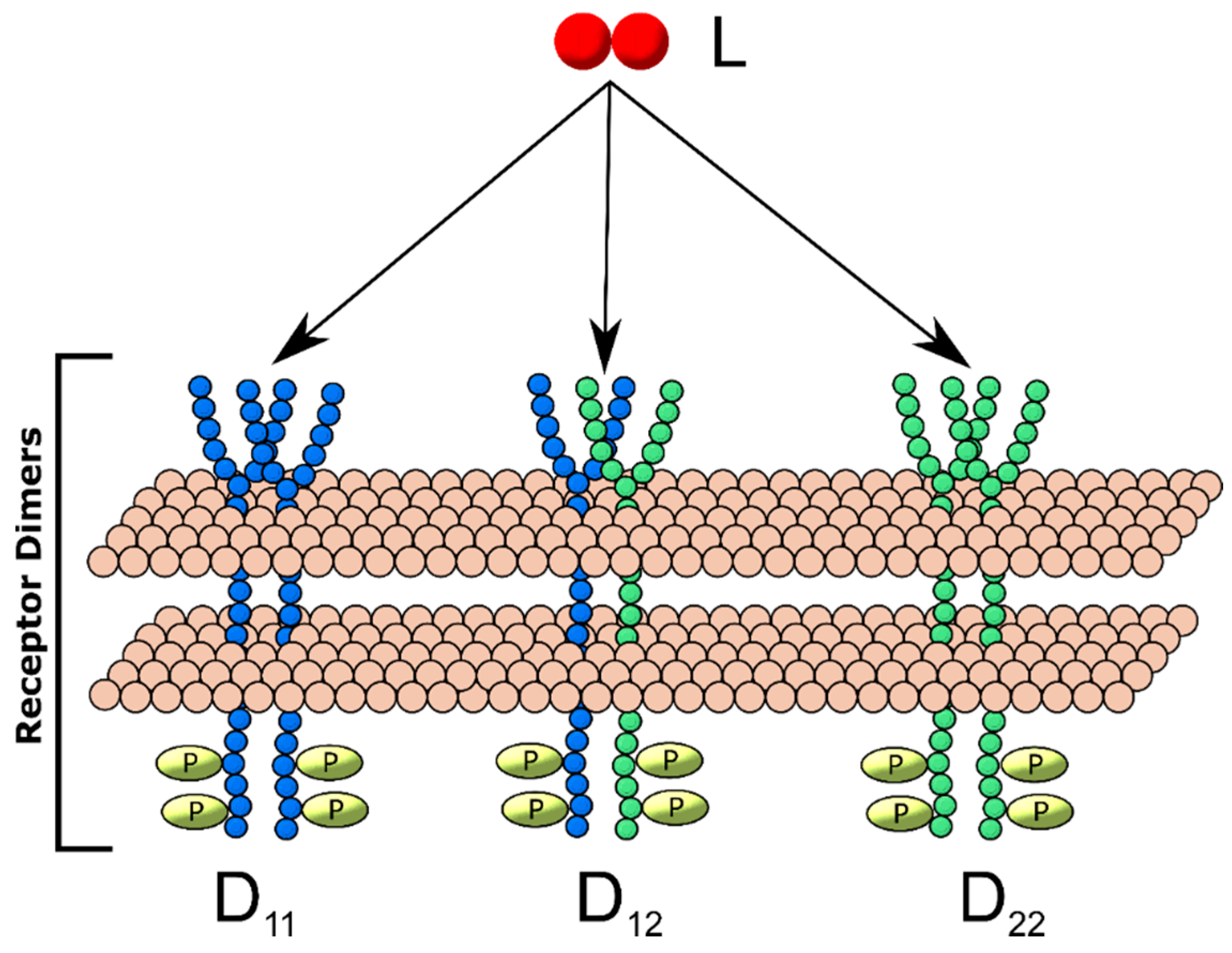
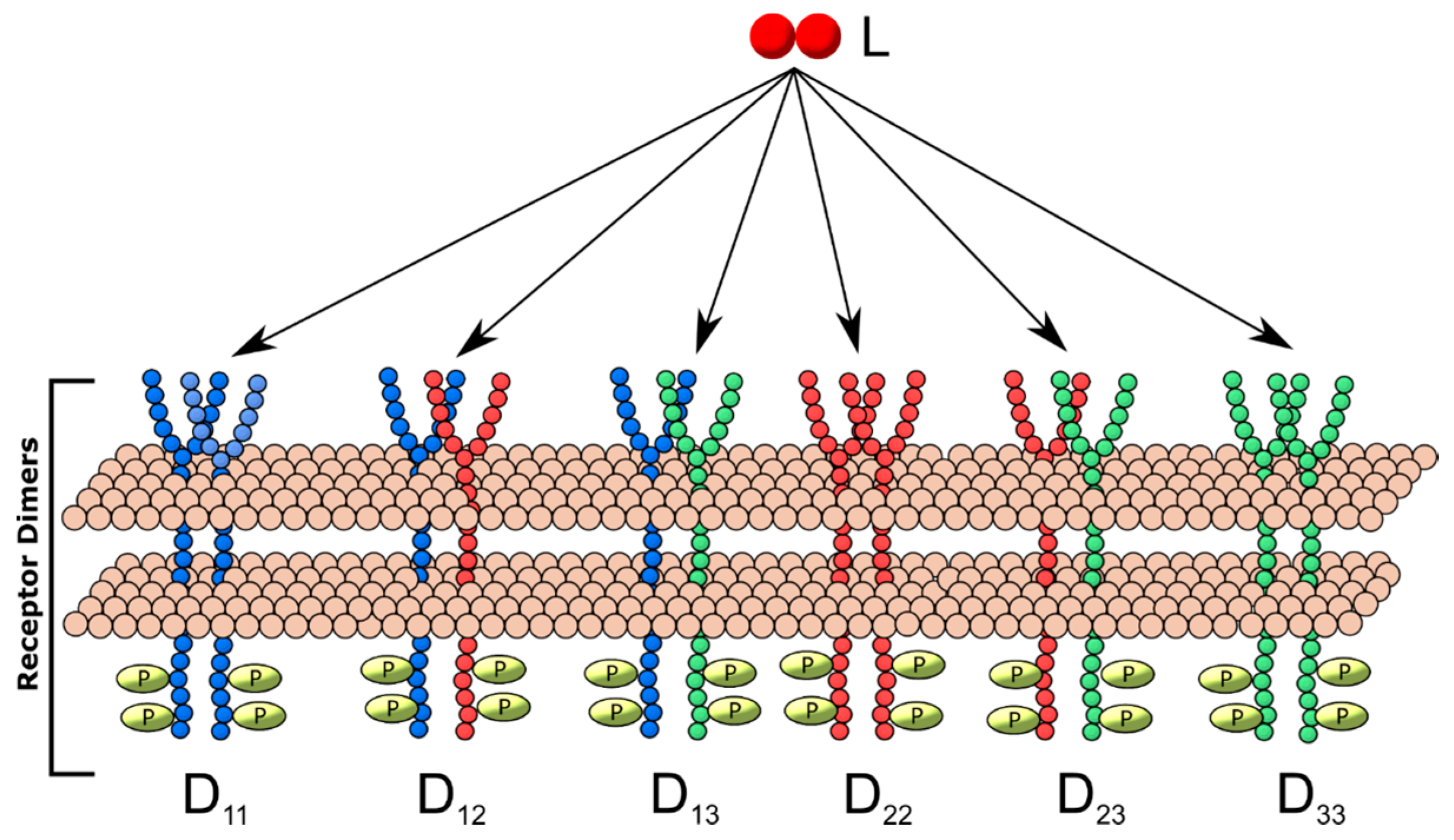
2.3. Generalized Multi-Receptor Models
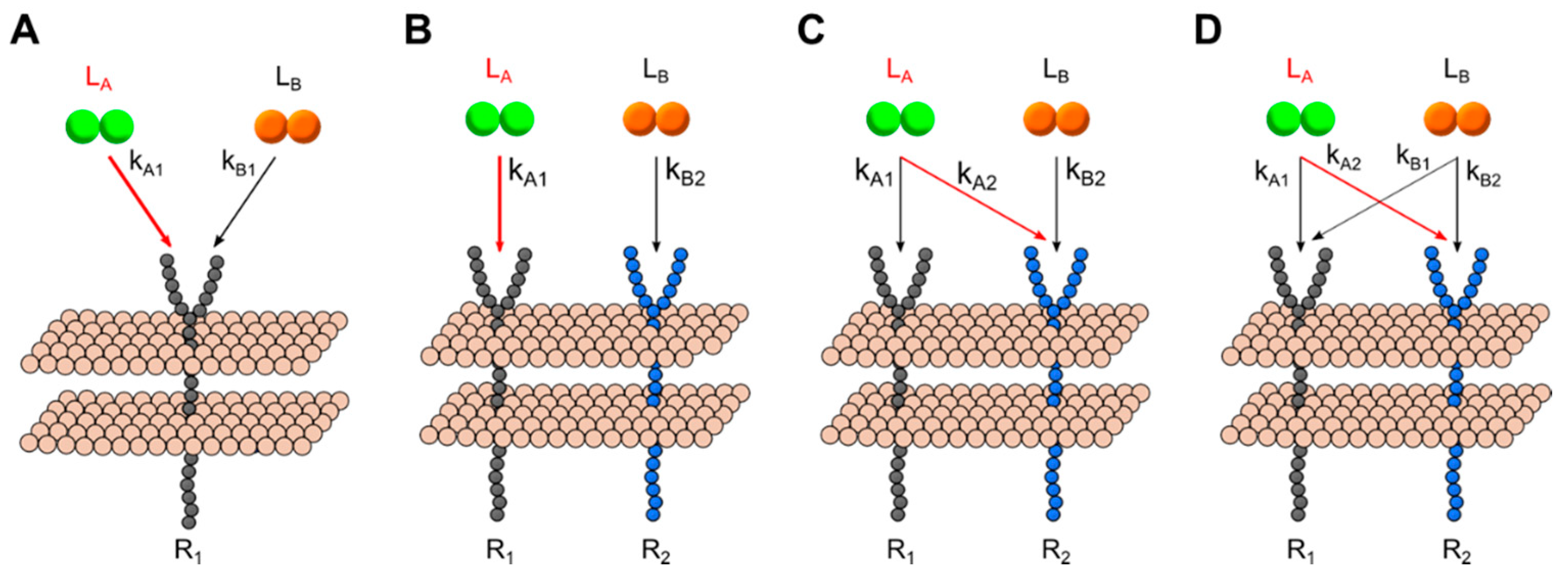
2.4. Generalized Multi-Ligand Models
3. Results
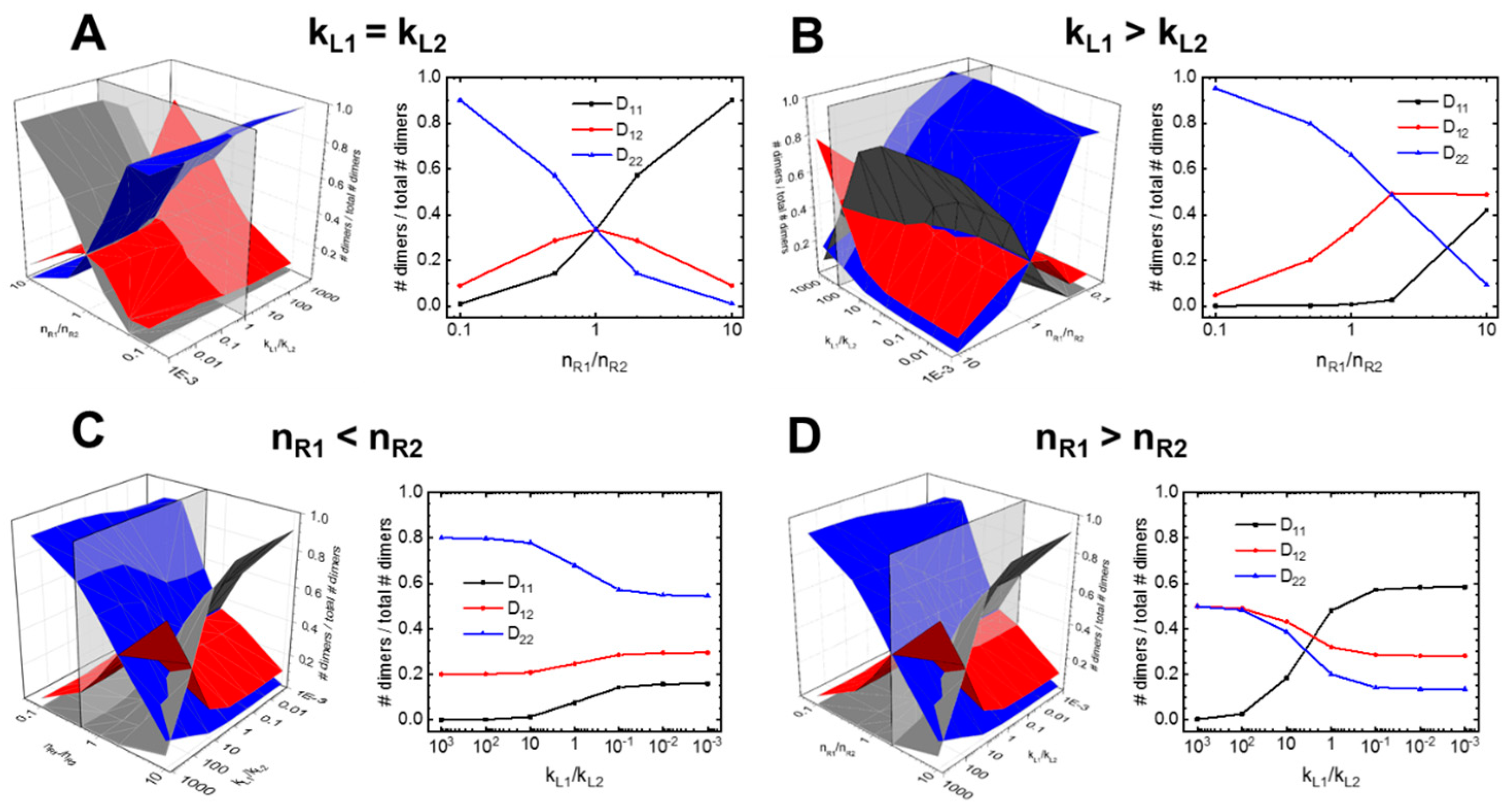
3.1. A guided Tour Across RTKs: Generalized Two-Receptor Model Demonstrates How Cross-Family Heterodimerization Complicates the Ligand:Receptor Binding Distributions
3.2. [R]-kL1 Relationships Mapped for Higher-Order Models
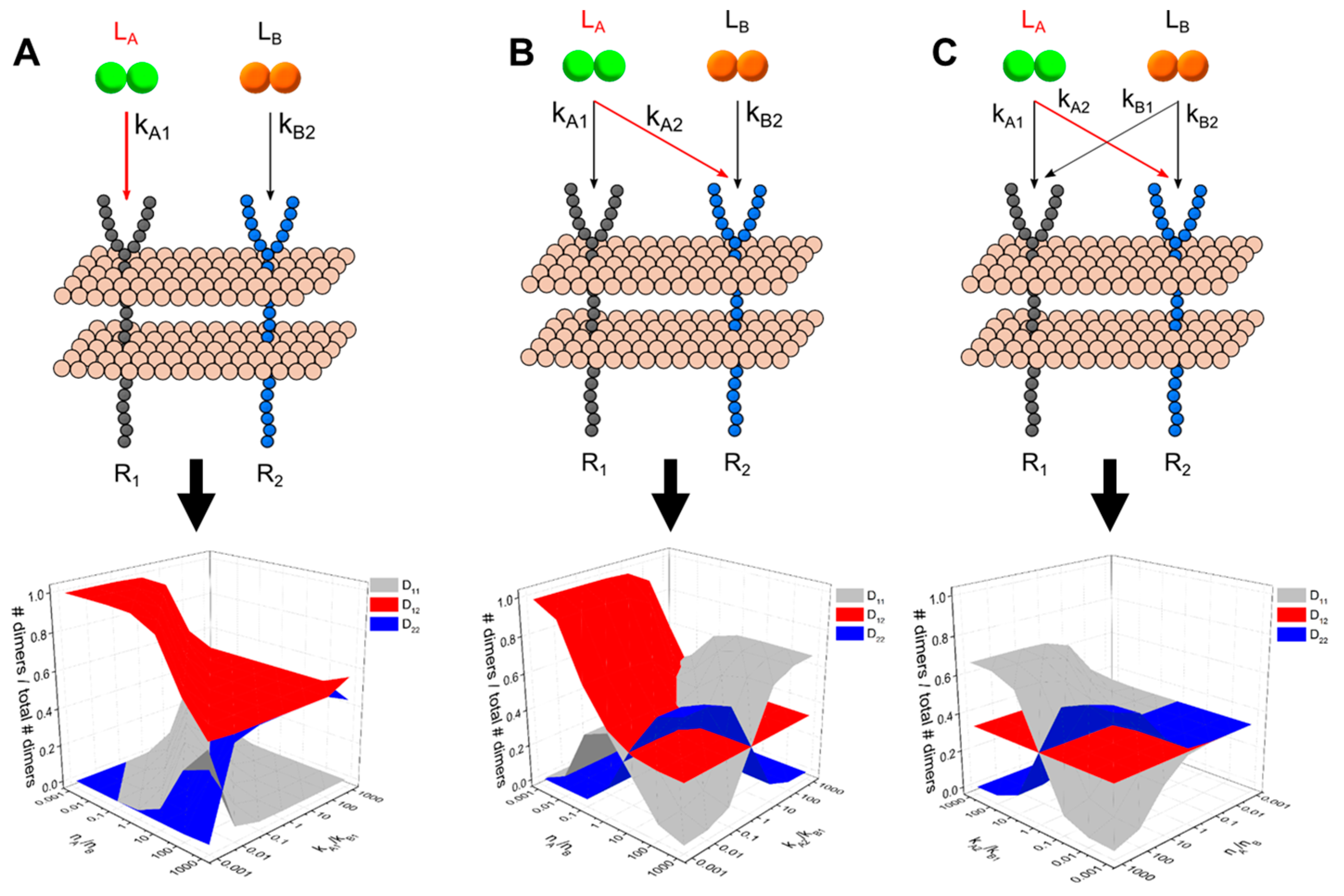
3.3. Dimerization Predictions Expanded for Cross-Family Ligand: Receptor Interaction Systems
4. Discussion
4.1. Generalized Modeling Predictions Guide Exploration of Specific RTK Systems
4.2. Generalized Models Predict Dimerization Patterns Observed Across RTK Systems
4.3. Generalized Models Improve Model Reusability
4.4. Generalized Model Enables Exploration of Cross-Family Interactions in Human Disease
4.5. Simplifying Models Enables Larger Picture Insights of Complex Biological Systems
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Schlessinger, J.; Ullrich, A. Growth factor signaling by receptor tyrosine kinases. Neuron 1992, 9, 383–391. [Google Scholar] [CrossRef]
- Pierce, G.F.; Mustoe, T.A.; Altrock, B.W.; Deuel, T.F. Thomason, a Role of platelet-derived growth factor in wound healing. J. Cell. Biochem. 1991, 45, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Bennett, N.T.; Schultz, G.S. Growth factors and wound healing: Biochemical properties of growth factors and their receptors. Am. J. Surg. 1993, 165, 728–737. [Google Scholar] [CrossRef]
- Bowen-Pope, D.F.; Malpass, T.W.; Foster, D.M.; Ross, R. Platelet-derived growth factor in vivo: Levels, activity and rate of clearance. Blood 1984, 64, 458–469. [Google Scholar] [PubMed]
- Schierling, W.; Troidl, K.; Troidl, C.; Schmitz-Rixen, T.; Schaper, W.; Eitenmüller, I.K. The role of angiogenic growth factors in arteriogenesis. J. Vasc. Res. 2009, 46, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Taylor, P.; Peterman, S.M.; Prakash, A.; Moran, M.F. Epidermal Growth Factor Receptor Phosphorylation Sites Ser(991) and Tyr(998) Are Implicated in the Regulation of Receptor Endocytosis and Phosphorylations at Ser(1039) and Thr(1041). Mol. Cell. Proteom. MCP 2009, 8, 2131–2144. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Hristova, K. Receptor tyrosine kinase transmembrane domains: Function, dimer structure and dimerization energetics. Cell Adhes. Migr. 2010, 4, 249–254. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Sarabipour, S.; Ballmer-Hofer, K.; Hristova, K. VEGFR-2 conformational switch in response to ligand binding. eLife 2016, 5, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.D.; Hristova, K. The RTK Interactome: Overview and Perspective on RTK Heterointeractions. Chem. Rev. 2018. [Google Scholar] [CrossRef]
- Carmeliet, P. Angiogenesis in life, disease and medicine. Nature 2005, 438, 932–936. [Google Scholar] [CrossRef]
- Danis, R.P.; Ciulla, T.A.; Criswell, M.; Pratt, L. Anti-angiogenic therapy of proliferative diabetic retinopathy. Expert Opin. Pharmacother. 2001, 2, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Nakatsu, M.N.; Sainson, R.C.A.; Aoto, J.N.; Taylor, K.L.; Aitkenhead, M.; Pérez-del-Pulgar, S.; Carpenter, P.M.; Hughes, C.C.W. Angiogenic sprouting and capillary lumen formation modeled by human umbilical vein endothelial cells (HUVEC) in fibrin gels: The role of fibroblasts and Angiopoietin-1. Microvasc. Res. 2003, 66, 102–112. [Google Scholar] [CrossRef]
- Li, X.; Pontén, A.; Aase, K.; Karlsson, L.; Abramsson, A.; Uutela, M.; Bäckström, G.; Hellström, M.; Boström, H.; Li, H.; et al. PDGF-C is a new protease-activated ligand for the PDGF α-receptor. Nat. Cell Biol. 2000, 2, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Bråkenhielm, E.; Li, X.; Pietras, K.; Widenfalk, J.; Ostman, A.; Eriksson, U.; Cao, Y. Angiogenesis stimulated by PDGF-CC, a novel member in the PDGF family, involves activation of PDGFR-alphaalpha and -alphabeta receptors. FASEB J. 2002, 16, 1575–1583. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Kumar, A.; Zhang, F.; Lee, C.; Li, Y.; Tang, Z.; Arjuna, P. VEGF-independent angiogenic pathways induced by PDGF-C. Oncotarget 2010, 1, 309–314. [Google Scholar] [PubMed]
- Meadows, K.L.; Hurwitz, H.I. Anti-VEGF therapies in the clinic. Cold Spring Harb. Perspect. Med. 2012, 2, a006577. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [Green Version]
- Heldin, C.H. Targeting the PDGF signaling pathway in tumor treatment. Cell Commun. Signal. 2013, 11, 97. [Google Scholar] [CrossRef]
- Neufeld, G.; Cohen, T.; Gengrinovitch, S.; Poltorak, Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999, 13, 9–22. [Google Scholar] [CrossRef]
- Olsson, A.-K.; Dimberg, A.; Kreuger, J.; Claesson-Welsh, L. VEGF receptor signalling—In control of vascular function. Nat. Rev. Mol. Cell Biol. 2006, 7, 359–371. [Google Scholar] [CrossRef]
- Benedito, R.; Rocha, S.F.; Woeste, M.; Zamykal, M.; Radtke, F.; Casanovas, O.; Duarte, A.; Pytowski, B.; Adams, R.H. Notch-dependent VEGFR3 upregulation allows angiogenesis without VEGF–VEGFR2 signalling. Nature 2012, 484, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Cudmore, M.J.; Hewett, P.W.; Ahmad, S.; Wang, K.-Q.; Cai, M.; Al-Ani, B.; Fujisawa, T.; Ma, B.; Sissaoui, S.; Ramma, W.; et al. The role of heterodimerization between VEGFR-1 and VEGFR-2 in the regulation of endothelial cell homeostasis. Nat. Commun. 2012, 3, 972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixelius, J.; Mäkinen, T.; Wirzenius, M.; Karkkainen, M.J.; Wernstedt, C.; Alitalo, K.; Claesson-Welsh, L. Ligand-induced Vascular Endothelial Growth Factor Receptor-3 (VEGFR-3) Heterodimerization with VEGFR-2 in Primary Lymphatic Endothelial Cells Regulates Tyrosine Phosphorylation Sites. J. Biol. Chem. 2003, 278, 40973–40979. [Google Scholar] [CrossRef] [Green Version]
- Kovalenko, M.V.M.; Kazlauskas, A.; William, J.L.; Lane, M.D. Platelet-Derived Growth Factor Receptor Family. Encycl. Biol. Chem. 2004, 3, 399–406. [Google Scholar]
- Chen, P.P.-H.H.; Chen, X.; He, X. Platelet-derived growth factors and their receptors: Structural and functional perspectives. Biochim. Biophys. Acta (BBA)-Proteins 2012, 1834, 2176–2186. [Google Scholar] [CrossRef] [PubMed]
- Fretto, L.J.L.J.; Snape, A.J.A.J.; Tomlinson, J.E.J.; Seroogy, J.J.; Wolf, D.L.; LaRochelle, W.J.; Giese, N.A. Mechanism of platelet-derived growth factor (PDGF) AA, AB and BB binding to alpha and beta PDGF receptor. J. Biol. Chem. 1993, 268, 3625–3631. [Google Scholar] [PubMed]
- Claesson-Welsh, L. Signal transduction by the PDGF receptors. Prog. Growth Factor Res. 1994, 5, 37–54. [Google Scholar] [CrossRef]
- Kazlauskas, A. PDGFs and their receptors. Gene 2017, 614, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Ball, S.G.; Shuttleworth, C.A.; Kielty, C.M. Vascular endothelial growth factor can signal through platelet-derived growth factor receptors. J. Cell Biol. 2007, 177, 489–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamer, S.B.; Chen, S.; Weddell, J.C.; Palasz, A.; Wittenkeller, A.; Kumar, M.; Imoukhuede, P.I. Discovery of High-Affinity PDGF-VEGFR Interactions: Redefining RTK Dynamics. Sci. Rep. 2017, 7, 16439. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Pennock, S.D.; Chen, X.; Kazlauskas, A.; Wang, Z. Platelet-derived Growth Factor Receptor-mediated Signal Transduction from Endosomes. J. Biol. Chem. 2004, 279, 8038–8046. [Google Scholar] [CrossRef] [PubMed]
- Horn, F.; Jackson, R. General mass action kinetics. Arch. Ration. Mech. Anal. 1972, 47, 81–116. [Google Scholar] [CrossRef]
- Mac Gabhann, F.; Popel, A.S.; Gabhann, F.M. Model of competitive binding of vascular endothelial growth factor and placental growth factor to VEGF receptors on endothelial cells. Am. J. Physiol. 2004, 286, H153–H164. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, A.; Schlessinger, J. Signal transduction by receptors with tyrosine kinase activity. Cell 1990, 61, 203–212. [Google Scholar] [CrossRef]
- Hubbard, S.R.; Till, J.H. Protein tyrosine kinase structure and function. Annu. Rev. Biochem. 2000, 69, 373–398. [Google Scholar] [CrossRef]
- Cross, M.J.; Dixelius, J.; Matsumoto, T.; Claesson-Welsh, L. VEGF-receptor signal transduction. Trends Biochem. Sci. 2003, 28, 488–494. [Google Scholar] [CrossRef]
- Heldin, C.H.; Wasteson, A.; Westermark, B. Interaction of platelet-derived growth factor with its fibroblast receptor. Demonstration of ligand degradation and receptor modulation. J. Biol. Chem. 1982, 257, 4216–4221. [Google Scholar] [PubMed]
- Larrivee, B.; Karsan, A. Signaling pathways induced by vascular endothelial growth factor (review). Int. J. Mol. Med. 2000, 5, 447–456. [Google Scholar] [CrossRef]
- Roy, H.; Bhardwaj, S.; Ylä-Herttuala, S. Biology of vascular endothelial growth factors. FEBS Lett. 2006, 580, 2879–2887. [Google Scholar] [CrossRef] [Green Version]
- Waltenberger, J.; Claesson-Welsh, L.; Siegbahn, A.; Shibuya, M.; Heldin, C.H. Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor. J. Biol. Chem. 1994, 269, 26988–26995. [Google Scholar] [PubMed]
- Socinski, M.A. Multitargeted receptor tyrosine kinase inhibition: An antiangiogenic strategy in non-small cell lung cancer. Cancer Treat. Rev. 2011, 37, 611–617. [Google Scholar] [CrossRef]
- Antebi, Y.E.; Linton, J.M.; Klumpe, H.; Bintu, B.; Gong, M.; Su, C.; McCardell, R.; Elowitz, M.B. Combinatorial Signal Perception in the BMP Pathway. Cell 2017, 170, 1184–1196. [Google Scholar] [CrossRef]
- Hendriks, B.S.; Orr, G.; Wells, A.; Wiley, H.S.; Lauffenburger, D.A. Parsing ERK activation reveals quantitatively equivalent contributions from epidermal growth factor receptor and HER2 in human mammary epithelial cells. J. Biol. Chem. 2005, 280, 6157–6169. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, B.S.; Opresko, L.K.; Wiley, H.S.; Lauffenburger, D. Quantitative analysis of HER2-mediated effects on HER2 and epidermal growth factor receptor endocytosis. Distribution of homo- and heterodimers depends on relative HER2 levels. J. Biol. Chem. 2003, 278, 23343–23351. [Google Scholar] [CrossRef]
- Kozer, N.; Barua, D.; Orchard, S.; Nice, E.C.; Burgess, A.W.; Hlavacek, W.S.; Clayton, A.H. Exploring higher-order EGFR oligomerisation and phosphorylation—A combined experimental and theoretical approach. Mol. bioSyst. 2013, 9, 1849–1863. [Google Scholar] [CrossRef]
- Edwards, J.S.; Wilson, B.S.; Hanien, D.; Lidke, D.S.; Xu, X.-P.; Zahoransky-Kohalmi, G.; Swift, M.; Steinkamp, M.P.; Volkmann, N.; Chen, Y.; et al. Orchestration of ErbB3 signaling through heterointeractions and homointeractions. Mol. Biol. Cell 2015, 26, 4109–4123. [Google Scholar] [Green Version]
- Chen, L.; Merzlyakov, M.; Cohen, T.; Shai, Y.; Hristova, K. Energetics of ErbB1 transmembrane domain dimerization in lipid bilayers. Biophys. J. 2009, 96, 4622–4630. [Google Scholar] [CrossRef]
- Mac Gabhann, F.; Popel, A.S. Dimerization of VEGF receptors and implications for signal transduction: A computational study. Biophys. Chem. 2007, 128, 125–139. [Google Scholar] [CrossRef]
- Bajikar, S.S.; Janes, K.A. Multiscale models of cell signaling. Ann. Biomed. Eng. 2012, 40, 2319–2327. [Google Scholar] [CrossRef]
- Bray, D. The cell as a thermostat: How much does it know? Adv. Syst. Biol. 2012, 736, 193–198. [Google Scholar]
- Chen, S.; Ansari, A.; Sterrett, W.; Hurley, K.; Kemball, J.; Weddell, J.C.; Imoukhuede, P.I. Current state-of-the-art and future directions in systems biology. Prog. Commun. Sci. 2014, 1, 12–26. [Google Scholar]
- Weddell, J.C.; Chen, S.; Imoukhuede, P.I. VEGFR1 promotes cell migration and proliferation through PLCγ and PI3K pathways. NPJ Syst. Biol. Appl. 2018, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Mac Gabhann, F.; Ji, J.W.; Popel, A.S. Multi-scale computational models of pro-angiogenic treatments in peripheral arterial disease. Ann. Biomed. Eng. 2007, 35, 982–994. [Google Scholar] [CrossRef] [PubMed]
- Mac Gabhann, F.; Ji, J.W.; Popel, A.S. VEGF gradients, receptor activation and sprout guidance in resting and exercising skeletal muscle. J. Appl. Physiol. (1985) 2007, 102, 722–734. [Google Scholar] [CrossRef] [PubMed]
- Vempati, P.; Popel, A.S.; Mac Gabhann, F. Formation of VEGF isoform-specific spatial distributions governing angiogenesis: Computational analysis. BMC Syst. Biol. 2011, 5, 59. [Google Scholar] [CrossRef]
- Hucka, M.; Finney, A.; Sauro, H.M.; Bolouri, H.; Doyle, J.C.; Kitano, H.; Bornstein, B.J.; Bray, D.; Cuellar, A.A.; Dronov, S.; et al. The systems biology markup language (SBML): A medium for representation and exchange of biochemical network models. Bioinformatics 2003, 19, 524–531. [Google Scholar] [CrossRef]
- Garny, A.; Nickerson, D.P.; Cooper, J.; Santos, R.W.D.; Miller, A.K.; McKeever, S.; Nielsen, P.M.F.; Hunter, P.J. CellML and associated tools and techniques. Philos. Trans. R. Soc. A 2008, 366, 3017–3043. [Google Scholar] [CrossRef] [Green Version]
- Neal, M.L.; König, M.; Nickerson, D.; Mısırlı, G.; Kalbasi, R.; Dräger, A.; Atalag, K.; Chelliah, V.; Cooling, M.T.; Cook, D.L.; et al. Harmonizing semantic annotations for computational models in biology. Brief. Bioinform. 2018, 20, 1–11. [Google Scholar] [CrossRef]
- Heath, A.P.; Kavraki, L.E. Computational challenges in systems biology. Comput. Sci. Rev. 2009, 3, 1–17. [Google Scholar] [CrossRef]
- Kuperstein, I.; Cohen, D.P.A.; Pook, S.; Viara, E.; Calzone, L. NaviCell: A web-based environment for navigation, curation and maintenance of large molecular interaction maps. BMC Syst. Biol. 2013, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Maus, C.; Rybacki, S.; Uhrmacher, A.M. Rule-based multi-level modeling of cell biological systems. BMC Syst. Biol. 2011, 5, 166. [Google Scholar] [CrossRef]
- Greenberg, J.I.; Shields, D.J.; Barillas, S.G.; Acevedo, L.M.; Murphy, E.; Huang, J.; Scheppke, L.; Stockmann, C.; Johnson, R.S.; Angle, N.; et al. A role for VEGF as a negative regulator of pericyte function and vessel maturation. Nature 2008, 456, 809–813. [Google Scholar] [CrossRef] [PubMed]
- Von Tiedemann, B.; Bilitewski, U. Characterization of the vascular endothelial growth factor-receptor interaction and determination of the recombinant protein by an optical receptor sensor. Biosens. Bioelectron. 2002, 17, 983–991. [Google Scholar] [CrossRef]
- Imoukhuede, P.I.; Dokun, A.O.; Annex, B.H.; Popel, A.S. Endothelial cell-by-cell profiling reveals the temporal dynamics of VEGFR1 and VEGFR2 membrane localization after murine hindlimb ischemia. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1085–H1093. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Le, T.; Harley, B.A.C.; Imoukhuede, P.I. Characterizing Glioblastoma Heterogeneity via Single-Cell Receptor Quantification. Front. Bioeng. Biotechnol. 2018, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chylek, L.A.; Harris, L.A.; Tung, C.S.; Faeder, J.R.; Lopez, C.F.; Hlavacek, W.S. Rule-based modeling: A computational approach for studying biomolecular site dynamics in cell signaling systems. Wiley Interdiscip. Rev. Syst. Biol. Med. 2014, 6, 13–36. [Google Scholar] [CrossRef]
- Hlavacek, W.S.; Faeder, J.R.; Blinov, M.L.; Perelson, A.S.; Goldstein, B. The Complexity of Complexes in Signal Transduction. Biotechnol. Bioeng. 2003, 84, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Hlavacek, W.S.; Faeder, J.R. Rules for modeling signal-transduction systems. Science 2006, 2006, re6. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mamer, S.B.; Palasz, A.A.; Imoukhuede, P.I. Mapping Tyrosine Kinase Receptor Dimerization to Receptor Expression and Ligand Affinities. Processes 2019, 7, 288. https://doi.org/10.3390/pr7050288
Mamer SB, Palasz AA, Imoukhuede PI. Mapping Tyrosine Kinase Receptor Dimerization to Receptor Expression and Ligand Affinities. Processes. 2019; 7(5):288. https://doi.org/10.3390/pr7050288
Chicago/Turabian StyleMamer, Spencer B., Alexandra A. Palasz, and P. I. Imoukhuede. 2019. "Mapping Tyrosine Kinase Receptor Dimerization to Receptor Expression and Ligand Affinities" Processes 7, no. 5: 288. https://doi.org/10.3390/pr7050288