
Hydrodeoxygenation of Guaiacol over Pd–Co and Pd–Fe Catalysts: Deactivation and Regeneration


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Catalyst Preparation and Characterization
2.3. HDO of Guaiacol
3. Results
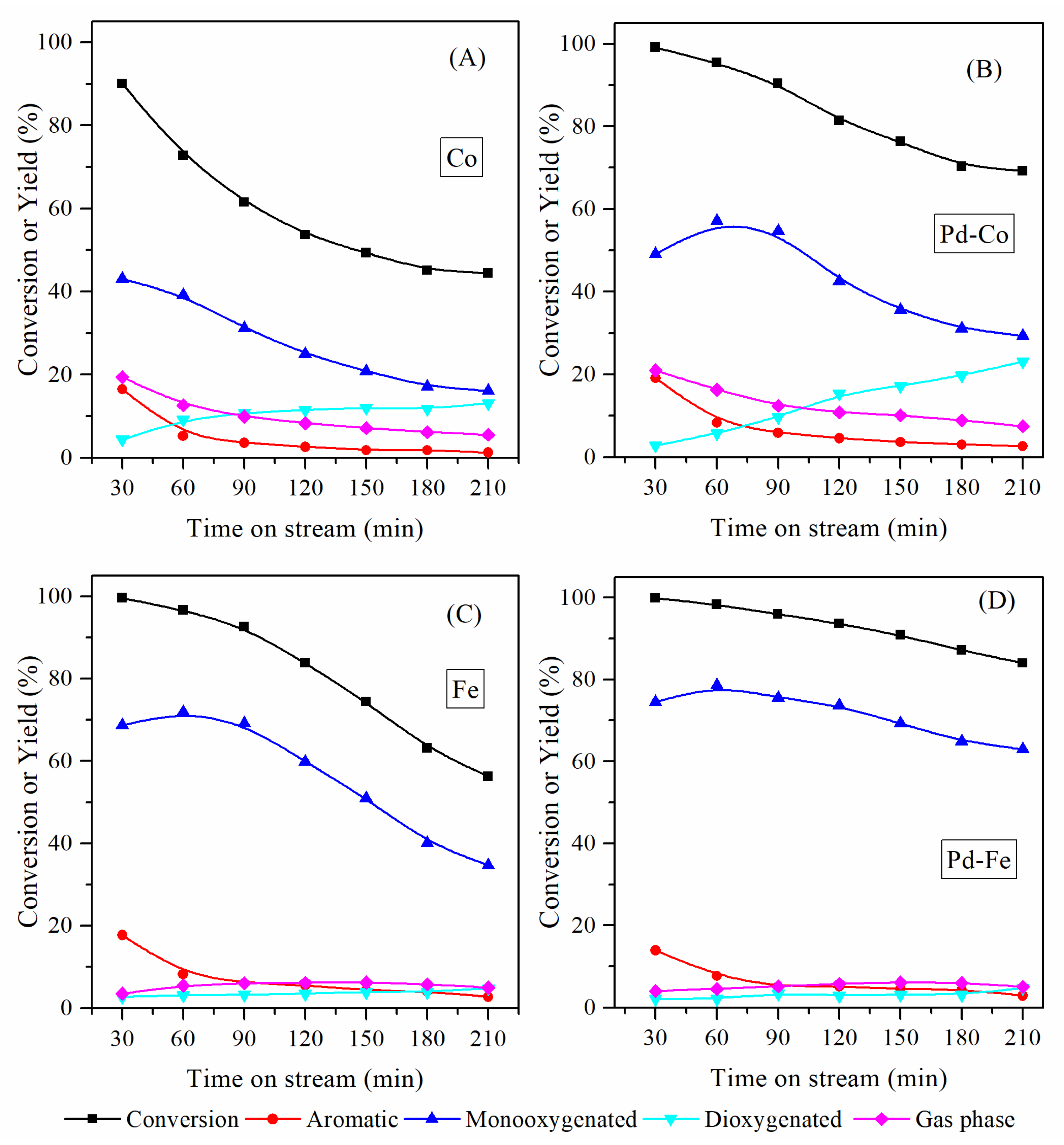
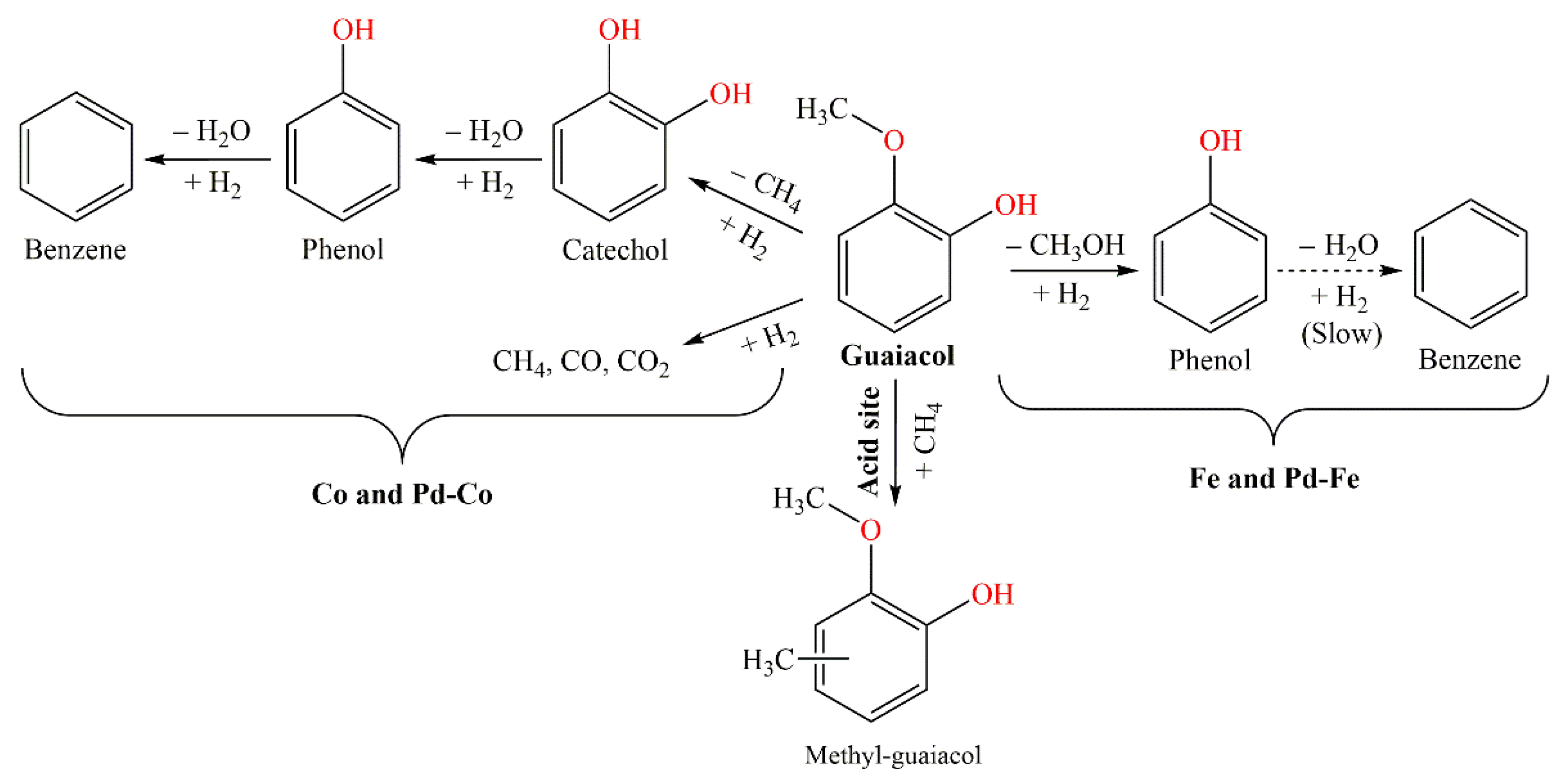
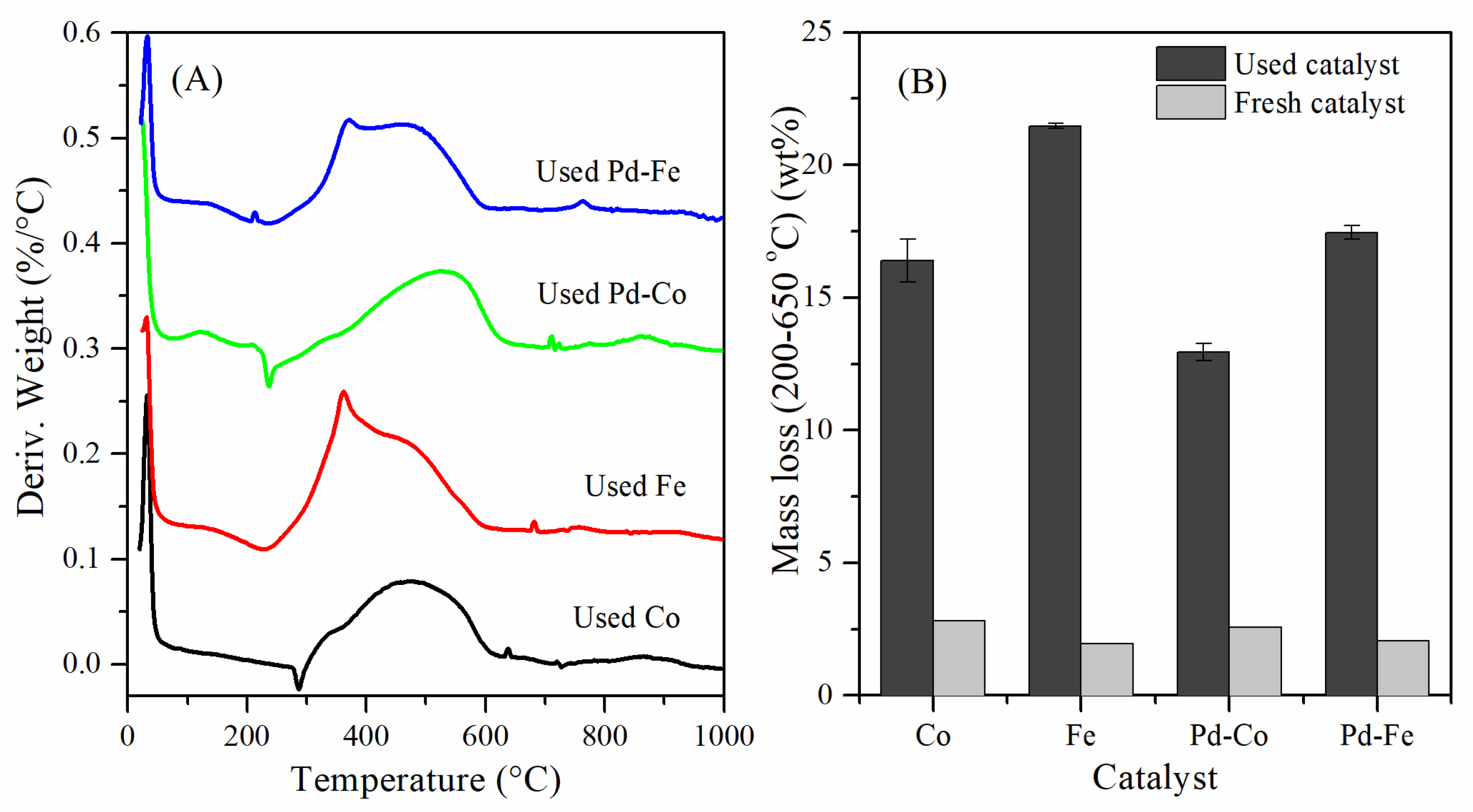
3.1. Catalytic Stability of Mono- and Bimetallic
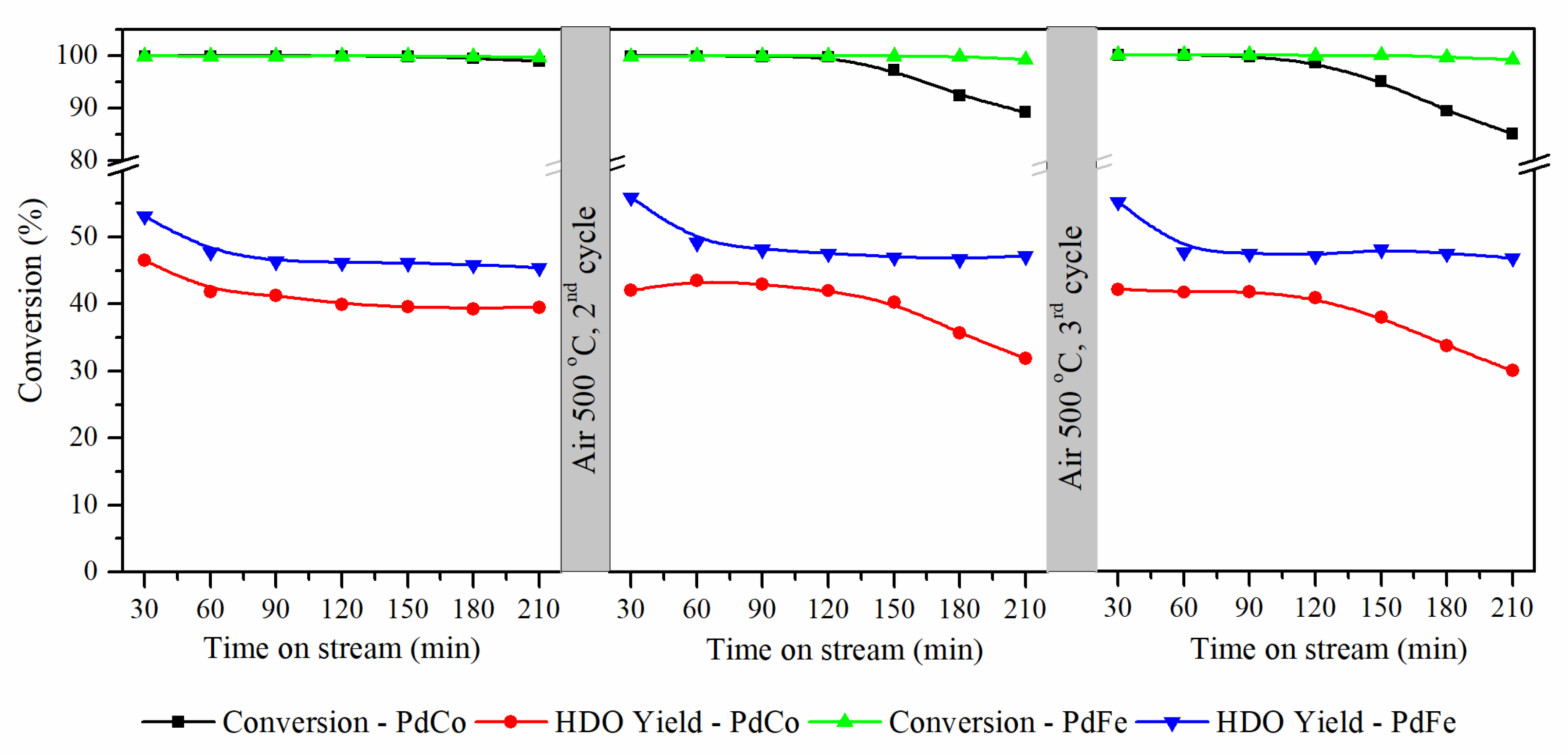
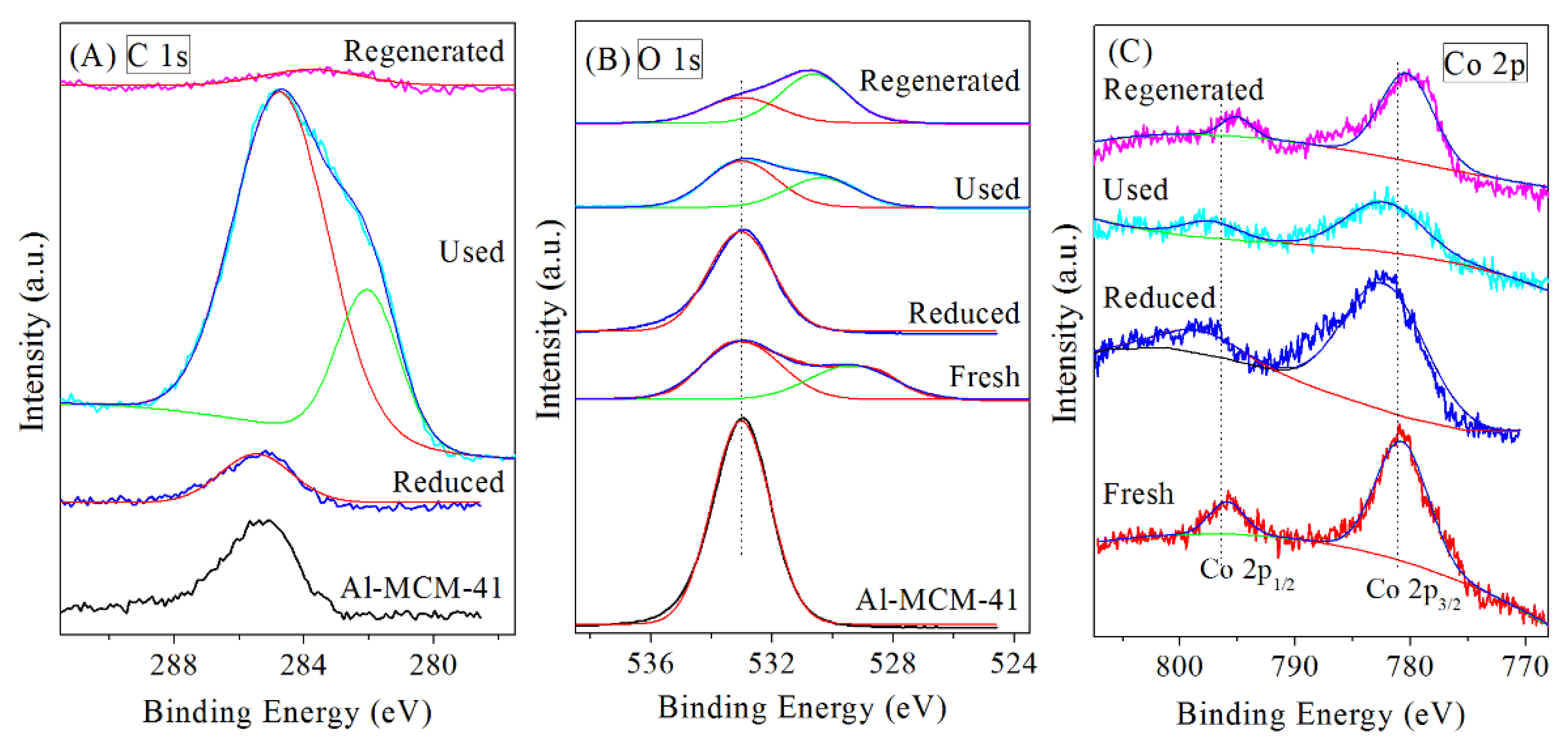
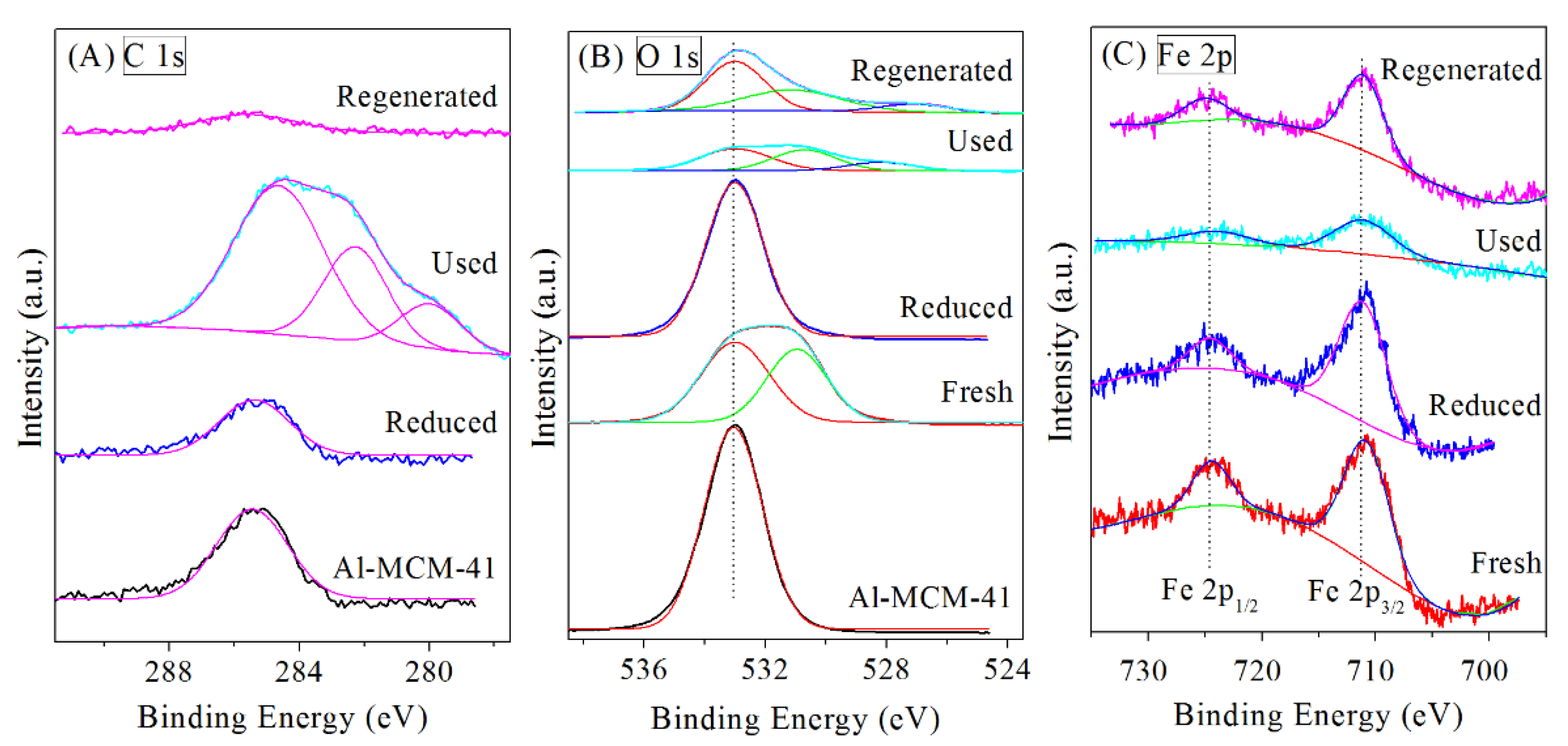
3.2. Regeneration of Bimetallic Catalysts
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bridgwater, A. Thermal Biomass Conversion and Utilization: Biomass Information System; EUR; 16863 EN.; Office for Official Publications of the European Communities: Luxembourg, 1996; ISBN 92-827-7207-1. [Google Scholar]
- Torri, I.D.V.; Paasikallio, V.; Faccini, C.S.; Huff, R.; Caramão, E.B.; Sacon, V.; Oasmaa, A.; Zini, C.A. Bio-Oil Production of Softwood and Hardwood Forest Industry Residues through Fast and Intermediate Pyrolysis and Its Chromatographic Characterization. Bioresour. Technol. 2016, 200, 680–690. [Google Scholar] [CrossRef]
- Koike, N.; Hosokai, S.; Takagaki, A.; Nishimura, S.; Kikuchi, R.; Ebitani, K.; Suzuki, Y.; Oyama, S.T. Upgrading of Pyrolysis Bio-Oil Using Nickel Phosphide Catalysts. J. Catal. 2016, 333, 115–126. [Google Scholar] [CrossRef] [Green Version]
- Olarte, M.V.; Zacher, A.H.; Padmaperuma, A.B.; Burton, S.D.; Job, H.M.; Lemmon, T.L.; Swita, M.S.; Rotness, L.J.; Neuenschwander, G.N.; Frye, J.G.; et al. Stabilization of Softwood-Derived Pyrolysis Oils for Continuous Bio-Oil Hydroprocessing. Top. Catal. 2015, 1–10. [Google Scholar] [CrossRef]
- Lehto, J.; Oasmaa, A.; Solantausta, Y.; Kytö, M.; Chiaramonti, D. Review of Fuel Oil Quality and Combustion of Fast Pyrolysis Bio-Oils from Lignocellulosic Biomass. Appl. Energy 2014, 116, 178–190. Available online: https://cris.vtt.fi/en/publications/fuel-oil-quality-and-combustion-of-fast-pyrolysis-bio-oils (accessed on 26 February 2021). [CrossRef]
- Uemura, Y.; Omar, W.N.; Razlan, S.; Afif, H.; Yusup, S.; Onoe, K. Mass and Energy Yields of Bio-Oil Obtained by Microwave-Induced Pyrolysis of Oil Palm Kernel Shell. J. Jpn. Inst. Energy 2012, 91, 954–959. [Google Scholar] [CrossRef] [Green Version]
- Meng, J.; Moore, A.; Tilotta, D.C.; Kelley, S.S.; Adhikari, S.; Park, S. Thermal and Storage Stability of Bio-Oil from Pyrolysis of Torrefied Wood. Energy Fuels 2015, 29, 5117–5126. [Google Scholar] [CrossRef]
- Mortensen, P.M.; Grunwaldt, J.-D.; Jensen, P.A.; Knudsen, K.G.; Jensen, A.D. A Review of Catalytic Upgrading of Bio-Oil to Engine Fuels. Appl. Catal. A Gen. 2011, 407, 1–19. [Google Scholar] [CrossRef]
- Sun, J.; Karim, A.M.; Zhang, H.; Kovarik, L.; Li, X.S.; Hensley, A.J.; McEwen, J.-S.; Wang, Y. Carbon-Supported Bimetallic Pd–Fe Catalysts for Vapor-Phase Hydrodeoxygenation of Guaiacol. J. Catal. 2013, 306, 47–57. [Google Scholar] [CrossRef]
- Nie, L.; de Souza, P.M.; Noronha, F.B.; An, W.; Sooknoi, T.; Resasco, D.E. Selective Conversion of m-Cresol to Toluene over Bimetallic Ni–Fe Catalysts. J. Mol. Catal. A Chem. 2014, 388–389, 47–55. [Google Scholar] [CrossRef]
- Bartholomew, C.H.; Farrauto, R.J. Fundamentals of Industrial Catalytic Processes; John Wiley & Son: Hoboken, NJ, USA, 2005; ISBN 978-0-471-45713-8. [Google Scholar]
- Saidi, M.; Samimi, F.; Karimipourfard, D.; Nimmanwudipong, T.; Gates, B.C.; Rahimpour, M.R. Upgrading of Lignin-Derived Bio-Oils by Catalytic Hydrodeoxygenation. Energy Environ. Sci. 2013, 7, 103–129. [Google Scholar] [CrossRef]
- Bridgwater, A.V. Review of Fast Pyrolysis of Biomass and Product Upgrading. Biomass-Bioenergy 2012, 38, 68–94. [Google Scholar] [CrossRef]
- Contreras, J.L.; Fuentes, G.A. Sintering of Supported Metal Catalysts, Sintering-Methods and Products, Volodymyr Shatokha; IntechOpen: London, UK, 2012. [Google Scholar] [CrossRef] [Green Version]
- Gao, D.; Schweitzer, C.; Hwang, H.T.; Varma, A. Conversion of Guaiacol on Noble Metal Catalysts: Reaction Performance and Deactivation Studies. Ind. Eng. Chem. Res. 2014, 53, 18658–18667. [Google Scholar] [CrossRef]
- Oh, S.; Hwang, H.; Choi, H.S.; Choi, J.W. The Effects of Noble Metal Catalysts on the Bio-Oil Quality during the Hydrodeoxygenative Upgrading Process. Fuel 2015, 153, 535–543. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, C.; Liu, Y.; Tang, S.; Chen, G.; Zhang, R.; Tang, X. Coke Formation on the Surface of Ni/HZSM-5 and Ni-Cu/HZSM-5 Catalysts during Bio-Oil Hydrodeoxygenation. Fuel 2017, 189, 23–31. [Google Scholar] [CrossRef]
- Remiro, A.; Valle, B.; Aguayo, A.T.; Bilbao, J.; Gayubo, A.G. Operating Conditions for Attenuating Ni/La2O3 –AAl2O3 Catalyst Deactivation in the Steam Reforming of Bio-Oil Aqueous Fraction. Fuel Process. Technol. 2013, 115, 222–232. [Google Scholar] [CrossRef]
- Wang, Y.; Fang, Y.; He, T.; Hu, H.; Wu, J. Hydrodeoxygenation of Dibenzofuran over Noble Metal Supported on Mesoporous Zeolite. Catal. Commun. 2011, 12, 1201–1205. [Google Scholar] [CrossRef]
- Li, W.; Li, F.; Wang, H.; Liao, M.; Li, P.; Zheng, J.; Tu, C.; Li, R. Hierarchical Mesoporous ZSM-5 Supported Nickel Catalyst for the Catalytic Hydrodeoxygenation of Anisole to Cyclohexane. Mol. Catal. 2020, 480, 110642. [Google Scholar] [CrossRef]
- Mortensen, P.M.; Gardini, D.; Damsgaard, C.D.; Grunwaldt, J.-D.; Jensen, P.A.; Wagner, J.B.; Jensen, A.D. Deactivation of Ni-MoS2 by Bio-Oil Impurities during Hydrodeoxygenation of Phenol and Octanol. Appl. Catal. A Gen. 2016, 523, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Jiang, E.; Du, Y.; Li, B. BTX from the Gas-Phase Hydrodeoxygenation and Transmethylation of Guaiacol at Room Pressure. Renew. Energy 2016, 96(Part A), 458–468. [Google Scholar] [CrossRef]
- Olcese, R.; Bettahar, M.M.; Malaman, B.; Ghanbaja, J.; Tibavizco, L.; Petitjean, D.; Dufour, A. Gas-Phase Hydrodeoxygenation of Guaiacol over Iron-Based Catalysts. Effect of Gases Composition, Iron Load and Supports (Silica and Activated Carbon). Appl. Catal. B Environ. 2013, 129, 528–538. [Google Scholar] [CrossRef]
- Zanuttini, M.S.; Costa, B.O.D.; Querini, C.A.; Peralta, M.A. Hydrodeoxygenation of m-Cresol with Pt Supported over Mild Acid Materials. Appl. Catal. A Gen. 2014, 482, 352–361. [Google Scholar] [CrossRef]
- Bi, P.; Yuan, Y.; Fan, M.; Jiang, P.; Zhai, Q.; Li, Q. Production of Aromatics through Current-Enhanced Catalytic Conversion of Bio-Oil Tar. Bioresour. Technol. 2013, 136, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Lobban, L.L.; Mallinson, R.G.; Resasco, D.E. Bifunctional Transalkylation and Hydrodeoxygenation of Anisole over a Pt/HBeta Catalyst. J. Catal. 2011, 281, 21–29. [Google Scholar] [CrossRef]
- Selvaraj, M.; Shanthi, K.; Maheswari, R.; Ramanathan, A. Hydrodeoxygenation of Guaiacol over MoO3-NiO/Mesoporous Silicates: Effect of Incorporated Heteroatom. Energy Fuels 2014, 28, 2598–2607. [Google Scholar] [CrossRef]
- Tran, N.T.T.; Uemura, Y.; Ramli, A.; Trinh, T.H. Vapor-Phase Hydrodeoxygenation of Lignin-Derived Bio-Oil over Al-MCM-41 Supported Pd-Co and Pd-Fe Catalysts. Mol. Catal. 2021, 111435. [Google Scholar] [CrossRef]
- Do, P.T.M.; Foster, A.J.; Chen, J.; Lobo, R.F. Bimetallic Effects in the Hydrodeoxygenation of Meta-Cresol on γ-Al2O3 Supported Pt–Ni and Pt–Co Catalysts. Green Chem. 2012, 14, 1388–1397. [Google Scholar] [CrossRef]
- Lai, Q.; Zhang, C.; Holles, J.H. Hydrodeoxygenation of Guaiacol over Ni@Pd and Ni@Pt Bimetallic Overlayer Catalysts. Appl. Catal. A Gen. 2016, 528, 1–13. [Google Scholar] [CrossRef]
- Tran, N.T.T.; Uemura, Y.; Chowdhury, S.; Ramli, A. Vapor-Phase Hydrodeoxygenation of Guaiacol on Al-MCM-41 Supported Ni and Co Catalysts. Appl. Catal. A Gen. 2016, 512, 93–100. [Google Scholar] [CrossRef]
- Yan, Y.; Jiang, S.; Zhang, H.; Zhang, X. Preparation of Novel Fe-ZSM-5 Zeolite Membrane Catalysts for Catalytic Wet Peroxide Oxidation of Phenol in a Membrane Reactor. Chem. Eng. J. 2015, 259, 243–251. [Google Scholar] [CrossRef]
- Bukallah, S.B.; Bumajdad, A.; Khalil, K.M.S.; Zaki, M.I. Characterization of Mesoporous VOx/MCM-41 Composite Materials Obtained via Post-Synthesis Impregnation. Appl. Surf. Sci. 2010, 256, 6179–6185. [Google Scholar] [CrossRef]
- Méndez, F.J.; Franco-López, O.E.; Bokhimi, X.; Solís-Casados, D.A.; Escobar-Alarcón, L.; Klimova, T.E. Dibenzothiophene Hydrodesulfurization with NiMo and CoMo Catalysts Supported on Niobium-Modified MCM-41. Appl. Catal. B Environ. 2017, 219, 479–491. [Google Scholar] [CrossRef]
- Misran, H.; Salim, M.A.; Ramesh, S. Effect of Ag Nanoparticles Seeding on the Properties of Silica Spheres. Ceram. Int. 2018, 44, 5901–5908. [Google Scholar] [CrossRef]
- Jiang, L.; Li, H.; Wang, Y.; Ma, W.; Zhong, Q. Characterization of Carbon Deposits on Coked Lithium Phosphate Catalysts for the Rearrangement of Propylene Oxide. Catal. Commun. 2015, 64, 22–26. [Google Scholar] [CrossRef]
- Tian, Y.-P.; Liu, X.-M.; Rood, M.J.; Yan, Z.-F. Study of Coke Deposited on a VOx-K2O/γ-Al2O3 Catalyst in the Non-Oxidative Dehydrogenation of Isobutane. Appl. Catal. A Gen. 2017, 545, 1–9. [Google Scholar] [CrossRef]
- Mao, A.; Wang, H.; Pan, R. Coke Deactivation of Activated Carbon-Supported Rubidium–Potassium Catalyst for C2F5I Gas-Phase Synthesis. J. Fluor. Chem. 2013, 150, 21–24. [Google Scholar] [CrossRef]
- Bai, T.; Zhang, X.; Wang, F.; Qu, W.; Liu, X.; Duan, C. Coking Behaviors and Kinetics on HZSM-5/SAPO-34 Catalysts for Conversion of Ethanol to Propylene. J. Energy Chem. 2016, 25, 545–552. [Google Scholar] [CrossRef]
- Gou, M.-L.; Cai, J.; Song, W.; Liu, Z.; Ren, Y.-L.; Pan, B.; Niu, Q. Coking and Deactivation Behavior of ZSM-5 during the Isomerization of Styrene Oxide to Phenylacetaldehyde. Catal. Commun. 2017, 98, 116–120. [Google Scholar] [CrossRef]
- Younis, A.; Chu, D.; Lin, X.; Lee, J.; Li, S. Bipolar Resistive Switching in P-Type Co3O4 Nanosheets Prepared by Electrochemical Deposition. Nanoscale Res. Lett. 2013, 8, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Q.; Zhang, Q.; Yang, G.; Chen, Q.; Li, J.; Wei, Q.; Tan, Y.; Wan, H.; Tsubaki, N. Insights into the Promotional Roles of Palladium in Structure and Performance of Cobalt-Based Zeolite Capsule Catalyst for Direct Synthesis of C5–C11 Iso-Paraffins from Syngas. J. Catal. 2016, 344, 378–388. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Pu, C.; Xu, L.; Cai, Y.; Guo, Y.; Guo, Y.; Lu, G. Effect of Supports over Pd/Fe2O3 on CO Oxidation at Low Temperature. Fuel Process. Technol. 2017, 160, 152–157. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tran, N.; Uemura, Y.; Trinh, T.; Ramli, A. Hydrodeoxygenation of Guaiacol over Pd–Co and Pd–Fe Catalysts: Deactivation and Regeneration. Processes 2021, 9, 430. https://doi.org/10.3390/pr9030430
Tran N, Uemura Y, Trinh T, Ramli A. Hydrodeoxygenation of Guaiacol over Pd–Co and Pd–Fe Catalysts: Deactivation and Regeneration. Processes. 2021; 9(3):430. https://doi.org/10.3390/pr9030430
Chicago/Turabian StyleTran, Nga, Yoshimitsu Uemura, Thanh Trinh, and Anita Ramli. 2021. "Hydrodeoxygenation of Guaiacol over Pd–Co and Pd–Fe Catalysts: Deactivation and Regeneration" Processes 9, no. 3: 430. https://doi.org/10.3390/pr9030430