Tandem-Homodimer of a β-Sheet-Forming Short Peptide Inhibits Random-to-β Structural Transition of Its Original Monomer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. General
2.2. Noncommercial Compounds
2.3. Sample Preparation
2.4. Circular Dichroism (CD) spectroscopy
2.5. Transmission Electron Microscopy (TEM)
2.6. Atomic Force Microscopy (AFM)
2.7. Proteolytic Degradation
3. Results and Discussion
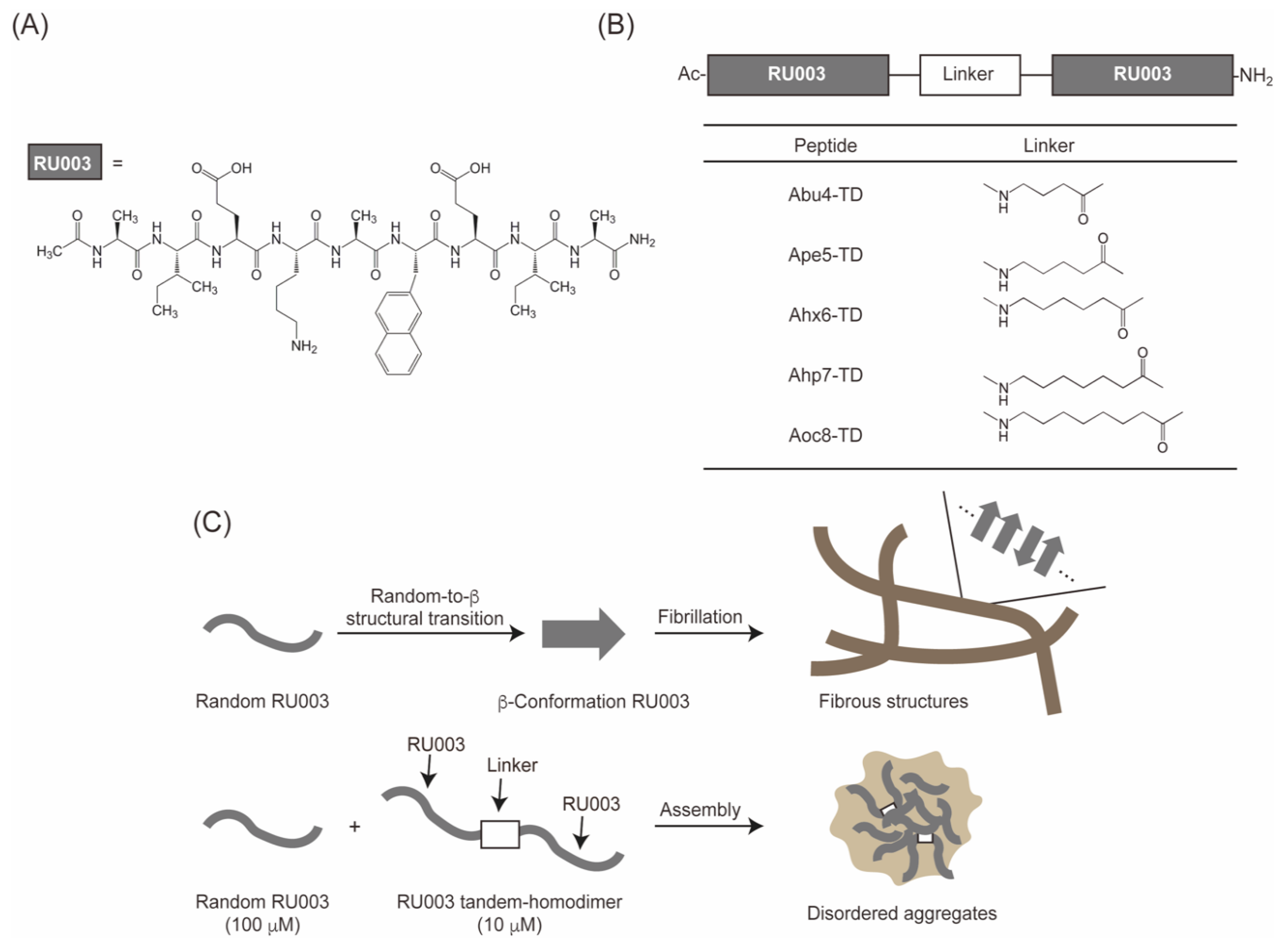
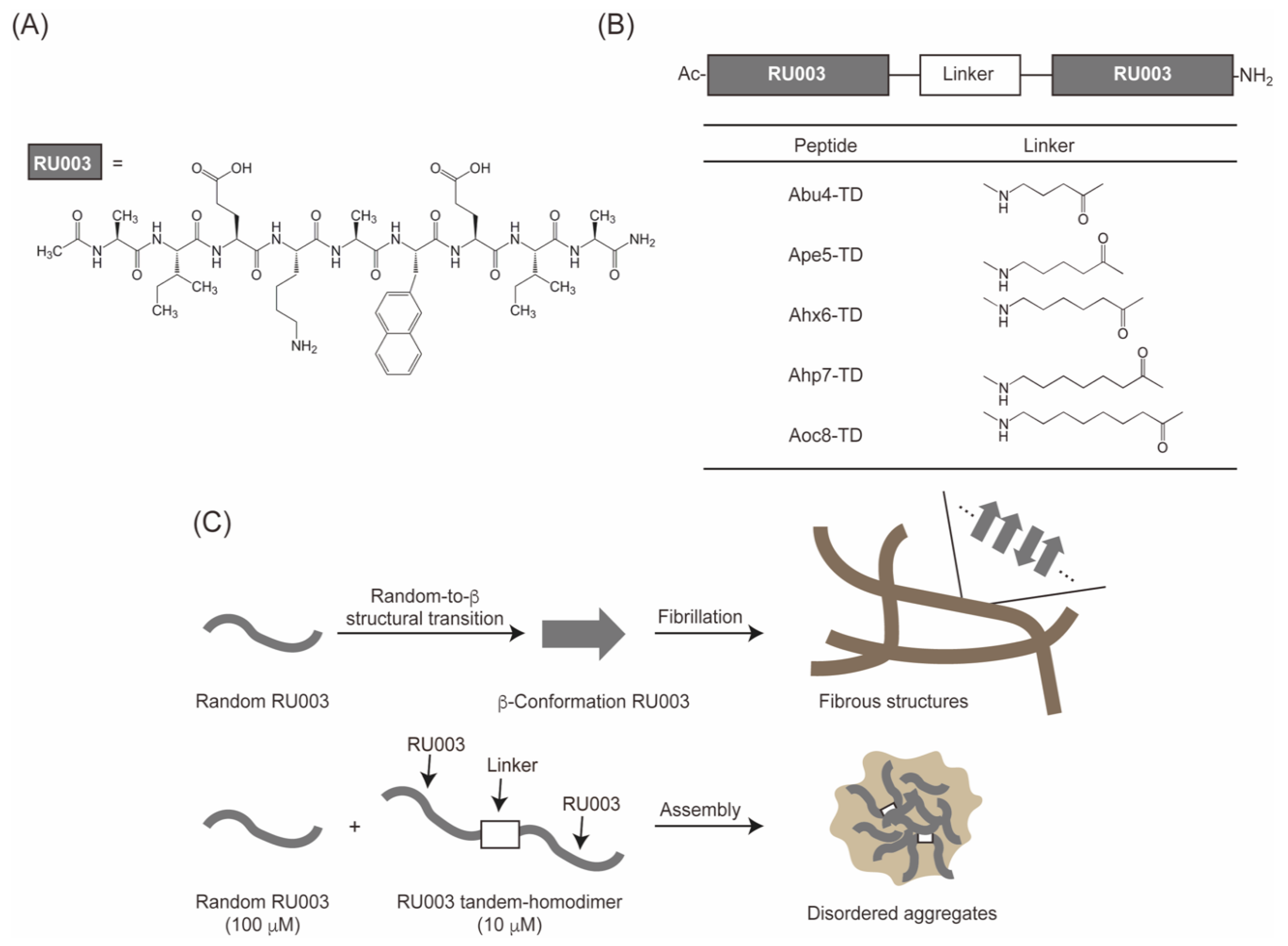
3.1. Design, Synthesis, and Characterization of the Peptides
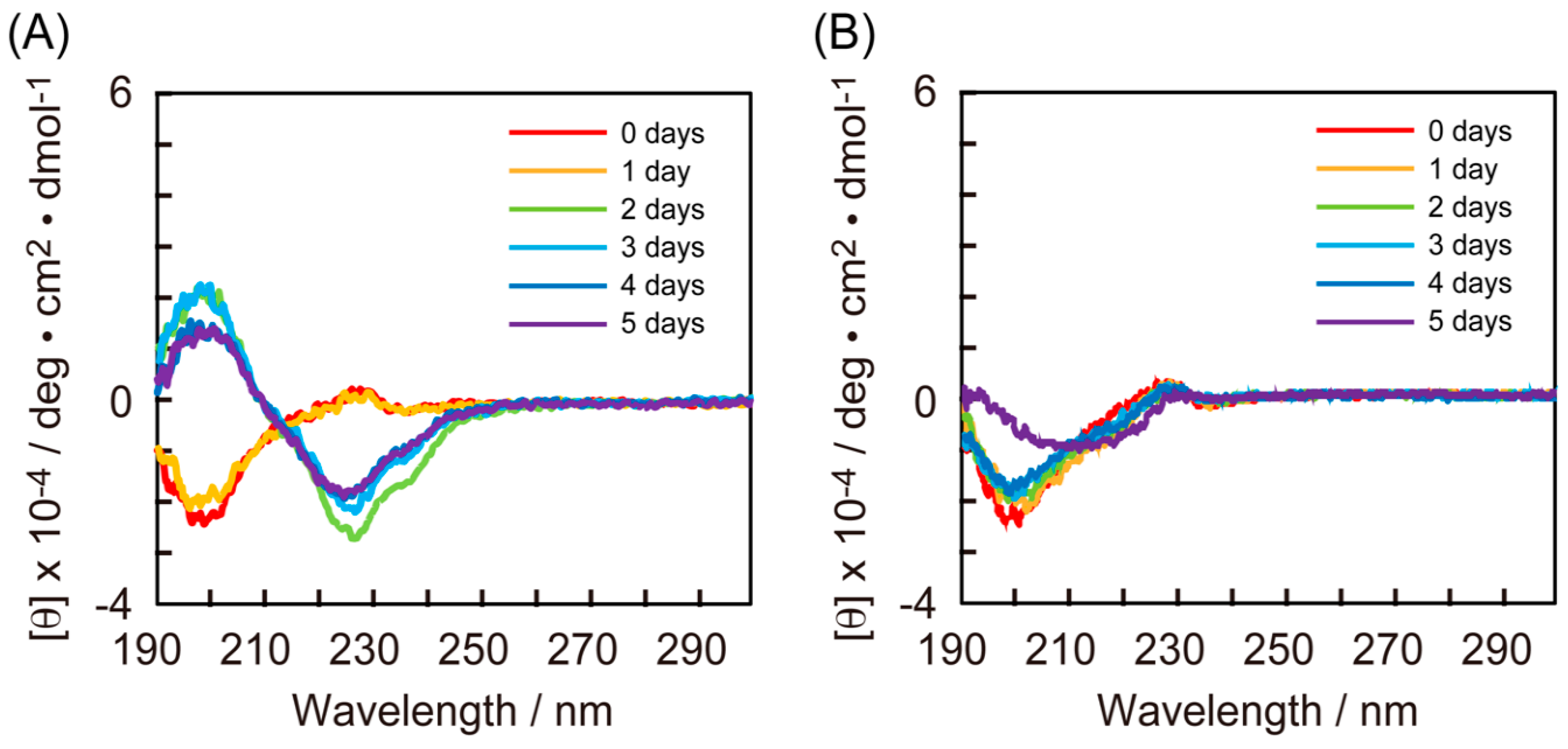
3.2. CD Measurements of RU003 Monomer and RU003 TDs alone
3.3. CD Measurements of the Mixtures of RU003 Monomer and Ape-TD
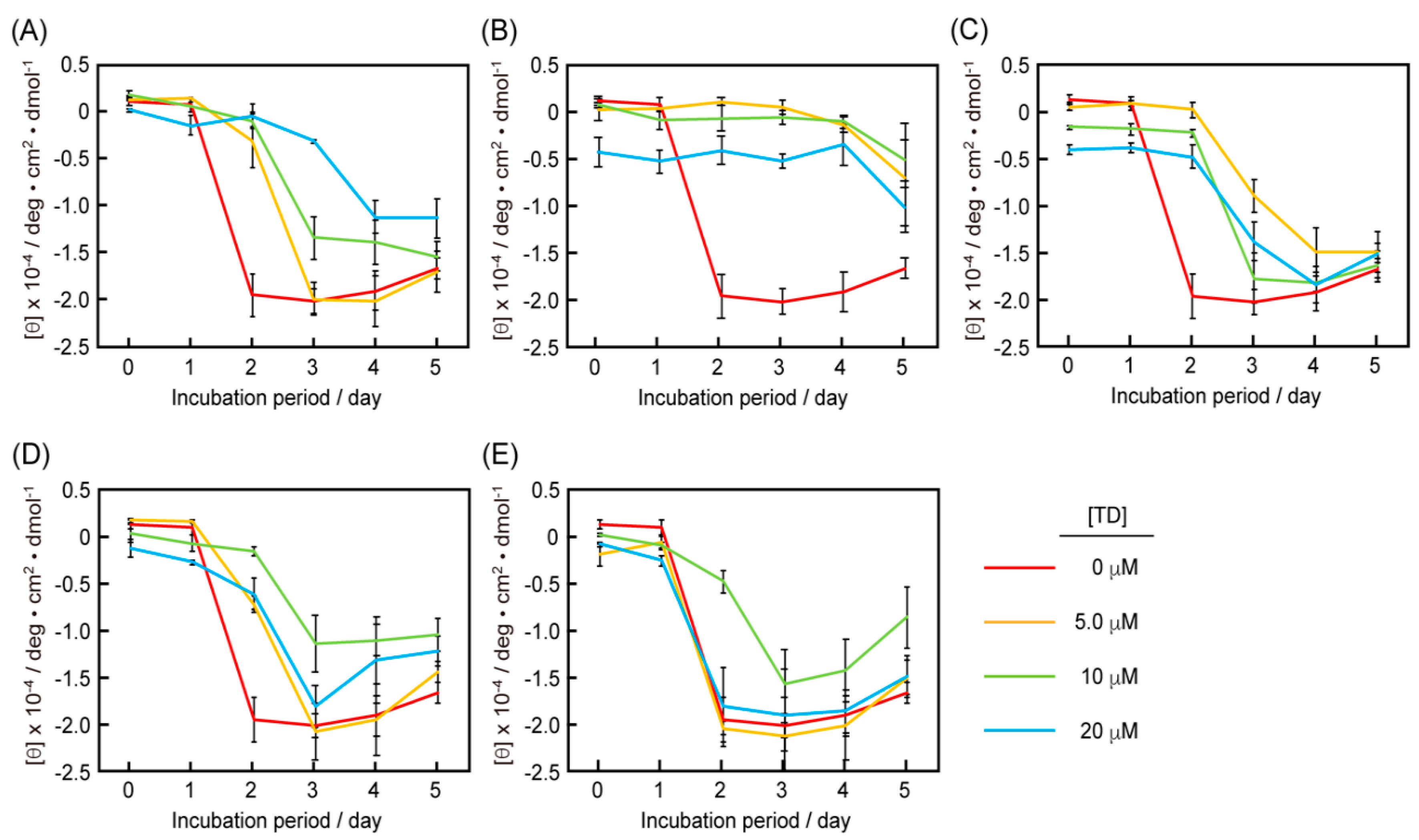
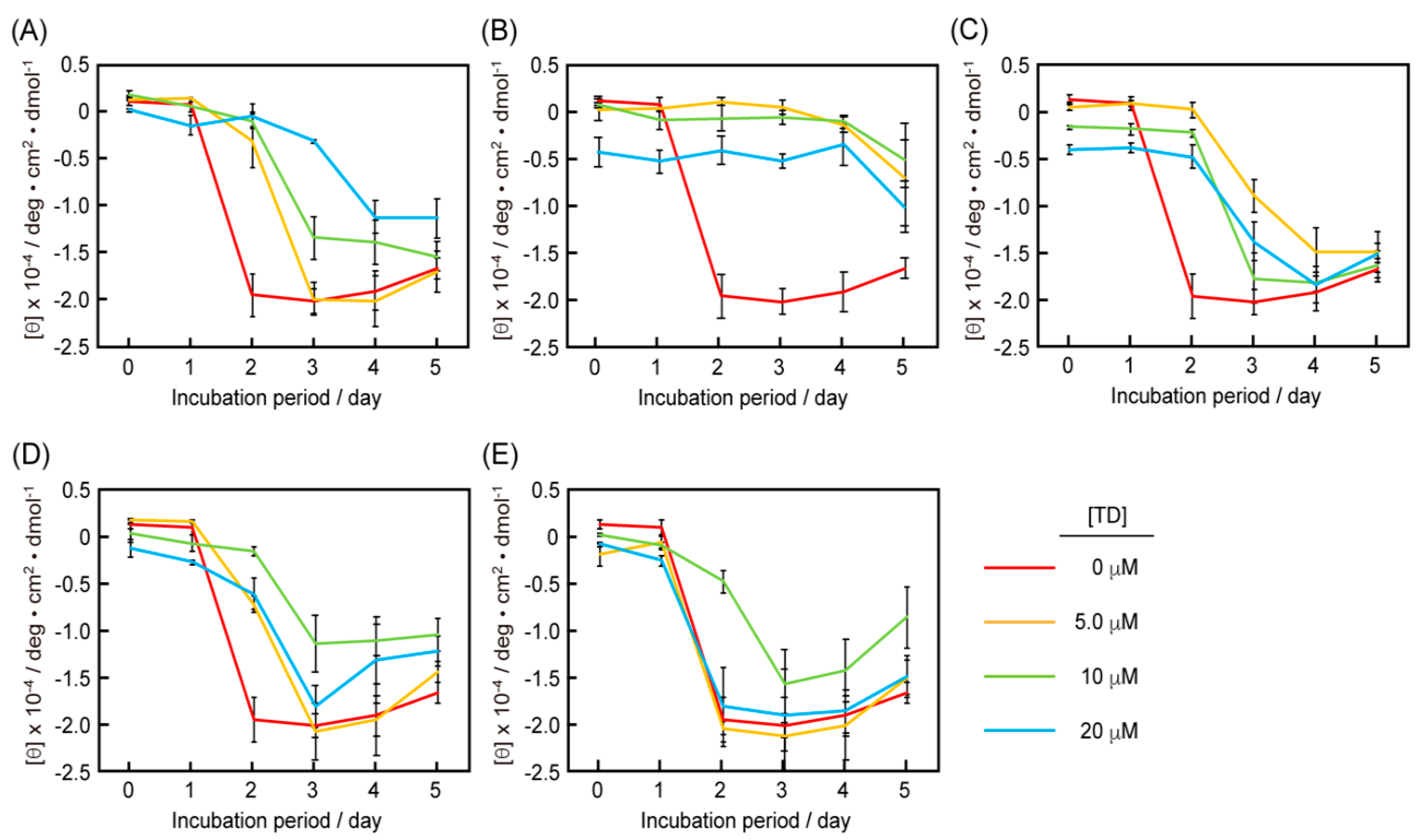
3.4. CD Measurements of the Mixtures of RU003 Monomer and RU003 TDs
3.5. Comparison of the Half-life Periods for the Random Peptide Structures
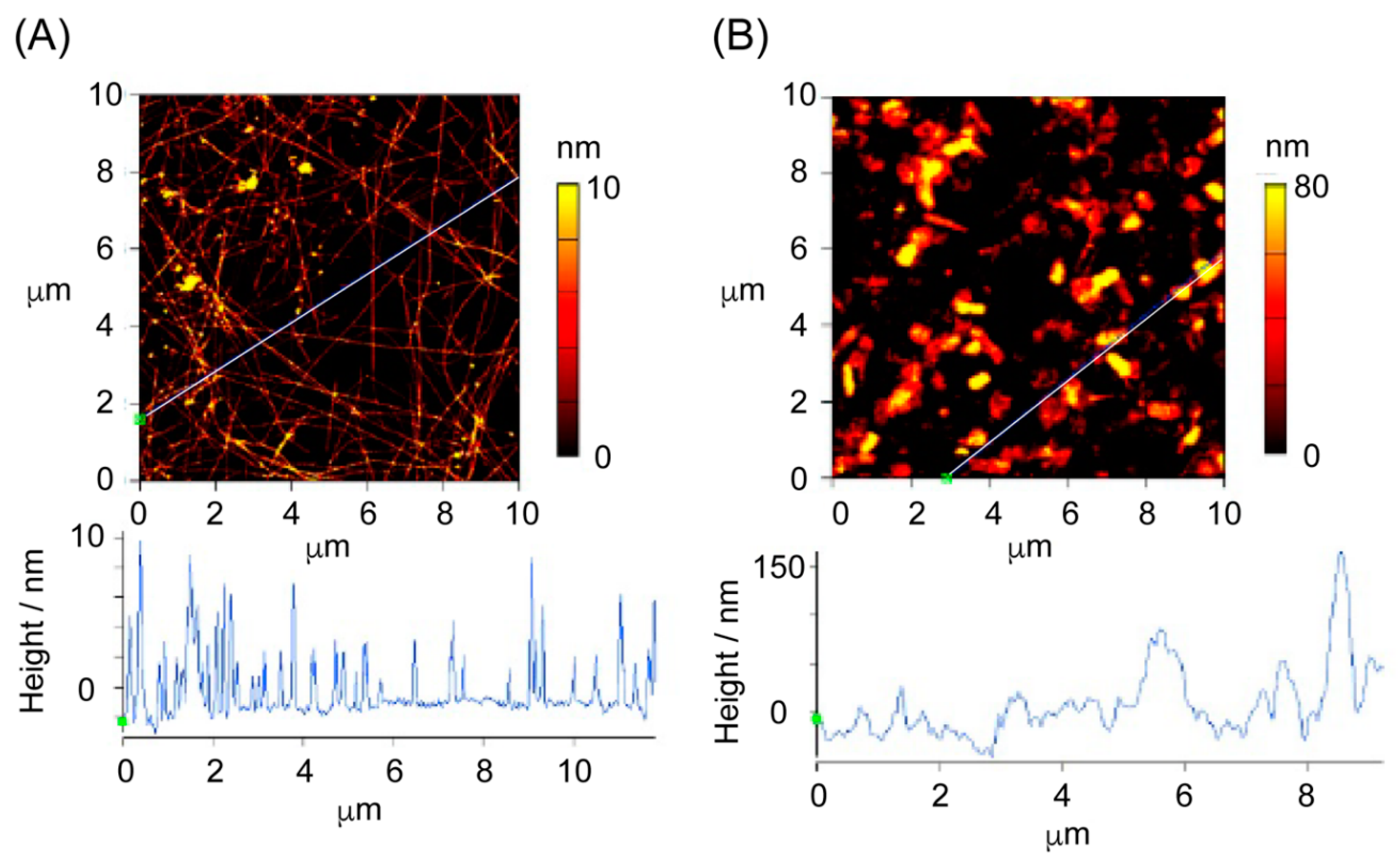
3.6. TEM and AFM Measurements of the Peptide Self-Assemblies
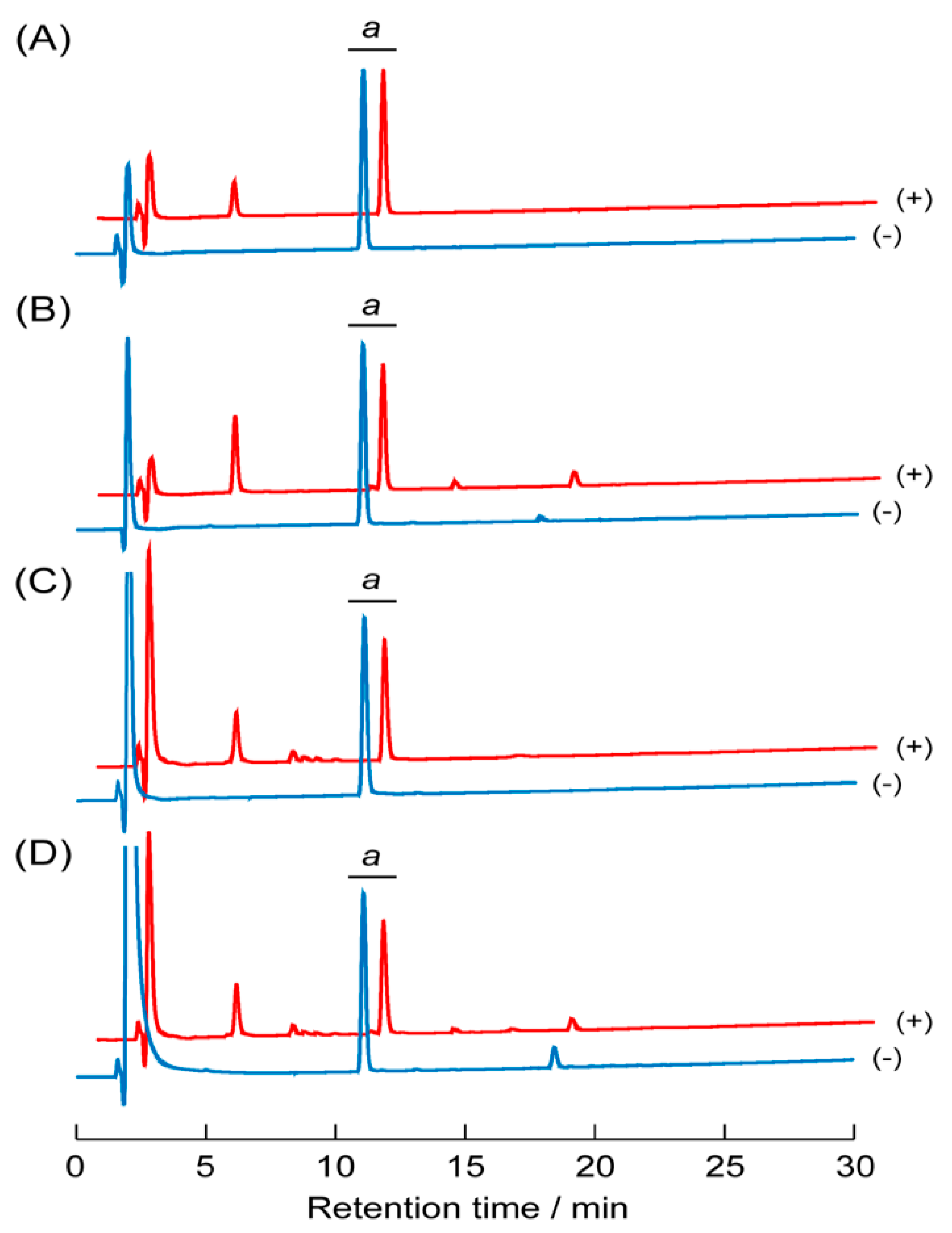
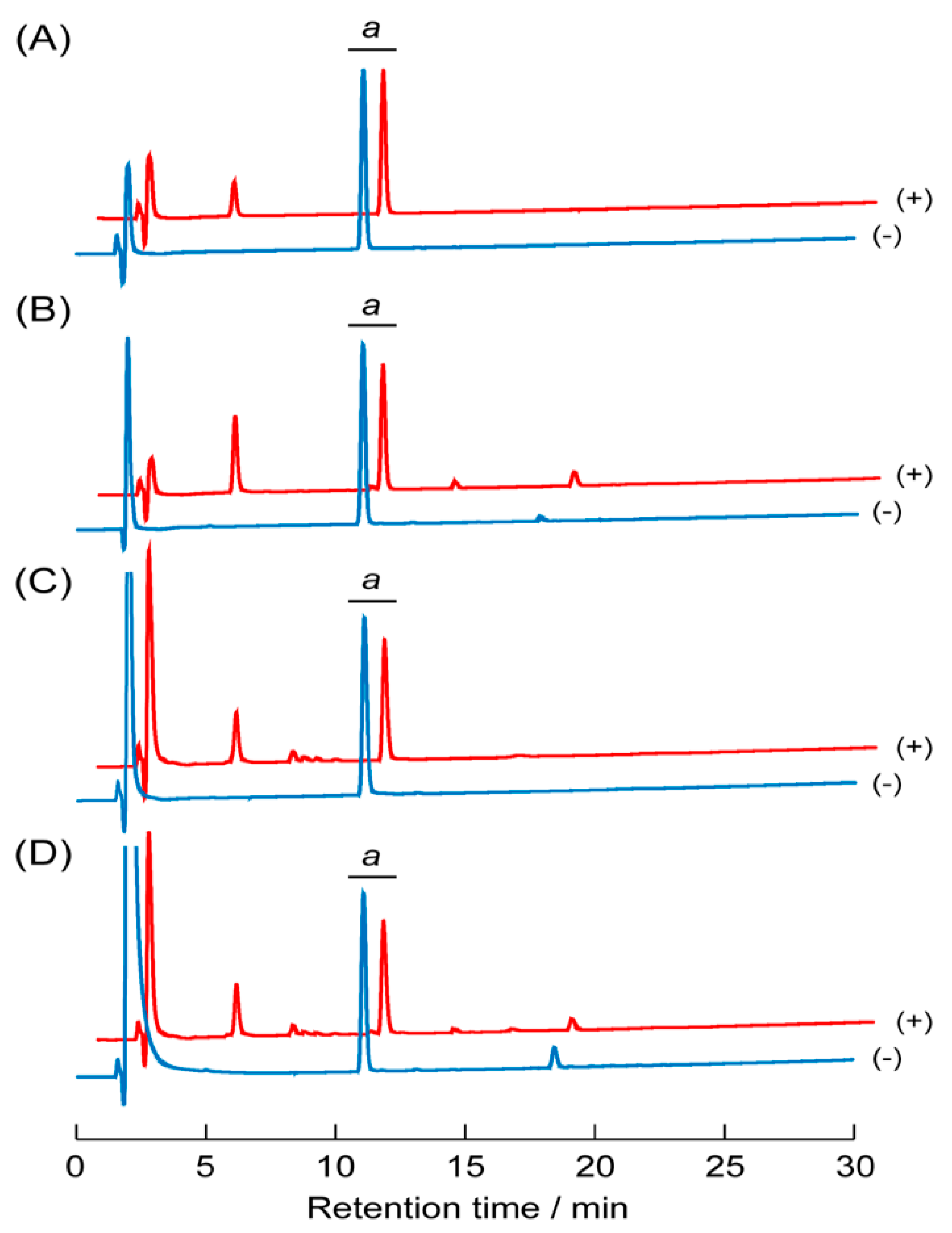
3.7. Proteolytic Stability of the Peptide Self-Assemblies
3.8. Possible Inhibitory Process
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Selkoe, D.J. Folding proteins in fatal ways. Nat. Cell Biol. 2003, 426, 900–904. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.X.; Qiang, W.; Yau, W.M.; Schwieters, C.D.; Meredith, S.C.; Tycko, R. Molecular structure of β-amyloid fibrils in Alzheimer’s disease brain tissue. Cell 2013, 154, 1257–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lansbury, P.T.; Lashuel, H.A. A century-old debate on protein aggregation and neurodegeneration enters the clinic. Nat. Cell Biol. 2006, 443, 774–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s disease and type II diabetes. Chem. Soc. Rev. 2012, 8, 101–112. [Google Scholar]
- Hamley, I.W. The Amyloid Beta Peptide: A Chemist’s Perspective. Role in Alzheimer’s and Fibrillization. Chem. Rev. 2012, 112, 5147–5192. [Google Scholar] [CrossRef] [PubMed]
- Hamley, I.W.; Castelletto, V. Self-Assembly of Peptide Bioconjugates: Selected Recent Research Highlights. Bioconjugate Chem. 2016, 28, 731–739. [Google Scholar] [CrossRef] [Green Version]
- Powers, E.T.; Powers, D.L. Mechanisms of Protein Fibril Formation: Nucleated Polymerization with Competing Off-Pathway Aggregation. Biophys. J. 2007, 94, 379–391. [Google Scholar] [CrossRef] [Green Version]
- Findeis, M.A. Approaches to discovery and characterization of inhibitors of amyloid β-peptide polymerization. Biochim. Biophys. Acta 2000, 1502, 76–84. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.; Moore, S.; Mayers, J.; Parkin, E.; Beeg, M.; Canovi, M.; Gobbi, M.; Mann, D.M.A.; Allsop, D. Development of a proteplytically stable retro-inverso peptide inhibitor of β-amyloid oligomerization as a potential novel treatment for Alzheimer’s disease. Biochemistry 2010, 49, 3261–3272. [Google Scholar] [CrossRef]
- Castelletto, V.; Ryumin, P.; Cramer, R.; Hamley, I.W.; Taylor, M.; Allsop, D.; Reza, M.; Ruokolainen, J.; Arnold, T.; Hermida-Merino, D.; et al. Self-Assembly and Anti-Amyloid Cytotoxicity Activity of Amyloid beta Peptide Derivatives. Sci. Rep. 2017, 7, 43637. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Park, J.; Wu, W.; Malagon, A.; Wang, L.; Vargas, E.; Wikramanayake, A.; Houk, K.N.; Leblanc, R.M. A resorcinrene for inhibition of Aβ fibrillation. Chem. Sci. 2017, 8, 2003–2009. [Google Scholar] [CrossRef] [Green Version]
- Jia, L.; Zhao, W.; Sang, J.; Wang, W.; Wei, W.; Wang, Y.; Zhao, F.; Lu, F.; Liu, F. Inhibitory Effect of a Flavonoid Dihydromyricetin against Aβ40 Amyloidogenesis and Its Associated Cytotoxicity. ACS Chem. Neurosci. 2019, 10, 4696–4703. [Google Scholar] [CrossRef]
- Sagnou, M.; Mavroidi, B.; Kaminari, A.; Boukos, N.; Pelecanou, M. Novel isatin thiosemicarbazone derivatives as potent inhibitors of β-amyloid peptide aggregation and toxicity. ACS Chem. Neurosci. 2020, 11, 266–2276. [Google Scholar] [CrossRef]
- Kumar, S.; Hamilton, A.D. α-Helix Mimetics as Modulators of Aβ Self-Assembly. J. Am. Chem. Soc. 2017, 139, 5744–5755. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Yu, C.-H.; Yu, H.; Borstnar, R.; Kamerlin, S.C.; Gräslund, A.; Abrahams, J.P.; Wärmländer, S.K. Cellular polyaminespromote amyloid-beta (Aβ) peptide fibrillation and modulate the aggregation pathways. ACS Chem. Neurosci. 2013, 4, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Ojha, B.; Morris, C.; Jiang, M.; Wojcikiewicz, E.P.; Rao, P.P.N.; Du, D. Positively Charged Chitosan and N-Trimethyl Chitosan Inhibit Aβ40 Fibrillogenesis. Biomacromolecules 2015, 16, 2363–2373. [Google Scholar] [CrossRef] [PubMed]
- Bucciantini, M.; Giannoni, E.; Chiti, F.; Baroni, F.; Formigli, L.; Zurdo, J.; Taddei, N.; Ramponi, G.; Dobson, C.M.; Stefani, M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nat. Cell Biol. 2002, 416, 507–511. [Google Scholar] [CrossRef]
- Kirkitadze, M.D.; Bitan, G.; Teplow, D.B. Paradigm shifts in Alzheimer’s disease and other neurodegenerative disorders: The emerging role of oligomeric assemblies. J. Neurosci. Res. 2002, 69, 567–577. [Google Scholar] [CrossRef] [Green Version]
- Goedert, M.; Spillantini, M.G. A Century of Alzheimer’s Disease. Science 2006, 314, 777–781. [Google Scholar] [CrossRef] [Green Version]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef]
- Taneja, V.; Verma, M.; Vats, A. Toxic species in amyloid disorders: Oligomers or mature fibrils. Ann. Indian Acad. Neurol. 2015, 18, 138–145. [Google Scholar] [CrossRef]
- Mroczko, B.; Groblewska, M.; Litman-Zawadska, A.; Kornhuber, J.; Lewczuk, P. Amyloid β oligomers (AβOs) in Alzheimer’s disease. J. Neural Transm. 2018, 125, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Bitan, G.; kirkitadze, M.D.; Lomakin, A.; Vollers, S.S.; Benedek, G.B.; Teplow, D.B. Amyloid β-protein (Aβ) assembly: Aβ40 and Aβ42 oligomerize through distinct pathways. Proc. Natl. Acad. Sci. USA 2003, 100, 330–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urbanc, B.; Cruz, L.; Yun, S.; Buldyrev, S.V.; Bitan, G.; Teplow, D.B.; Stanley, H.E. In silico study, of amyloid β-protein folding and oligomerization. Proc. Natl. Acad. Sci. USA 2004, 101, 17345–17350. [Google Scholar] [CrossRef] [Green Version]
- Murakami, K. Conformation-specific antibodies to target amyloid β oligomers and their application to immunotherapy for Alzheimer’s disease. Biosci. Biotechnol. Biochem. 2014, 78, 1293–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Rahimi, F.; Bitan, G. Modulation of amyloid β-protein assembly by homologous C-terminal fragments as a strategy for inhibiting Aβ toxicity. ACS Chem. Neurosci. 2016, 7, 845–856. [Google Scholar] [CrossRef] [Green Version]
- Deike, S.; Rothemund, S.; Voigt, B.; Samantray, S.; Strodel, B.; Binder, W.H. β-Turn mimetis synthetic peptides as amyloid-β aggregation inhibitors. Bioorg. Chem. 2020, 101, 104013. [Google Scholar] [CrossRef]
- Mustafi, S.M.; Garai, K.; Crick, S.L.; Baban, B.; Frieden, C. Substoichiometric inhibition of Aβ1-40 aggregation by a tandem Aβ40-1-Gly8-Aβ1-40 peptide. Biochim. Biophys. Res. Commun. 2010, 397, 509–512. [Google Scholar] [CrossRef] [Green Version]
- Klein, A.N.; Ziehm, T.; Tusche, M.; Buitenhuis, J.; Bartnik, D.; Böddrich, A.; Wiglenda, T.; Wanker, E.E.; Funke, S.A.; Brener, O.; et al. Optimization of the All-D Peptide D3 for Aβ Oligomer Elimination. PLoS ONE 2016, 11, e0153035. [Google Scholar] [CrossRef] [Green Version]
- Riek, R.; Güntert, P.; Döbeli, H.; Wipf, B.; Wüthrich, K. NMR studies in aqueous solution fail to identify significant conformational differences between the monomeric forms of two Alzheimer peptides with widely different plaque-competence, Aβ(1–40)ox and Aβ(1–42)ox. JBIC J. Biol. Inorg. Chem. 2001, 268, 5930–5936. [Google Scholar] [CrossRef] [Green Version]
- Swanekamp, R.J.; Weich, J.J.; Nilsson, B.L. Proteolytic stability of amphipathic peptide hydrogels composed of self-assembled pleated β-sheet or coassembled rippled β-sheet fibrils. Chem. Commun. 2014, 50, 10133–10136. [Google Scholar] [CrossRef]
- Tomizaki, K.-Y.; Kubo, S.; Ahn, S.-A.; Satake, M.; Imai, T. Biomimetic Alignment of Zinc Oxide Nanoparticles along a Peptide Nanofiber. Langmuir 2012, 28, 13459–13466. [Google Scholar] [CrossRef]
- Tomizaki, K.-Y.; Tanaka, A.; Shimada, H.; Nishizawa, K.; Wada, T.; Imai, T. The alkyl linkers in tandem-homodimers of a β-sheet-forming nonapeptide affect the self-assembled nanostructures. Bioorg. Med. Chem. Lett. 2016, 26, 2659–2662. [Google Scholar] [CrossRef]
- Diaferia, C.; Gianolio, E.; Sibillano, T.; Mercurio, F.A.; Leone, M.; Giannini, C.; Balasco, N.; Vitagliano, L.; Morelli, G.; Accardo, A. Cross-beta nanostructures based on dinaphthylalanine Gd-conjugates loaded with doxorubicin. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadqi, M.; Lapidus, L.J.; Muñoz, V. How fast is protein hydrophobic collapse? Proc. Natl. Acad. Sci. USA 2003, 100, 12117–12122. [Google Scholar] [CrossRef] [Green Version]
- Chan, W.C.; White, P.D. Fmoc Solid Phase Peptide Synthesis: A Practical Approach; Oxford University Press: New York, NY, USA, 2000; pp. 41–76. [Google Scholar]
- Manavalan, P.; Jr, W.C.J. Sensitivity of circular dichroism to protein tertiary structure class. Nat. Cell Biol. 1983, 305, 831–832. [Google Scholar] [CrossRef]
- Bowerman, C.J.; Liyanage, W.; Federation, A.J.; Nilsson, B.L. Tuning β-Sheet Peptide Self-Assembly and Hydrogelation Behavior by Modification of Sequence Hydrophobicity and Aromaticity. Biomacromolecules 2011, 12, 2735–2745. [Google Scholar] [CrossRef]
- Lee, N.R.; Bowerman, C.J.; Nilsson, B.L. Effects of Varied Sequence Pattern on the Self-Assembly of Amphipathic Peptides. Biomacromolecules 2013, 14, 3267–3277. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomizaki, K.-y.; Iori, T.; Fukushima, H.; Nakabayashi, Y.; Matsumoto, Y.; Imai, T. Tandem-Homodimer of a β-Sheet-Forming Short Peptide Inhibits Random-to-β Structural Transition of Its Original Monomer. Processes 2020, 8, 1421. https://doi.org/10.3390/pr8111421
Tomizaki K-y, Iori T, Fukushima H, Nakabayashi Y, Matsumoto Y, Imai T. Tandem-Homodimer of a β-Sheet-Forming Short Peptide Inhibits Random-to-β Structural Transition of Its Original Monomer. Processes. 2020; 8(11):1421. https://doi.org/10.3390/pr8111421
Chicago/Turabian StyleTomizaki, Kin-ya, Tomomi Iori, Hideyasu Fukushima, Yasuhiro Nakabayashi, Yoshiki Matsumoto, and Takahito Imai. 2020. "Tandem-Homodimer of a β-Sheet-Forming Short Peptide Inhibits Random-to-β Structural Transition of Its Original Monomer" Processes 8, no. 11: 1421. https://doi.org/10.3390/pr8111421
APA StyleTomizaki, K.-y., Iori, T., Fukushima, H., Nakabayashi, Y., Matsumoto, Y., & Imai, T. (2020). Tandem-Homodimer of a β-Sheet-Forming Short Peptide Inhibits Random-to-β Structural Transition of Its Original Monomer. Processes, 8(11), 1421. https://doi.org/10.3390/pr8111421