
Structural Insights into a Fusion Protein between a Glutaredoxin-like and a Ferredoxin-Disulfide Reductase Domain from an Extremophile Bacterium

 ,
,  ,
, 
Abstract
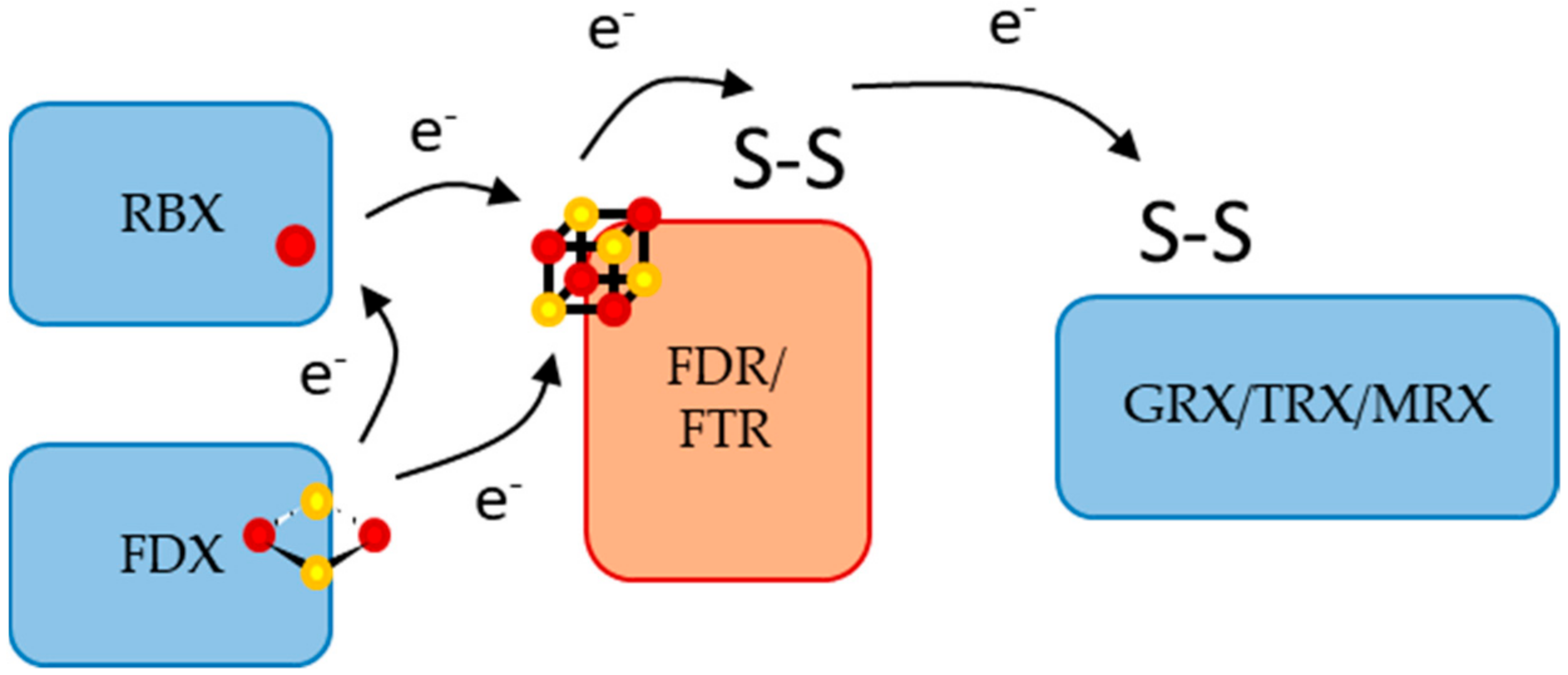
:1. Introduction
2. Results
2.1. Several Natural Hybrid Proteins Contain a Ferredoxin–Thioredoxin Reductase-like Domain Fused to Redox Active Domains
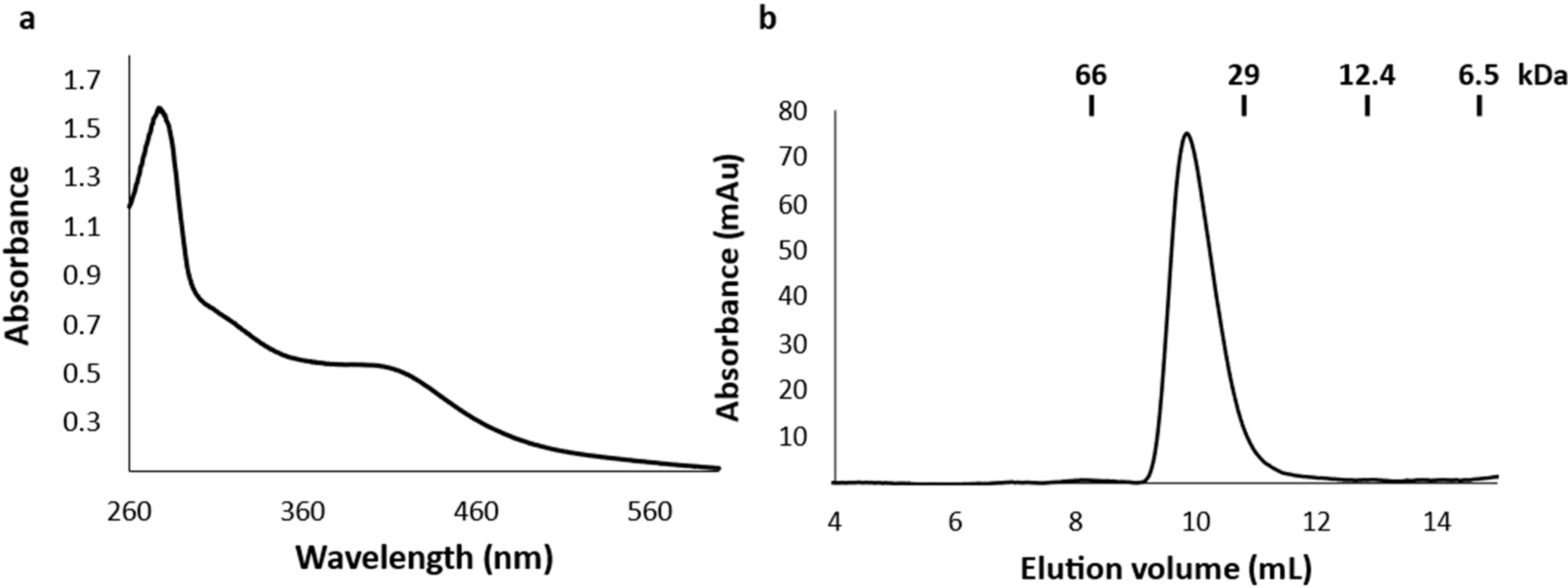
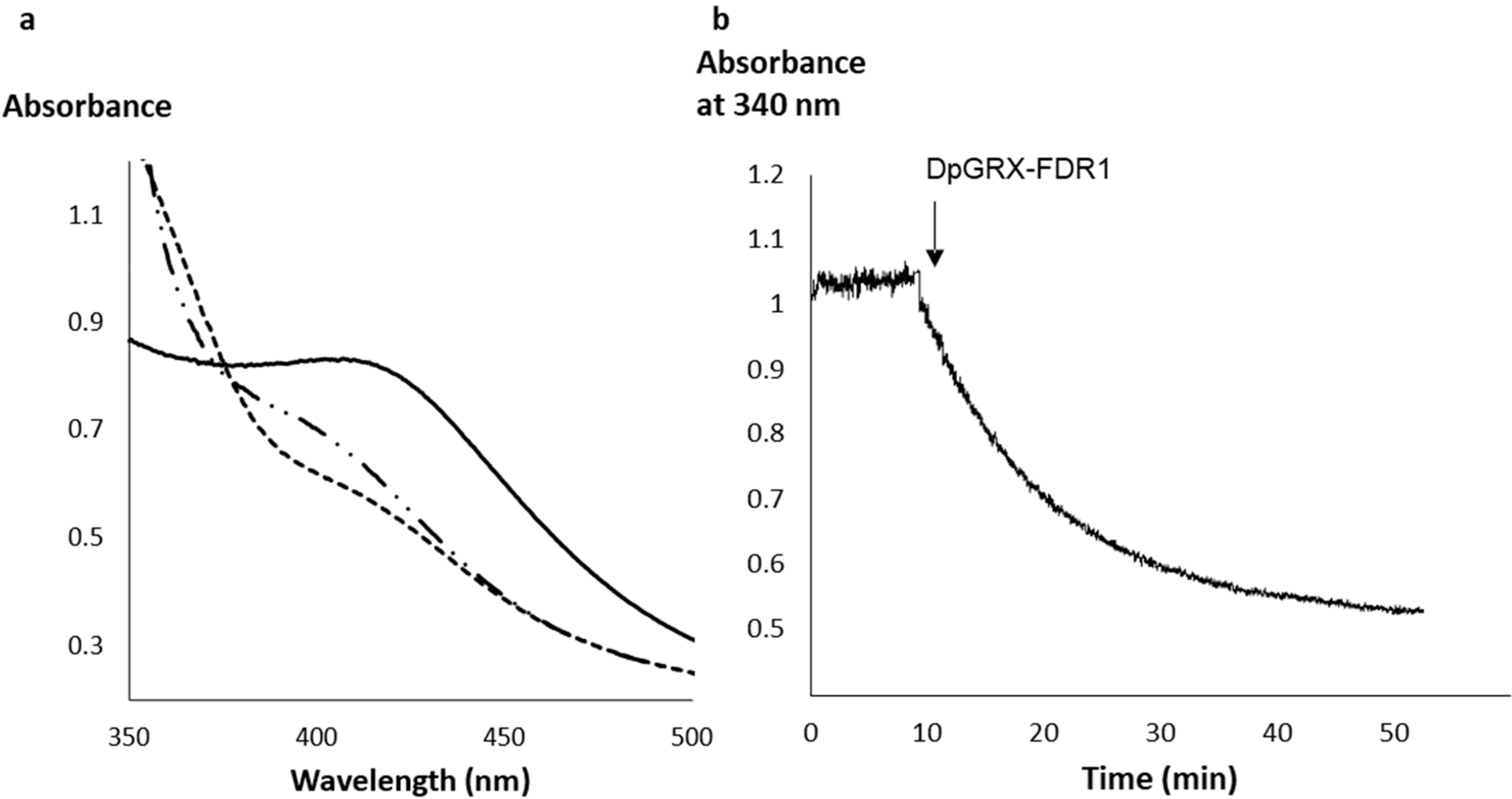
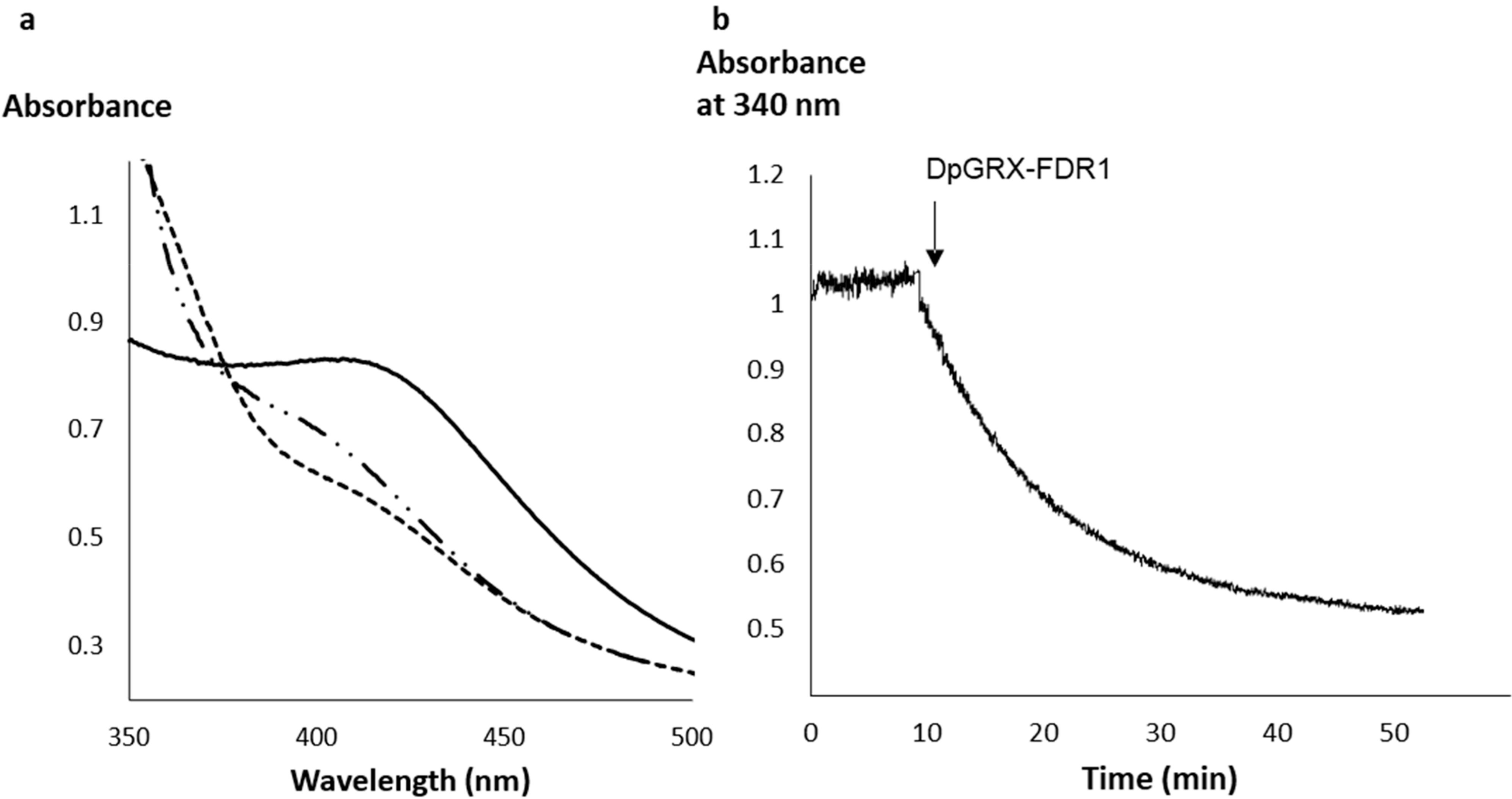
2.2. The Recombinant DpGRX-FDR1 Expressed in Escherichia coli Is a [4Fe–4S] Cluster-Bound Homodimer
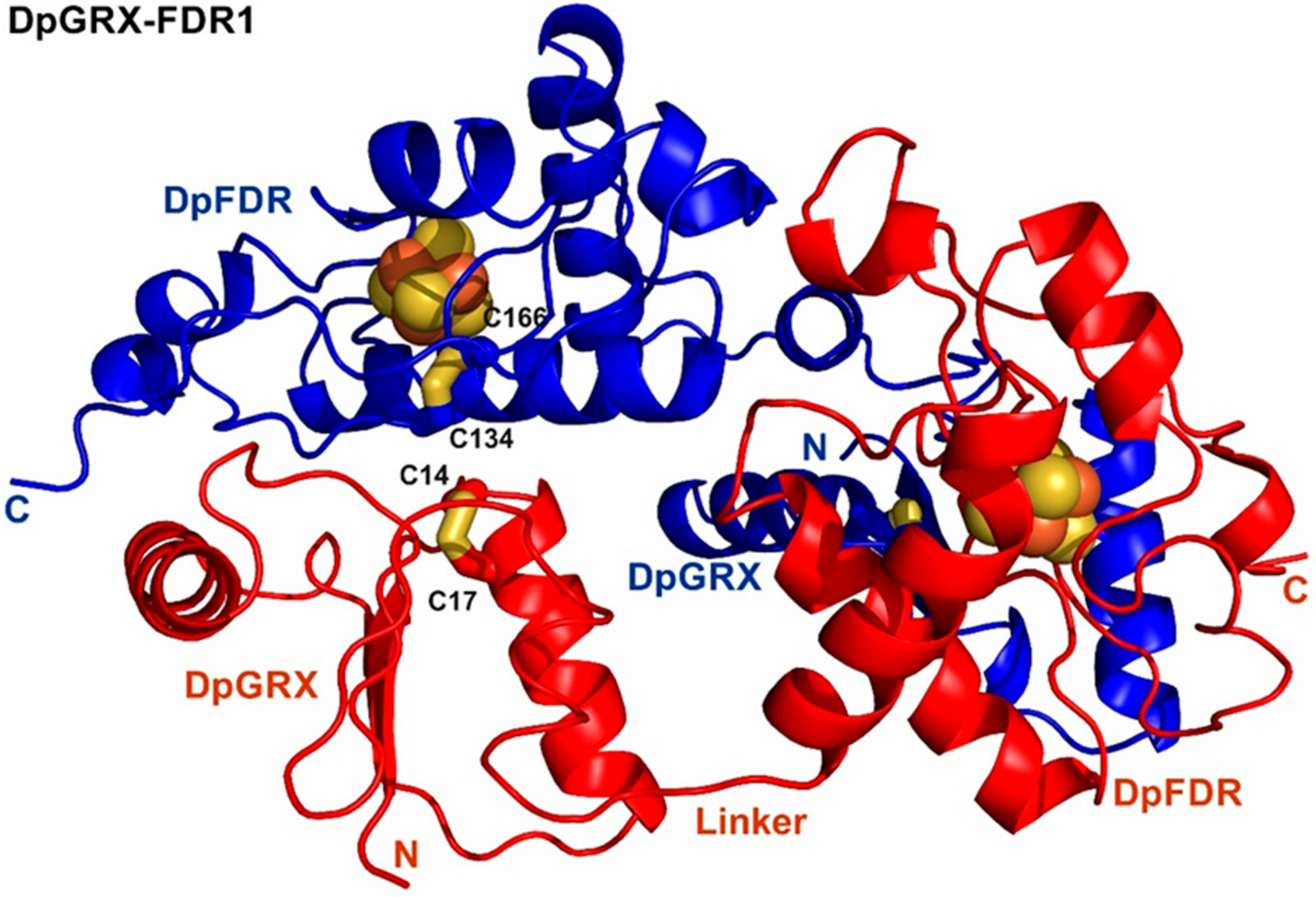
2.3. Crystal Structure of DpGRX-FDR1
3. Discussion
4. Materials and Methods
4.1. PCR Cloning
4.2. Expression in E. coli and Purification of the Recombinant Proteins
4.3. Quantification of Iron and Acid-Labile Sulfide Bound to Proteins
4.4. Reduction of DpGRX-FDR1
4.5. Crystallography
4.6. Sequence Analysis and 3D Structure Modelling
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chibani, K.; Couturier, J.; Selles, B.; Jacquot, J.-P.; Rouhier, N. The Chloroplastic Thiol Reducing Systems: Dual Functions in the Regulation of Carbohydrate Metabolism and Regeneration of Antioxidant Enzymes, Emphasis on the Poplar Redoxin Equipment. Photosyn. Res. 2010, 104, 75–99. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Hisabori, T. Distinct Electron Transfer from Ferredoxin-Thioredoxin Reductase to Multiple Thioredoxin Isoforms in Chloroplasts. Biochem. J. 2017, 474, 1347–1360. [Google Scholar] [CrossRef] [PubMed]
- Jacquot, J.-P.; Eklund, H.; Rouhier, N.; Schürmann, P. Structural and Evolutionary Aspects of Thioredoxin Reductases in Photosynthetic Organisms. Trends Plant. Sci. 2009, 14, 336–343. [Google Scholar] [CrossRef]
- Dai, S.; Schwendtmayer, C.; Schürmann, P.; Ramaswamy, S.; Eklund, H. Redox Signaling in Chloroplasts: Cleavage of Disulfides by an Iron-Sulfur Cluster. Science 2000, 287, 655–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, S.; Friemann, R.; Glauser, D.A.; Bourquin, F.; Manieri, W.; Schürmann, P.; Eklund, H. Structural Snapshots along the Reaction Pathway of Ferredoxin-Thioredoxin Reductase. Nature 2007, 448, 92–96. [Google Scholar] [CrossRef]
- Xu, X.; Schürmann, P.; Chung, J.-S.; Hass, M.A.S.; Kim, S.-K.; Hirasawa, M.; Tripathy, J.N.; Knaff, D.B.; Ubbink, M. Ternary Protein Complex of Ferredoxin, Ferredoxin:Thioredoxin Reductase, and Thioredoxin Studied by Paramagnetic NMR Spectroscopy. J. Am. Chem. Soc. 2009, 131, 17576–17582. [Google Scholar] [CrossRef]
- Walters, E.M.; Garcia-Serres, R.; Jameson, G.N.L.; Glauser, D.A.; Bourquin, F.; Manieri, W.; Schürmann, P.; Johnson, M.K.; Huynh, B.H. Spectroscopic Characterization of Site-Specific [Fe4S4] Cluster Chemistry in Ferredoxin:Thioredoxin Reductase: Implications for the Catalytic Mechanism. J. Am. Chem. Soc. 2005, 127, 9612–9624. [Google Scholar] [CrossRef] [Green Version]
- Jameson, G.N.L.; Walters, E.M.; Manieri, W.; Schürmann, P.; Johnson, M.K.; Huynh, B.H. Spectroscopic Evidence for Site Specific Chemistry at a Unique Iron Site of the [4Fe−4S] Cluster in Ferredoxin:Thioredoxin Reductase. J. Am. Chem. Soc. 2003, 125, 1146–1147. [Google Scholar] [CrossRef] [PubMed]
- Balsera, M.; Uberegui, E.; Susanti, D.; Schmitz, R.A.; Mukhopadhyay, B.; Schürmann, P.; Buchanan, B.B. Ferredoxin:Thioredoxin Reductase (FTR) Links the Regulation of Oxygenic Photosynthesis to Deeply Rooted Bacteria. Planta 2013, 237, 619–635. [Google Scholar] [CrossRef]
- Wei, Y.; Li, B.; Prakash, D.; Ferry, J.G.; Elliott, S.J.; Stubbe, J. A Ferredoxin Disulfide Reductase Delivers Electrons to the Methanosarcina barkeri Class III Ribonucleotide Reductase. Biochemistry 2015, 54, 7019–7028. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.K.; Kumar, R.S.S.; Yennawar, N.H.; Yennawar, H.P.; Ferry, J.G. Structural and Biochemical Characterization of a Ferredoxin: Thioredoxin Reductase-like Enzyme from Methanosarcina acetivorans. Biochemistry 2015, 54, 3122–3128. [Google Scholar] [CrossRef] [PubMed]
- Prakash, D.; Walters, K.A.; Martinie, R.J.; McCarver, A.C.; Kumar, A.K.; Lessner, D.J.; Krebs, C.; Golbeck, J.H.; Ferry, J.G. Toward a Mechanistic and Physiological Understanding of a Ferredoxin:Disulfide Reductase from the Domains Archaea and Bacteria. J. Biol. Chem. 2018, 293, 9198–9209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yenugudhati, D.; Prakash, D.; Kumar, A.K.; Kumar, R.S.S.; Yennawar, N.H.; Yennawar, H.P.; Ferry, J.G. Structural and Biochemical Characterizations of Methanoredoxin from Methanosarcina acetivorans, a Glutaredoxin-Like Enzyme with Coenzyme M-Dependent Protein Disulfide Reductase Activity. Biochemistry 2016, 55, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Rabus, R.; Ruepp, A.; Frickey, T.; Rattei, T.; Fartmann, B.; Stark, M.; Bauer, M.; Zibat, A.; Lombardot, T.; Becker, I.; et al. The Genome of Desulfotalea psychrophila, a Sulfate-Reducing Bacterium from Permanently Cold Arctic Sediments. Environ. Microbiol. 2004, 6, 887–902. [Google Scholar] [CrossRef] [PubMed]
- Couturier, J.; Koh, C.S.; Zaffagnini, M.; Winger, A.M.; Gualberto, J.M.; Corbier, C.; Decottignies, P.; Jacquot, J.-P.; Lemaire, S.D.; Didierjean, C.; et al. Structure-Function Relationship of the Chloroplastic Glutaredoxin S12 with an Atypical WCSYS Active Site. J. Biol. Chem. 2009, 284, 9299–9310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mesecke, N.; Mittler, S.; Eckers, E.; Herrmann, J.M.; Deponte, M. Two Novel Monothiol Glutaredoxins from Saccharomyces cerevisiae Provide Further Insight into Iron-Sulfur Cluster Binding, Oligomerization, and Enzymatic Activity of Glutaredoxins. Biochemistry 2008, 47, 1452–1463. [Google Scholar] [CrossRef] [PubMed]
- Chibani, K.; Tarrago, L.; Schürmann, P.; Jacquot, J.-P.; Rouhier, N. Biochemical Properties of Poplar Thioredoxin z. FEBS Lett. 2011, 585, 1077–1081. [Google Scholar] [CrossRef] [Green Version]
- Couturier, J.; Przybyla-Toscano, J.; Roret, T.; Didierjean, C.; Rouhier, N. The Roles of Glutaredoxins Ligating Fe–S Clusters: Sensing, Transfer or Repair Functions? Biochim. Biophys. Acta 2015, 1853, 1513–1527. [Google Scholar] [CrossRef]
- Berndt, C.; Christ, L.; Rouhier, N.; Mühlenhoff, U. Glutaredoxins with Iron-Sulphur Clusters in Eukaryotes—Structure, Function and Impact on Disease. Biochim. Biophys. Acta Bioenerg. 2020, 1862, 148317. [Google Scholar] [CrossRef]
- Rouhier, N.; Lemaire, S.D.; Jacquot, J.-P. The Role of Glutathione in Photosynthetic Organisms: Emerging Functions for Glutaredoxins and Glutathionylation. Annu. Rev. Plant. Biol. 2008, 59, 143–166. [Google Scholar] [CrossRef]
- Nathaniel, C.; Wallace, L.A.; Burke, J.; Dirr, H.W. The Role of an Evolutionarily Conserved Cis-Proline in the Thioredoxin-like Domain of Human Class Alpha Glutathione Transferase A1-1. Biochem. J. 2003, 372, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Holm, L.; Laakso, L.M. Dali Server Update. Nucleic Acids Res. 2016, 44, W351–W355. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.; Henrick, K. Secondary-Structure Matching (SSM), a New Tool for Fast Protein Structure Alignment in Three Dimensions. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2256–2268. [Google Scholar] [CrossRef] [PubMed]
- Røhr, Å.K.; Hammerstad, M.; Andersson, K.K. Tuning of Thioredoxin Redox Properties by Intramolecular Hydrogen Bonds. PLoS ONE 2013, 8, e69411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stehr, M.; Schneider, G.; Aslund, F.; Holmgren, A.; Lindqvist, Y. Structural Basis for the Thioredoxin-like Activity Profile of the Glutaredoxin-like NrdH-Redoxin from Escherichia coli. J. Biol. Chem. 2001, 276, 35836–35841. [Google Scholar] [CrossRef] [Green Version]
- Van Laer, K.; Dziewulska, A.M.; Fislage, M.; Wahni, K.; Hbeddou, A.; Collet, J.-F.; Versées, W.; Mateos, L.M.; Tamu Dufe, V.; Messens, J. NrdH-Redoxin of Mycobacterium Tuberculosis and Corynebacterium Glutamicum Dimerizes at High Protein Concentration and Exclusively Receives Electrons from Thioredoxin Reductase. J. Biol. Chem. 2013, 288, 7942–7955. [Google Scholar] [CrossRef] [Green Version]
- Susanti, D.; Wong, J.H.; Vensel, W.H.; Loganathan, U.; DeSantis, R.; Schmitz, R.A.; Balsera, M.; Buchanan, B.B.; Mukhopadhyay, B. Thioredoxin Targets Fundamental Processes in a Methane-Producing Archaeon, Methanocaldococcus jannaschii. Proc. Natl. Acad. Sci. USA 2014, 111, 2608–2613. [Google Scholar] [CrossRef] [Green Version]
- Jung, H.-C.; Lim, J.K.; Yang, T.-J.; Kang, S.G.; Lee, H.S. Direct Electron Transfer between the FrhAGB-Encoded Hydrogenase and Thioredoxin Reductase in the Nonmethanogenic Archaeon Thermococcus onnurineus NA1. Appl. Environ. Microbiol. 2020, 86, e02630-19. [Google Scholar] [CrossRef]
- Juniar, L.; Tanaka, H.; Yoshida, K.; Hisabori, T.; Kurisu, G. Structural Basis for Thioredoxin Isoform-Based Fine-Tuning of Ferredoxin-Thioredoxin Reductase Activity. Protein Sci. 2020, 29, 2538–2545. [Google Scholar] [CrossRef]
- McCarver, A.C.; Lessner, D.J. Molecular Characterization of the Thioredoxin System from Methanosarcina acetivorans. FEBS J. 2014, 281, 4598–4611. [Google Scholar] [CrossRef] [Green Version]
- Séry, A.; Housset, D.; Serre, L.; Bonicel, J.; Hatchikian, C.; Frey, M.; Roth, M. Crystal Structure of the Ferredoxin I from Desulfovibrio africanus at 2.3 A Resolution. Biochemistry 1994, 33, 15408–15417. [Google Scholar] [CrossRef] [PubMed]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold—Making Protein Folding Accessible to All. bioRxiv 2021, 1–8. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liebschner, D.; Afonine, P.V.; Baker, M.L.; Bunkóczi, G.; Chen, V.B.; Croll, T.I.; Hintze, B.; Hung, L.W.; Jain, S.; McCoy, A.J.; et al. Macromolecular Structure Determination Using X-rays, Neutrons and Electrons: Recent Developments in Phenix. Acta Crystallogr. D Struct. Biol. 2019, 75, 861–877. [Google Scholar] [CrossRef] [Green Version]
- Cowtan, K. The Buccaneer Software for Automated Model Building. 1. Tracing Protein Chains. Acta Crystallogr. D Biol. Crystallogr. 2006, 62, 1002–1011. [Google Scholar] [CrossRef] [Green Version]
- Smart, O.S.; Womack, T.O.; Flensburg, C.; Keller, P.; Paciorek, W.; Sharff, A.; Vonrhein, C.; Bricogne, G. Exploiting Structure Similarity in Refinement: Automated NCS and Target-Structure Restraints in BUSTER. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 368–380. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and Development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Collection | Soleil Proxima 1 |
|---|---|
| Space group | C2 |
| Cell constants a, b, c, α, β, γ | 123.6 Å 49.3 Å 86.0 Å 90.0°116.5°90.0° |
| Resolution limits a (Å) | 41.42–2.08 (2.13–2.08) |
| Rmeas a,b (%) | 3.7 (23.4) |
| CC1/2 (%) a,c | 99.9 (97.2) |
| Nr. of observations a,d | 162,975 (11,975) |
| Nr. unique reflections a,d | 54,222 (3975) |
| Mean((I)/sd(I)) a,d | 23.0 (5.9) |
| Average redundancy a,d (%) | 98.7 (97.8) |
| Multiplicity a | 3.0 (3.0) |
| Mean anomalous difference a,e | 1.42 (0.82) |
| Refinement | |
| Rwork/Rfree (%) | 19.1/22.3 (21.9/36.1) |
| Nr test set reflections | 1353 (4.8%) |
| Atoms: protein, ligands, water | 3229, 46, 328 |
| B-wilson/B-average (Å2) | 29/37 |
| r.m.s.d.bonds (Å)/angles (°) | 0.008/3.16 |
| Ramachandran: Fav./All. (%) | 99/1 |
| PDB ENTRY | 7PWE |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zannini, F.; Mathiot, S.; Couturier, J.; Didierjean, C.; Rouhier, N. Structural Insights into a Fusion Protein between a Glutaredoxin-like and a Ferredoxin-Disulfide Reductase Domain from an Extremophile Bacterium. Inorganics 2022, 10, 24. https://doi.org/10.3390/inorganics10020024
Zannini F, Mathiot S, Couturier J, Didierjean C, Rouhier N. Structural Insights into a Fusion Protein between a Glutaredoxin-like and a Ferredoxin-Disulfide Reductase Domain from an Extremophile Bacterium. Inorganics. 2022; 10(2):24. https://doi.org/10.3390/inorganics10020024
Chicago/Turabian StyleZannini, Flavien, Sandrine Mathiot, Jérémy Couturier, Claude Didierjean, and Nicolas Rouhier. 2022. "Structural Insights into a Fusion Protein between a Glutaredoxin-like and a Ferredoxin-Disulfide Reductase Domain from an Extremophile Bacterium" Inorganics 10, no. 2: 24. https://doi.org/10.3390/inorganics10020024
APA StyleZannini, F., Mathiot, S., Couturier, J., Didierjean, C., & Rouhier, N. (2022). Structural Insights into a Fusion Protein between a Glutaredoxin-like and a Ferredoxin-Disulfide Reductase Domain from an Extremophile Bacterium. Inorganics, 10(2), 24. https://doi.org/10.3390/inorganics10020024