Abstract
Novel hexafluoroisopropylboranes (CF3)(CF2H)CFBH2·L and -borate anions [(CF3)(CF2H)CFBH2X]− with Lewis basic heterocyclic ligands L and the anionic substituents X = F− and CN− were obtained. The syntheses were accomplished by substitution reactions of the dimethyl sulfide adduct (CF3)(CF2H)CFBH2·SMe2, which was synthesized on a large scale. The hexafluoroisopropylboranes and -borates were characterized by NMR and vibrational spectroscopy, elemental analysis, and single-crystal X-ray diffraction. In addition, the thermal and electrochemical stabilities were investigated by DSC measurements and cyclic voltammetry and selected experimental data and trends are compared with theoretical ones.
1. Introduction
The synthesis, characterization, and investigation of new (per)fluoroalkyl boranes and borates has been the subject of chemical research for many years, which in part is due to their versatile properties that have led to various applications [1,2,3,4]. Especially noteworthy are the (per)fluoroalkyl borate anions, which have been studied as weakly coordinating anions (WCAs) [3,4,5,6,7,8]. The anionic charge of WCAs should be efficiently delocalized and they should be hardly polarizable. Thus, most WCAs contain fluorine atoms at their surface and comprise comparably large molecular volumes [5,6]. Perfluoroalkyl groups in particular have proven to be very efficient at stabilizing borate anions by delocalizing the negative charge. The tetrakis(trifluoromethyl)borate anion [B(CF3)4]− [3,9,10] belongs to the most weakly coordinating anions and salts with reactive cations such as N5+ [11], NO+ [12], {H(OEt2)2}+ [13], Ph3C+ [13], and [Co(CO)2(NO)2]+ [14] have been isolated. The [(CF3)3BF]− anion [15] has weakly coordinating properties and has been used, for example, in the stabilization of the [Co(CO)5]+ [14,16] and [Mn(CO)6]+ cations [17]. However, the chemical stability of trifluoromethyl groups at boron is limited and strong Brønsted and Lewis acids are known to remove a fluoride ion from a B–CF3 group, resulting either in a complete depletion of the CF3 group or in its transformation into a different C-based substituent [3,18]. The [B(CF3)4]− anion, for example, has been converted into the borane carbonyl (CF3)3BCO in concentrated sulfuric acid [19,20]. This borane carbonyl has been used for the preparation of unprecedented tris(trifluoromethyl)borate anions including the series [(CF3)3BCX]− (I = N, P, As) [21,22] and the aforementioned WCA [(CF3)3BF]− [14,16,17,18], highlighting the stabilizing effect of the borane (CF3)3B [1,2,3] in particular and of related perfluoroalkylboranes in general.
In addition to their application as WCAs, perfluoroalkylborate anions have been used as building blocks for hydrophobic ionic liquids (ILs). In particular, monoperfluoroalkyltrifluoroborate anions [CnF2n+1BF3]− (n = 1–4) have been intensively studied in IL chemistry [23,24,25,26,27,28,29]. Related mixed monoperfluoroalkyl(cyano)(fluoro)borate anions [CnF2n+1B(CN)3–xFx]− (n = 1–4; x = 0–2) have also been used as anionic components of ILs [30,31]. Some of these perfluoroalkylcyanoborate ILs and related metal salts have been employed for the design of lanthanide complexes and hybrid materials [32]. In addition, a few of these ILs have been studied as electrolyte components, e.g., for dye-sensitized solar cells (Grätzel cells, DSSCs) [30]. A further example for perfluoroalkylborate ILs are low-viscosity and low-melting organic salts with the pentafluoroethyltrihydridoborate anion [C2F5BH3]− [33] and related mixed pentafluoroethyl(cyano)(hydrido)borate, trifluoromethyl(cyano)(hydrido)borate anions, and the [CF3CH2B(CN)3]− anion [31].
Due to their weakly coordinating nature, perfluoroalkyl- and mixed perfluoroalkyl(cyano)(fluoro)borate anions have been discussed as counterions for metal-ion batteries, e.g., lithium-ion batteries LIBs [34], and perfluoroalkylfluoroborate ILs have been suggested as electrolyte components for such batteries [35]. For example, the lithium salts Li[RFBF3] [36,37,38] and Li[B(CF3)4] [39] have been studied and other lithium perfluoroalkylborates have been investigated by quantum chemical methods [40].
In addition to perfluoroalkylborates, (per)fluoroalkylboranes have attracted interest and amine-stabilized tris(trifluoromethyl)boranes [1,2,3] and the borane carbonyls (CnF2n+1)BCO (n = 1–3) [3,19,20,41] have been especially intensively investigated. Some of these perfluoroalkylborane adducts have been used for the preparation of weakly coordinating borate anions (vide supra) and ligands for transition metal and lanthanide complexes. In the course of the synthesis of pentafluoroalkylhydridoborate anions, borane adducts of the general type C2F5BH2·L (L = CH3CN, THF, 4-cyanopyridine, PPh3) were synthesized for the first time starting from the borate anion [C2F5BH3]− via hydride abstraction [33]. The [C2F5BH3]− anion has been obtained by reaction of BH3·THF and pentafluoroethyl lithium [33].
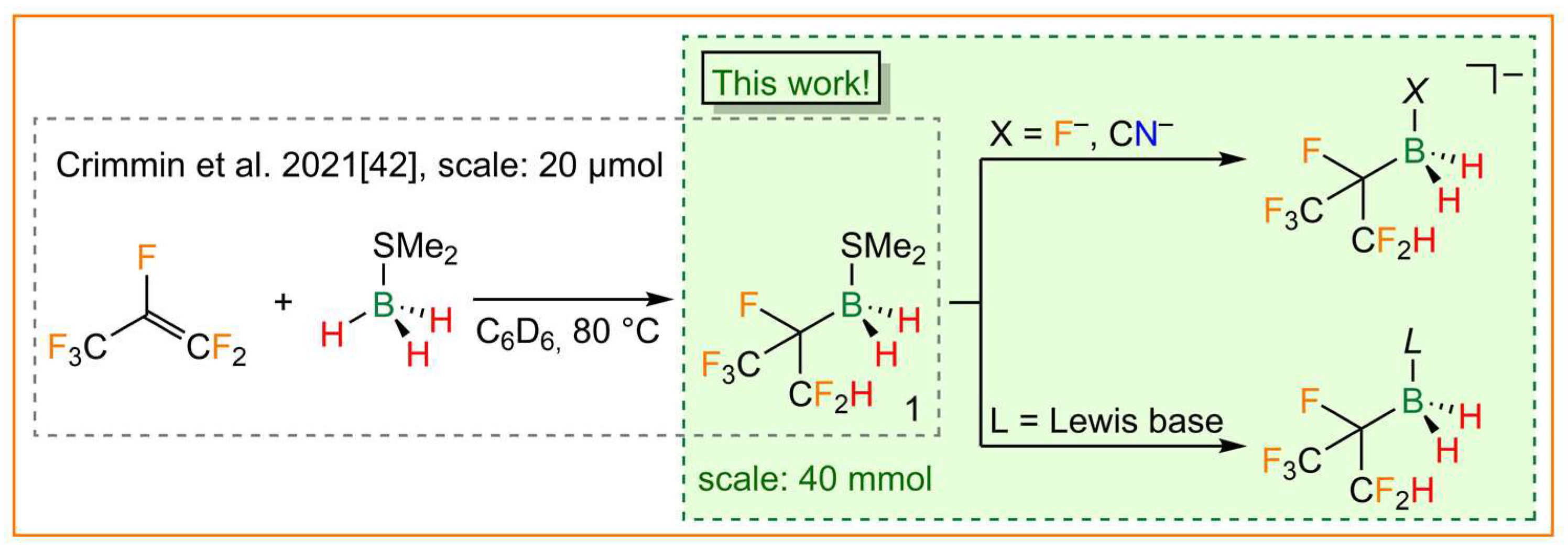
A different approach was described for the synthesis of novel partially fluorinated alkylhydridoborane adducts in 2019. These adducts were obtained as unexpected products in an attempt to hydrodefluorinate hexafluoropropene with the hydridoborane adducts BH3·THF and BH3·SMe2 (Figure 1) [42]. The hexafluoroisopropyldihydridoborane adducts, which are easily accessible using the aforementioned route, are the first selectively accessible boranes described that bear polyfluoroalkyl substituents that contain a single hydrogen atom. Furthermore, the polyfluoroalkyl group is branched, which is rarely found in per- and polyfluoroalkylboron chemistry [18].
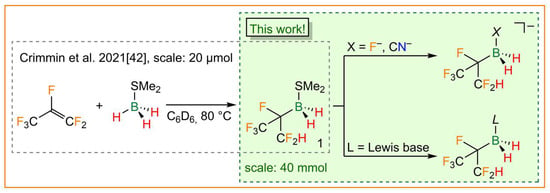
Figure 1.
Synthesis of the hexafluoroisopropylborane dimethyl sulfide adduct 1 [42] (left) and the hexafluoroisopropylborate and -borane adducts presented in this work (right).
Herein, we report on the convenient large-scale synthesis of the hexafluoroisopropylborane dimethyl sulfide adduct (CF3)(CF2H)CFBH2·SMe2 (1) and its follow-up chemistry to yield novel hexafluoroisopropylborate anions and related borane adducts (Figure 1). Selected structural, spectroscopic, and electrochemical properties of the neutral borane adducts and borate anions are presented and the data are compared with those derived from DFT calculations.
2. Results and Discussion
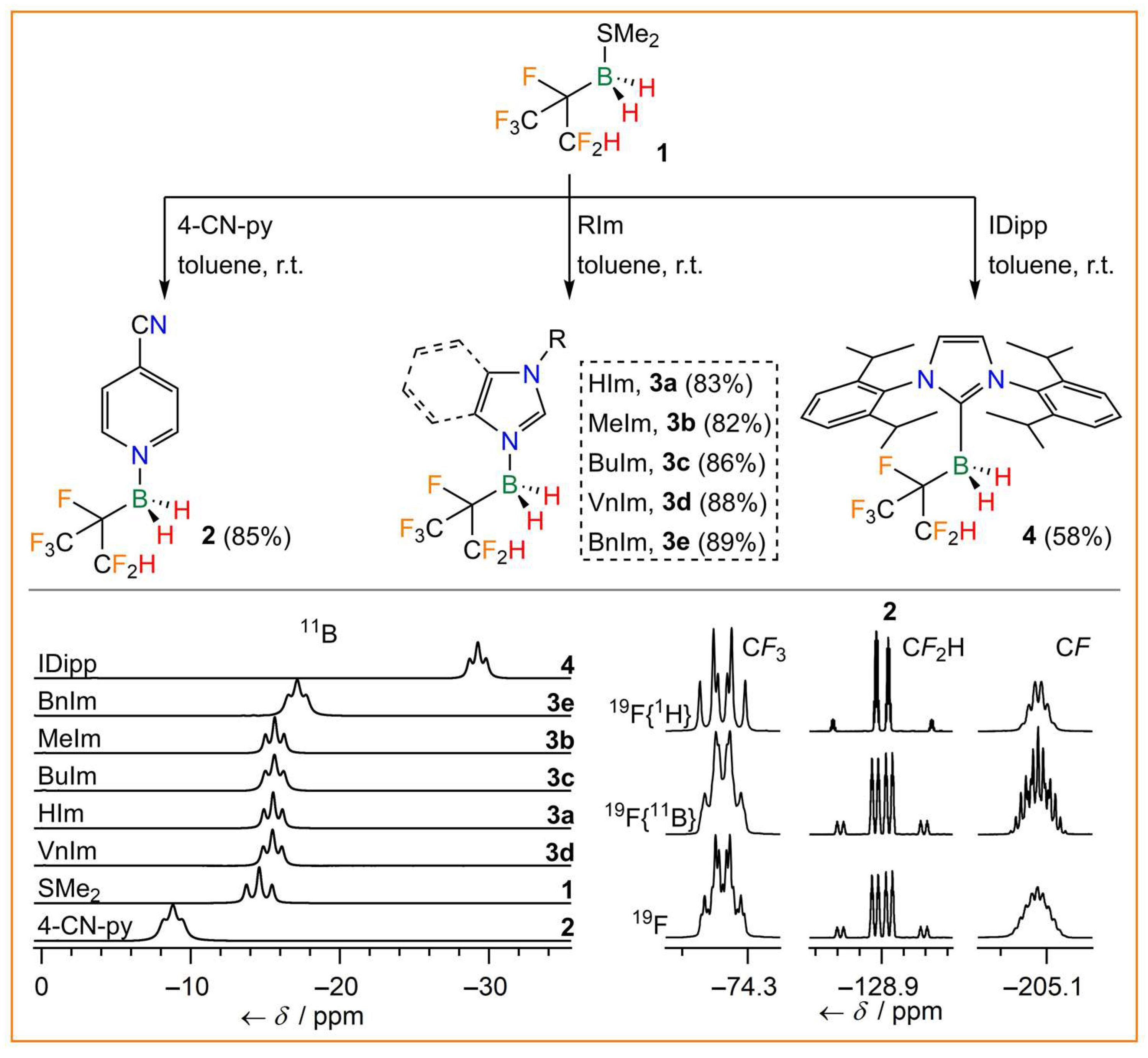
Hexafluoroisopropylborane dimethyl sulfide adduct 1 was prepared as a racemate similar to the method described by Crimmin et al. in 2019 (Figure 1) [42]. However, the initial nuclear magnetic resonance (NMR) experiment (20 µmol) was scaled up to 40 mmol. In contrast to the deuterobenzene that was used in the literature procedure, toluene was employed as a solvent and only 1.05 equivalents of hexafluoroisopropene were required. In summary, the 0.4 M solutions of 1 have become easily accessible using a cost-effective method via an almost quantitative hydroboration reaction of BH3·SMe2 and hexafluoroisopropene. With 1 in hand, several Lewis-base-stabilized hexafluoroisopropylboranes were synthesized, as depicted in Figure 2.
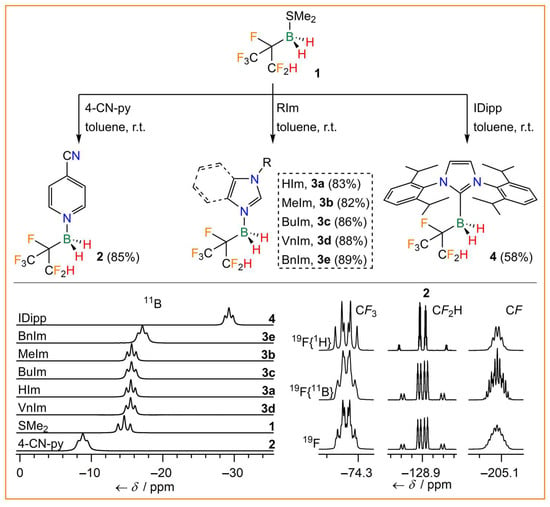
Figure 2.
Synthesis of hexafluoroisopropylborane adducts 2, 3a–e, and 4 (top), 11B NMR spectra (bottom left), and 19F NMR spectra of 2 (bottom right).
The 4-cyanopyridine (4-CN-py) adduct 2 was synthesized from 1 and 4-CN-py in a yield of 85%. Borane adducts of pyridine and 4-cyanopyridine are valuable starting compounds for imidazole and related N-heterocyclic derivatives with boranes attached to one or two nitrogen atoms of the central N-heterocyclic ring. Adducts of this type with the borane Lewis acids B(CN)3 [43] and C2F5BF2 have been deprotonated to obtain mono- and dianionic N-heterocyclic carbenes (NHCs) as well as anionic cyclic (alkyl)(amino)carbenes (Ani-cAACs) [44,45,46]. However, it was found to be even more convenient to react 1 directly with imidazole derivatives to furnish the corresponding imidazole adducts. Complexes of hexafluoroisopropylborane 1 with imidazole (HIm, 3a), 1-methylimidazole (MeIm, 3b), 1-butylimidazole (BuIm, 3c), 1-vinylimidazole (VnIm, 3d), and benzimidazole (BnIm, 3e) were isolated in high yields of 82–89%. Furthermore, stabilization of hexafluoroisopropylborane was achieved by attachment of 1 to the carbene C atom of a N-heterocyclic carbene, i.e., by the reaction of 1 with 1,3-bis(2,6-diisopropylphenyl)-imidazolin-2-ylidene (IDipp, 4). Since 1 was obtained as a racemate via the hydroboration reaction, 2, 3a–e, and 4 were also obtained as racemates. The borane adducts 2, 3a–e, and 4 were found to be stable against hydrolysis, which underlines the stabilizing effect of the electron-withdrawing (CF3)(CF2H)CF group. Hence, borane adducts of 1 are more stable than, for example, related adducts of fluorodicyanoborane, e.g., BF(CN)2·4-CN-py undergoes rapid hydrolysis [43]. All hexafluoroisopropylborane adducts were characterized by elemental analysis and NMR and vibrational spectroscopy (Table 1, Figure 2). The 11B NMR signals are in the region typical for tetravalent boron and their shift depends on the Lewis base coordinated to boron. The signals are split into triplets, due to 1J(11B,1H) couplings of 88.1–100.2 Hz. The 19F NMR signals reveal complex coupling patterns as a result of mJ(19F,19F) (m = 3,4), nJ(19F,1H) (n = 2–4), and 2J(19F,11B) couplings (Figure 2). The IR and Raman spectra of the adducts 2, 3a–e, and 4 display characteristic B−H stretching bands in the region of 2403–2471 cm−1 (Table 1). Furthermore, the thermal properties of the hexafluoroisopropylborane adducts were investigated by differential scanning calorimetry (DSC). 2, 3b, and 4 are solids at room temperature, whereas 3a, 3c, 3d, and 3e are liquids with glass transitions in the range of −87 to −37 °C. All adducts are very thermally robust with decomposition temperatures in the range of 182–256 °C.

Table 1.
Selected experimental and calculated 1 spectroscopic and thermal properties of 2, 3a–e, and 4 2.
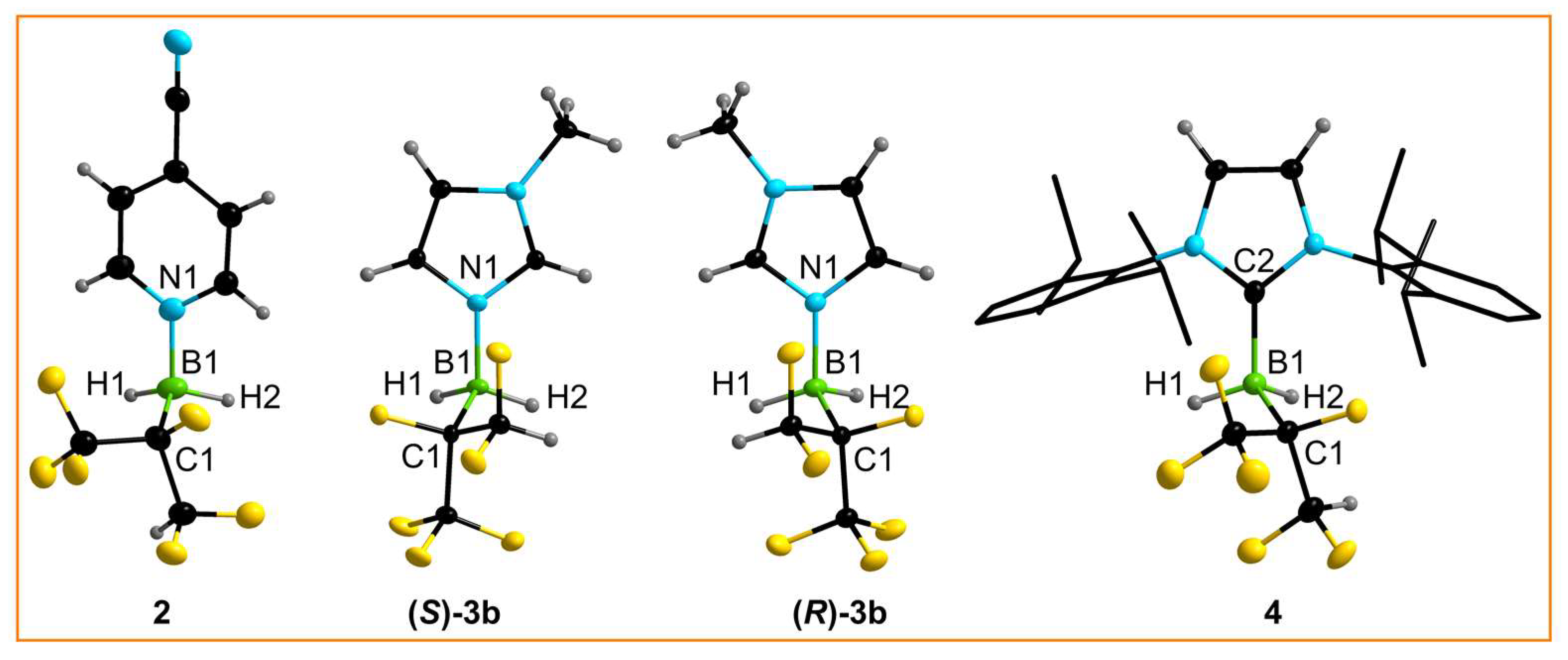
Single crystals of 2, 3b, and 4 were studied by SC-XRD (single-crystal X-ray diffraction). Whereas 2 and 4 crystallized as racemate in the centrosymmetric space group P, single crystals of the two enantiomers (S)-3b and (R)-3b were obtained and the structures were refined in the chiral space group P1 (Figure 3). The lengths of the dative B−N bonds in 2 and 3b (Table 2) are in the range of isoelectronic C−C single bonds and of the B−N bonds in the adducts of the Lewis superacid B(CN)3 with 4-CN-py (1.592(2) pm) [43] and MeIm (1.558(4) pm) [45]. The B−CNHC bond length of 4 is slightly longer than that of BH3·IDipp (1.585(4) pm) [47], which probably reflects the greater steric demand of the hexafluoroisopropylborane compared with BH3 (vide infra).
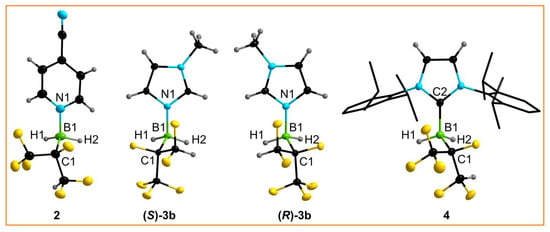
Figure 3.
Crystal structures of the borane adducts 2, (S)-3b, (R)-3b, and 4 (ellipsoids are drawn at the 35% probability level except for H atoms, which are depicted with arbitrary radii; disorder in the (CF3)(CF2H)CF groups of 2 and 4 and the H atoms in the Dipp units of 4 are omitted for clarity and the C atoms of the Dipp units are shown as wire and stick models).
Table 2.
Selected experimental and calculated 1 structural properties of 2, (S)-3b, (R)-3b, and 4 2.
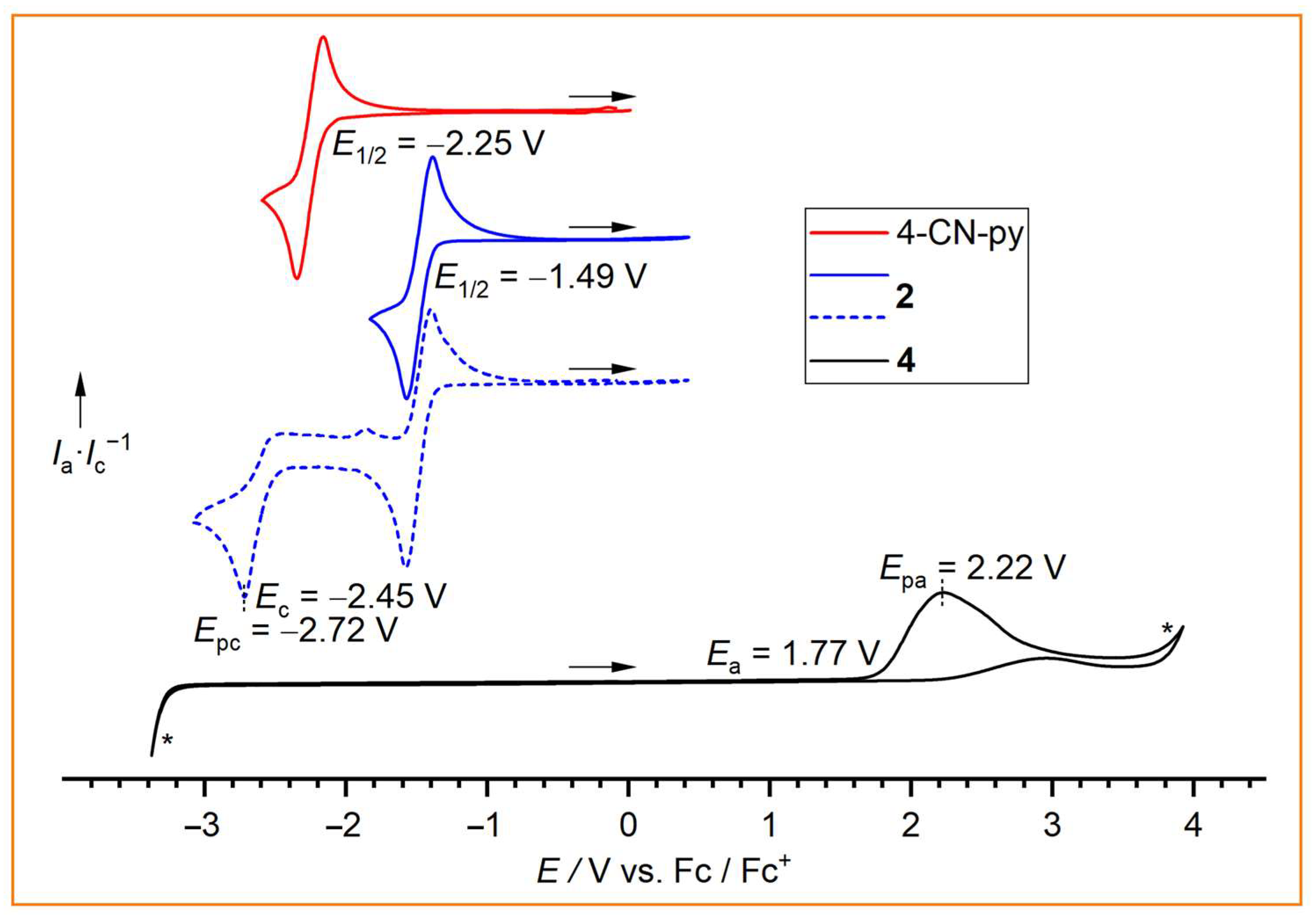
In order to study their electrochemical properties, 2 and 4 were investigated by cyclic voltammetry (Figure 4). 4-Cyanopyridine is known to form a relatively stable radical anion at a potential of E1/2 = −2.25 V (ipa/ipc = 0.82), which corresponds to a quasireversible reduction [43,48]. The 4-cyanopyridine adduct 2 exhibits a similar quasireversible reduction at E1/2 = −1.49 V (ipa/ipc = 0.88). Thus, 2 is reduced at a more negative potential than the related 4-CN-py adducts of B(CN)3 (−1.04 V), BF(CN)2 (−1.14), and BH(CN)2, (−1.25 V) [43]. Furthermore, an additional irreversible reduction was observed at −2.45 V for 2. NHC adducts of boranes are of particular interest as additives for LIB electrolytes, in which they serve as overvoltage protection [49,50,51]. Therefore, the electrochemical behavior of the NHC adduct 4 was investigated by cyclic voltammetry on a 0.01 M solution of 4 in acetonitrile with [nBu4N][PF6](0.1 M). NHC adduct 4 shows only a single irreversible oxidation in the electrochemical window of the solution at 1.77 V vs. Fc/Fc+ or 5.02 V vs. Li/Li+ [52], which is higher than the cut-off potential of the 1,3-dimethylimidazolidin-trifluoroborane adduct (4.4 V vs. Li/Li+) [51]. Since the cut-off potential of 4 is still clearly within the electrochemical window, 4 might be a promising additive for overvoltage protection in electrochemical devices.
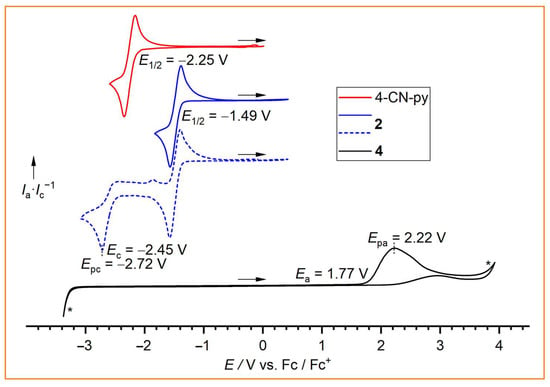
Figure 4.
Cyclic voltammograms of 4-CN-py, 2, and 4 (asterisks mark the boundaries of the electrochemical window) measured in CH3CN containing 0.1 mol·L−1 [nBu4N][PF6] with a Ag/Ag+ reference electrode versus Fc/Fc+ (scan rate 0.05 V·s−1).
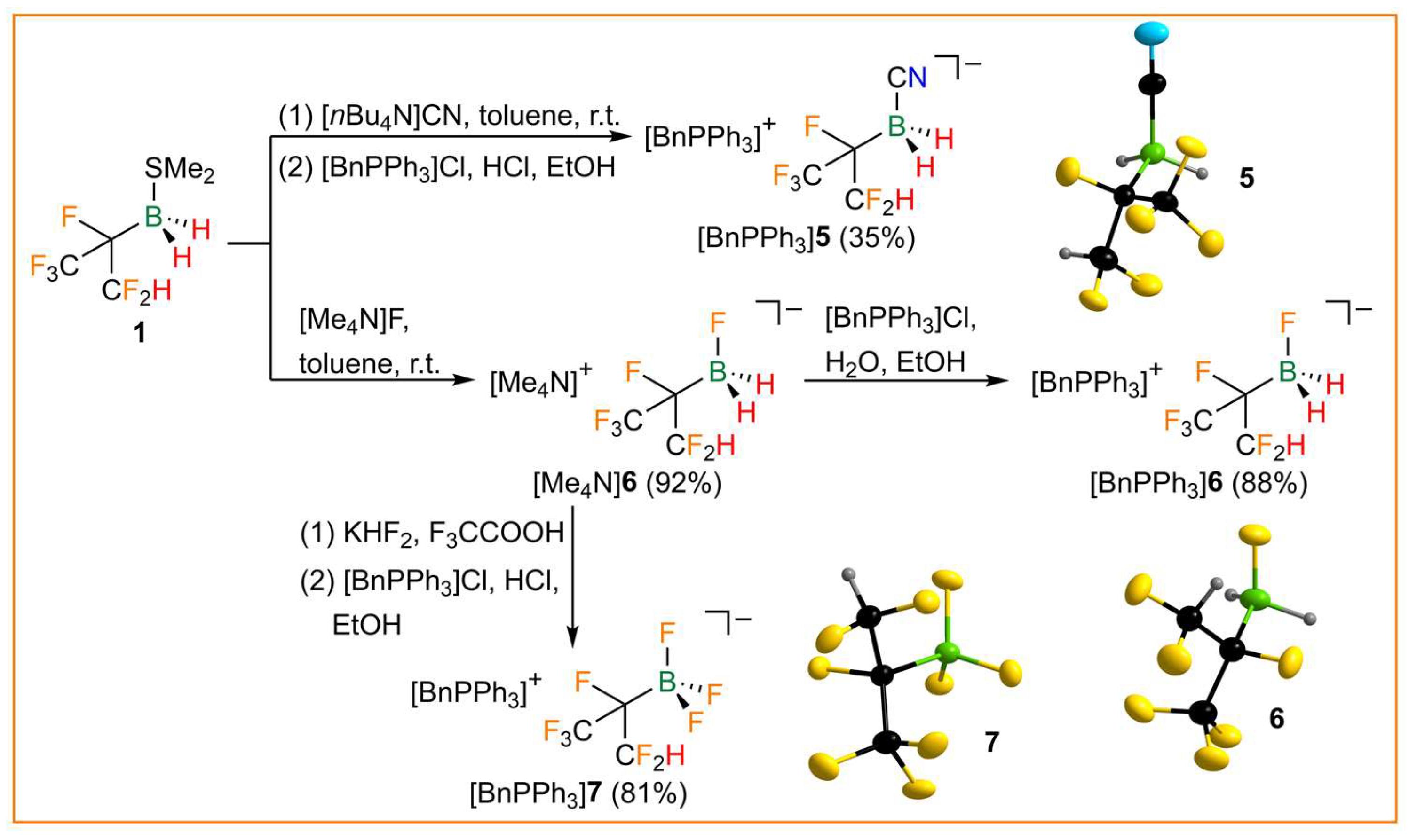
The reaction of 1 with the organic cyanide and fluoride salts [nBu4N]CN and [Me4N]F resulted in the formation of the respective tetraalkylammonium salt of the hexafluoroisopropyldihydridocyanoborate (5) and -fluoroborate anion (6). Both anions were isolated as benzyltriphenylphosphonium ([BnPPh3]+) salts after metathesis (Figure 5). Fluorination of [Me4N]6 with KHF2 in trifluoroacetic acid under evolution of hydrogen gave the corresponding trifluoroborate 7, which was also isolated as a [BnPPh3]+ salt. [BnPPh3]5, [BnPPh3]6, and [BnPPh3]7 were characterized by elemental analysis and NMR and vibrational spectroscopy (Table 3). The 11B NMR signals show complex coupling patterns due to 2J(19F,11B), 1J(11B,1H), and/or 1J(19F,11B) (Table 3, Figure S5 in the Supporting Information). The hexafluoroisopropylborates are very thermally robust, with meting points of 160–164 °C. Whereas [BnPPh3]6 melts under decomposition, [BnPPh3]5 and [BnPPh3]7 are stable up to more than 250 °C in the solid state. [BnPPh3]5, [BnPPh3]6, and [BnPPh3]7 are isostructural and crystallize as racemates in the monoclinic space group P21/n (Figure 5). Due to disorder of the anions, a comparison of bond parameters is obsolete. However, the structure of anion 6 is particularly of interest as it allows the assessment of the steric demand of hexafluoroisopropylborane (CF3)(CF2H)CFBH2 [53]. Since anion 6 is severely disordered over four positions in the crystal structure, the structural parameters derived from DFT calculations were used. The %VBur of (CF3)(CF2H)CFBH2 was determined to be 39.2%, which is in between the %VBur values of B(CN)3 (38.9%) and C2F5BF2 (39.7%) [53].
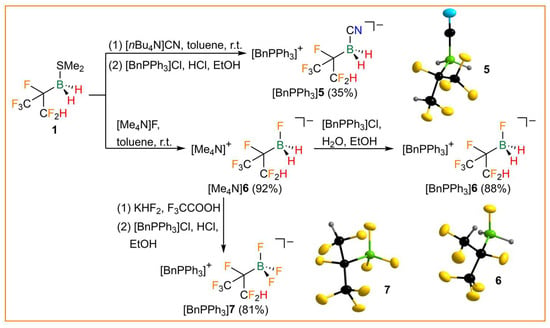
Figure 5.
Synthesis of the hexafluoroisopropylborates [BnPPh3]5, [BnPPh3]6, and [BnPPh3]7 and the anions in the respective crystal structures (ellipsoids are drawn at the 35% probability level except for H atoms, which are depicted with arbitrary radii; disorder in the (CF3)(CF2H)CF groups of 5 and 6 and disorder in 7 is omitted for clarity).
Table 3.
Selected experimental and calculated 1 spectroscopic, thermal, and structural properties of [BnPPh3]5, [BnPPh3]6, and [BnPPh3]7 2.
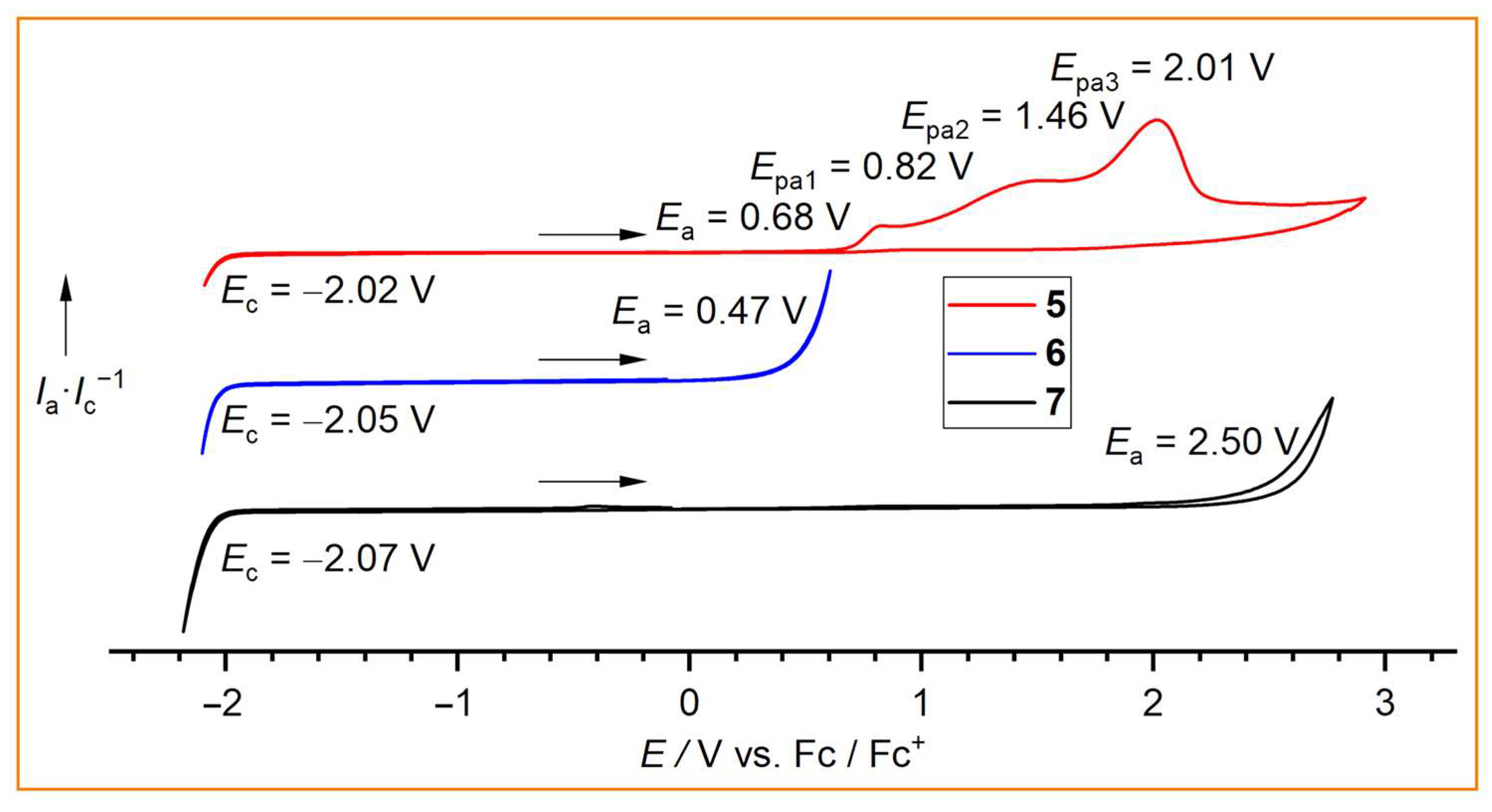
To test their use as conducting salts or electrolyte additives and to verify the electrochemical stabilities of the new borate anions 5, 6, and 7, cyclic voltammetry was performed on 0.05 M solutions in acetonitrile (Figure 6). Since the cathodic limit is predominantly defined by the reduction of the [BnPPh3]+ cation, almost identical limits were observed for all three borates. However, for the anodic limit, which is mainly determined by the electrochemical stability of the borate anion against oxidation, significant differences were found. The fluorodihydridoborate anion [(CF3)(CF2H)CFBH2F]− (6) is the least stable (Ea = 0.47 V) in the series, whereas the trifluoroborate anion [(CF3)(CF2H)CFBF3]− (7) is the most electrochemically robust anion, with Ea = 2.50 V. This value is similar to the anodic limits that have been reported for neat room temperature ionic liquids (RTILs) with related perfluoroalkylborate anions, e.g., Ea = 2.1 V for [EMIm][CF3BF3] and [EMIm][C2F5BF3] (EMIm = 1-ethyl-3-methylimidazolium) [30]. The cyanodihydridoborate anion [(CF3)(CF2H)CFBH2(CN)]− (5) exhibits a slightly higher stability than anion 6 (Ea = 0.68 V). It undergoes a complex irreversible multistep oxidation (Figure 6). Similar or even slightly higher oxidative stabilities are often observed for cyanoborate anions compared with analogous fluoroborate anions [30,31,54]. For example, the neat RTIL [BMPL][BF(CN)3] ([BMPL]MFB; BMPL = 1-butyl-1-methylpyrrolidinium) is more stable (Ea = 2.6 V) than the related RTIL [BMPL][BF2(CN)2] ([BMPL]DFB; Ea = 2.0 V) [54].
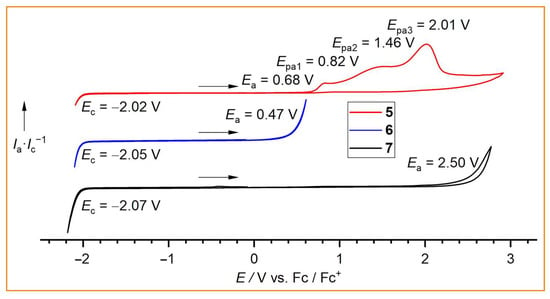
Figure 6.
Cyclic voltammograms of [BnPPh3]5, [BnPPh3]6, and [BnPPh3]7 as 0.05 M solutions in CH3CN with a Ag/Ag+ reference electrode versus Fc/Fc+ (scan rate 0.05 V·s−1).
3. Materials and Methods
3.1. General Synthetic Aspects
All reactions involving air-sensitive compounds were performed under an argon atmosphere using standard Schlenk line techniques in tubes equipped with PTFE stems (Rettberg, Göttingen and Young, London, UK).
3.2. Analytical Instruments and Details
1H, 11B, 13C, 19F, and 31P NMR spectra were recorded at 25 °C in CD2Cl2 and (CD3)2CO on a Bruker Avance 500 spectrometer, a Bruker Avance III 400 Nanobay spectrometer, or a Bruker Avance Neo 400 spectrometer. NMR signals were referenced against TMS (1H and 13C with Ξ (13C) = 25.145020 MHz), BF3·OEt2 in CDCl3 (Ξ (11B) = 32.083974 MHz), CCl3F (Ξ (19F) = 94.094011 MHz), and H3PO4 (Ξ (31P) = 40.480742 MHz) [55]. Chemical shifts were calibrated against the residual solvent signal, respectively (δ(1H): CD2Cl2 5.32, (CD2H)(CD3)CO 2.05 ppm; δ(13C): CD2Cl2 53.84, (CD3)2CO 206.26 and 29.84 ppm) [56]. HRMS-ESI spectra were recorded using an Exactive Plus mass spectrometer with Orbitrap (Thermo Scientific, Waltham, MA, USA) equipped with an ESI source (3.5 kV spray voltage) or an APCI probe (4.0 µA discharge current) (HRMS = high-resolution mass spectrometry, ESI = electrospray ionization). IR spectra of the solid samples were recorded at room temperature with a Bruker Alpha-FT-IR spectrometer (Karlsruhe, Germany) with an apodized resolution of 2 cm−1 in the attenuated total reflection (ATR) mode in the region of 4000–400 cm−1 using a setup with a diamond crystal. Raman spectra were measured at room temperature on a Bruker MultiRAM FT spectrometer with an apodized resolution of 2 cm−1 in the region of 4000–400 cm−1. The samples were contained in melting point capillaries and excited with the 1064 nm line of a Nd/YAG laser. Thermal analyses were performed with a DSC 204 F1 Phoenix (Netzsch, Selb, Germany) in the temperature range of −20 to 500 °C with a heating rate of 10 K min−1. Elemental analyses (C, H, N) were performed either with a Euro EA3000 instrument (HEKA-Tech, Wegberg, Germany) or with an Elementar Vario MICRO cube instrument (Elementar Analysensysteme, Langenselbold, Germany). Details of the DFT calculations and electrochemical measurements are included in the Supporting Information.
3.3. Chemicals
All standard chemicals were obtained from commercial sources and used without further purification. [nBu4N]CN was synthesized according to a known procedure [57]. Solvents were dried according to standard protocols [58] and stored under argon atmosphere in flasks with valves with PTFE stems (Rettberg, Göttingen and Young, London).
3.4. Syntheses
3.4.1. Hexafluoroisopropylborane-dimethylsulfide Adduct (1)
The synthesis was carried out following the procedure in the literature [42]; however, the amount of reactants used was increased by a factor of 2100, whereas the amount of solvent was only increased by a factor of 200.
Hexafluoropropene C3F6 (6.31 g, 42.0 mmol) was condensed into a solution of BH3·SMe2 (2.0 M in toluene, 20 mL, 40.0 mmol) in toluene (80 mL) at −78 °C. The reaction mixture was slowly heated to 100 °C in a closed reaction vessel under vigorous stirring and kept at this temperature for one hour. The hexafluoroisopropylborane-dimethylsulfide solution obtained was used without further purification. Yield: 100 mL of a 0.4 M solution of (CF3)(CF2H)CFBH2·SMe2 (40.0 mmol) in toluene.
11B NMR (128.43 MHz): δ = −14.6 (t, 1B, BH2, 1J(11B,1H) = 109.7 Hz) ppm; 19F NMR (376.66 MHz): δ = −75.1 (m, 3F, CF3), −128.5 (dm, 2F, CF2H, 2J(19F,1H) = 53.5 Hz), −199.6 (m, 1F, CF) ppm.
3.4.2. Hexafluoroisopropylborane-4-cyanopyridine Adduct (2)
A solution of 1 (0.2 M in toluene, 100 mL, 20.0 mmol) was added to 4-cyanopyridine (2.08 g, 20.0 mmol). After stirring for 14 h at room temperature, all volatiles were removed under reduced pressure. The solid residue was taken up into CH2Cl2 (30 mL) and washed with water (3 × 10 mL). Subsequently, the organic solvent was removed under reduced pressure and the product was dried under vacuum. Yield: 4.53 g (16.9 mmol, 85%, calculated for 1) of a light-yellow solid.
1H NMR (500.13 MHz, CD2Cl2): δ = 8.79 (d, 2H, CH, 3J(1H,1H) = 6.7 Hz), 8.79 (dm, 2H, CH, 3J(1H,1H) = 6.8 Hz), 5.95 (td, 1H, CF2H, 2J(19F,1H) = 54.6 Hz, 3J(19F,1H) = 9.2 Hz), 2.77(m, 2H, BH2) ppm; 1H{11B} NMR (500.13 MHz, CD2Cl2): δ = 8.79 (d, 2H, CH, 3J(1H,1H) = 6.7 Hz), 8.79 (d, 2H, CH, 3J(1H,1H) = 6.8 Hz)), 5.95 (td, 1H, CF2H, 2J(19F,1H) = 54.6 Hz, 3J(19F,1H) = 9.2 Hz), 2.77(d, 2H, BH2, 3J(19F,1H) = 24.7 Hz) ppm; 1H{19F} NMR (400.30 MHz, CD2Cl2): δ = 8.79 (d, 2H, CH, 3J(1H,1H) = 6.7 Hz), 8.79 (dm, 2H, CH, 3J(1H,1H) = 6.8 Hz), 5.95 (m, 1H, CF2H), 2.77(m, 2H, BH2) ppm; 11B NMR (160.46 MHz, CD2Cl2): δ = −8.8 (t, 1B, BH2, 1J(11B,1H) = 94.8 Hz) ppm; 11B{1H} NMR (160.46 MHz, CD2Cl2): δ = −8.8 (s, 1B, BH2) ppm; 11B{19F} NMR (160.46 MHz, CD2Cl2): δ = −8.8 (t, 1B, BH2, 1J(11B,1H) = 94.8 Hz) ppm; 13C{1H} NMR (125.76 MHz, CD2Cl2): δ = 150.3 (s, 2C, CH), 128.5 (s, 2C, CH), 125.9 (s, 1C, C−CN), 125.0 (qdt, 1C, CF3, 1J(19F,13C) = 284.1 Hz, 2J(19F,13C) = 25.7 Hz, 3J(19F,13C) = 5.4 Hz), 114.8 (s, 1C, CN), 114.9 (tdq, 1C, CF2H, 1J(19F,13C) = 246.3 Hz, 2J(19F,13C) = 27.2 Hz, 3J(19F,13C) = 2.9 Hz), 95.4 (d, br, 1C, CF, 1J(19F,13C) = 183.0 Hz) ppm; 13C{11B,1H} NMR (75.48 MHz, CD2Cl2): δ = 150.3 (s, 2C, CH), 128.5 (s, 2C, CH), 125.9 (s, 1C, C−CN), 125.0 (qdt, 1C, CF3, 1J(19F,13C) = 284.1 Hz, 2J(19F,13C) = 25.7 Hz, 3J(19F,13C) = 5.4 Hz), 114.9 (s, 1C, CN), 114.8 (tdq, 1C, CF2H, 1J(19F,13C) = 246.3 Hz, 2J(19F,13C) = 27.2 Hz, 3J(19F,13C) = 2.9 Hz), 95.4 (dqt, 1C, CF, 1J(19F,13C) = 183.0 Hz, 2J(19F,13C) = 28.8 Hz, 2J(19F,13C) = 25.1 Hz) ppm; 13C{19F} NMR (125.76 MHz, CD2Cl2): δ = 150.3 (d, 2C, CH, 1J(13C,1H) = 191.8 Hz), 128.5 (ddt, 2C, CH, 1J(13C,1H) = 176.2 Hz, 2J(13C,1H) = 6.5 Hz, 3J(13C,1H) = 2.7 Hz), 125.9 (t, 1C, C−CN, 3J(13C,1H) = 7.9 Hz), 125.0 (s, 1C, CF3), 114.9 (t, 1C, CN, 3J(13C,1H) = 5.3 Hz), 114.8 (d, 1C, CF2H, 1J(13C,1H) = 190.4 Hz), 95.4 (s, br, 1C, CF) ppm; 19F NMR (376.82 MHz, CD2Cl2): δ = −74.3 (dtt, 3F, CF3, 3J(19F,19F) = 10.3 Hz, 4J(19F,19F) = 7.9 Hz, 4J(19F,1H) = 2.2 Hz), −128.9 (dm, 2F, CF2H, 2J(19F,1H) = 54.2 Hz), −205.1 (tqm, 1F, CF, 3J(19F,19F) = 10.6 Hz, 3J(19F,19F) = 10.6 Hz) ppm; 19F{1H} NMR (376.82 MHz, CD2Cl2): δ = −74.3 (dt, 3F, CF3, 3J(19F,19F) = 10.3 Hz, 4J(19F,19F) = 7.9 Hz), −128.9 (m, 2F, CF2H), −205.1 (tq, 1F, CF, 3J(19F,19F) = 10.6 Hz, 3J(19F,19F) = 10.6 Hz) ppm; 19F{11B} NMR (470.59 MHz, CD2Cl2): δ = −74.3 (dtt, 3F, CF3, 3J(19F,19F) = 10.3 Hz, 4J(19F,19F) = 7.9 Hz, 4J(19F,1H) = 2.2 Hz), −128.9 (dm, 2F, CF2H, 2J(19F,1H) = 54.2 Hz), −205.1 (tqm, 1F, CF, 3J(19F,19F) = 10.6 Hz, 3J(19F,19F) = 10.6 Hz) ppm.
IR (ATR): 2464, 2430 cm−1 ((B–H)).
Raman: 2471, 2433 cm−1 ((B–H)), 2247 cm−1 ((C≡N)).
Elemental analysis: Calculated (%) for C9H7BF6N2, C 40.34, H 2.63, N 10.45; found, C 41.04, H 2.83, N 10.46.
HMRS ((–)-ESI): m/z (isotopic abundance) calculated for C9H7BF6N2: 268.06 (100.0%), 267.06 (24.8%), 269.06 (9.7%), 268.07 (2.4%); found, 268.06 (100.0%), 267.06 (25.2%), 269.06 (9.4%), 268.07 (1.0%).
Crystals of 2 suitable for an X-ray diffraction study were obtained via the slow evaporation of an acetone solution.
3.4.3. Hexafluoroisopropylborane-imidazole Adducts (3a–e)
A solution of 1 (0.4 M in toluene, 12.5 mL, 5.00 mmol) was added to the corresponding imidazole (5.00 mmol). After stirring for 48 h at room temperature, the reaction mixture was washed with water (3 × 5 mL). Subsequently, the organic phase was dried over Na2SO4, filtered, and the organic solvent was removed under reduced pressure. The product was dried under vacuum.
Hexafluoroisopropylborane-imidazole adduct 3a was prepared using imidazole (340 mg, 5.00 mmol). Yield: 963 mg (4.15 mmol, 85%, calculated for 1) of a colorless liquid.
Hexafluoroisopropylborane-imidazole-adduct 3a can also be prepared by using 1-(trimethylsilyl)imidazole (701 mg, 733 µL, 5.00 mmol). Yield: 1.03 g (4.44 mmol, 89%, calculated for 1) of a colorless liquid.
1H NMR (500.13 MHz, CD2Cl2): δ = 9.84 (s, br, 1H, NH), 8.00 (s, 1H, NCHN), 7.20 (s, br, 1H, CH), 7.15 (m, 1H, CH), 5.91 (tdq, 1H, CF2H, 2J(19F,1H) = 54.5 Hz, 3J(19F,1H) = 9.4 Hz, 4J(19F,1H) = 0.6 Hz), 2.46 (qd, 2H, BH2, 1J(11B,1H) = 99.8, 3J(19F,1H) = 26.8) ppm; 1H{11B} NMR (500.13 MHz, CD2Cl2): δ = 9.84 (s, br, 1H, NH), 8.00 (s, 1H, NCHN), 7.20 (s, br, 1H, CH), 7.15 (s, br, 1H, CH), 5.91 (td, 1H, CF2H, 2J(19F,1H) = 54.5 Hz, 3J(19F,1H) = 9.4 Hz), 2.46 (dq, 2H, BH2, 3J(19F,1H) = 26.8, 4J(19F,1H) = 2.7) ppm; 1H{19F} NMR (400.30 MHz, CD2Cl2): δ = 9.84 (s, br, 1H, NH), 8.00 (s, 1H, NCHN), 7.20 (s, br, 1H, CH), 7.15 (m, 1H, CH), 5.91 (m, 1H, CF2H), 2.46(q, 2H, BH2, 1J(11B,1H) = 99.2) ppm; 11B NMR (160.46 MHz, CD2Cl2): δ = −15.5 (t, 1B, BH2, 1J(11B,1H) = 99.8 Hz) ppm; 11B{1H} NMR (160.46 MHz, CD2Cl2): δ = −15.5 (s, 1B, BH2) ppm; 11B{19F} NMR (160.46 MHz, CD2Cl2): δ = −15.5 (t, 1B, BH2, 1J(11B,1H) = 99.8 Hz) ppm; 13C{1H} NMR (125.76 MHz, CD2Cl2): δ = 136.7 (s, 1C, NCHN), 127.9 (s, 1C, CH), 125.5 (qdt, 1C, CF3, 1J(19F,13C) = 283.6 Hz, 2J(19F,13C) = 25.9 Hz, 3J(19F,13C) = 4.9 Hz), 117.4 (s, 1C, CH), 115.3 (tdq, 1C, CF2H, 1J(19F,13C) = 246.6 Hz, 2J(19F,13C) = 27.0 Hz, 3J(19F,13C) = 2.8 Hz), 96.0 (d, br, 1C, CF, 1J(19F,13C) = 180.5 Hz) ppm; 13C{11B,1H} NMR (75.48 MHz, CD2Cl2): δ = 136.9 (s, 1C, NCHN), 125.4 (s, 1C, CH), 125.5 (qdt, 1C, CF3, 1J(19F,13C) = 283.6 Hz, 2J(19F,13C) = 25.9 Hz, 3J(19F,13C) = 4.9 Hz), 117.8 (s, 1C, CH), 115.5 (tdq, 1C, CF2H, 1J(19F,13C) = 246.6 Hz, 2J(19F,13C) = 27.0 Hz, 3J(19F,13C) = 2.8 Hz), 96.0 (dqt, 1C, CF, 1J(19F,13C) = 180.5 Hz, 2J(19F,13C) = 29.1 Hz, 2J(19F,13C) = 23.4 Hz) ppm; 13C{19F} NMR (125.76 MHz, CD2Cl2): δ = 136.7 (d, 1C, NCHN, 1J(13C,1H) = 217.8 Hz), 127.9 (dm, 1C, CH, 1J(13C,1H) = 200.3 Hz), 125.5 (s, 1C, CF3), 117.4 (dm, 1C, CH, 1J(13C,1H) = 199.3 Hz), 115.3 (dt, 1C, CF2H, 1J(13C,1H) = 188.7 Hz, 3J(13C,1H) = 3.3 Hz), 96.0 (m, br, 1C, CF) ppm; 19F NMR (376.82 MHz, CD2Cl2): δ = −73.9 (dtt, 3F, CF3, 3J(19F,19F) = 10.3 Hz, 4J(19F,19F) = 7.9 Hz, 4J(19F,1H) = 2.2 Hz), −129.5 (dm, 2F, CF2H, 2J(19F,1H) = 54.4 Hz), −205.3 (m, 1F, CF) ppm; 19F{1H} NMR (376.82 MHz, CD2Cl2): δ = −73.9 (dt, 3F, CF3, 3J(19F,19F) = 10.3 Hz, 4J(19F,19F) = 7.9 Hz), −129.5 (m, 2F, CF2H), −205.3 (m, 1F, CF) ppm; 19F{11B} NMR (470.59 MHz, CD2Cl2): δ = −73.9 (dtt, 3F, CF3, 3J(19F,19F) = 10.3 Hz, 4J(19F,19F) = 7.9 Hz, 4J(19F,1H) = 2.2 Hz), −129.5 (dm, 2F, CF2H, 2J(19F,1H) = 54.4 Hz), −205.3 (m, 1F, CF) ppm.
IR (ATR): 3447 cm−1 ((N–H)), 2414 cm−1 ((B–H)).
Raman: 2418 cm−1 ((B–H)).
Elemental analysis: Calculated (%) for C6H7BF6N2, C 31.07, H 3.04, N 12.06; found, C 31.09, H 3.07, N 12.85.
HMRS ((-)-APCI): m/z (isotopic abundance) calculated for deprotonated 3a C6H6BF6N2−: 231.05 (100.0%), 230.06 (24.8%), 232.06 (6.5%), 231.06 (1.6%); found, 231.05 (100.0%), 230.06 (23.7%), 232.06 (5.8%).
Hexafluoroisopropylborane-1-methylimidazole adduct 3b was prepared using 1-methylimidazole (410 mg, 396 µL, 5.00 mmol). Yield: 1.01 g (4.11 mmol, 82%, calculated for 1) of a colorless solid.
1H NMR (500.13 MHz, CD2Cl2): δ = 7.79 (s, 1H, NCHN), 7.10 (s, br, 1H, CH), 6.96 (m, 1H, CH), 5.88 (tdq, 1H, CF2H, 2J(19F,1H) = 54.5 Hz, 3J(19F,1H) = 9.4 Hz, 4J(19F,1H) = 0.6 Hz), 3.76 (s, 3H, CH3), 2.39 (qm, 2H, BH2, 1J(11B,1H) = 100.2) ppm; 1H{11B} NMR (500.13 MHz, CD2Cl2): δ = 7.79 (s, 1H, NCHN), 7.10 (s, br, 1H, CH), 6.96 (s, br, 1H, CH), 5.91 (td, 1H, CF2H, 2J(19F,1H) = 54.5 Hz, 3J(19F,1H) = 9.4 Hz), 3.76 (s, 3H, CH3), 2.39 (dq, 2H, BH2, 3J(19F,1H) = 27.1, 4J(19F,1H) = 2.8) ppm; 1H{19F} NMR (400.30 MHz, CD2Cl2): δ = 7.79 (s, 1H, NCHN), 7.10 (s, br, 1H, CH), 6.96 (m, 1H, CH), 5.88 (m, 1H, CF2H), 3.76 (s, 3H, CH3), 2.39 (q, 2H, BH2, 1J(11B,1H) = 100.2) ppm; 11B NMR (160.46 MHz, CD2Cl2): δ = −15.6 (t, 1B, BH2, 1J(11B,1H) = 100.2 Hz) ppm; 11B{1H} NMR (160.46 MHz, CD2Cl2): δ = −15.6 (s, 1B, BH2) ppm; 11B{19F} NMR (160.46 MHz, CD2Cl2): δ = −15.6 (t, 1B, BH2, 1J(11B,1H) = 100.2 Hz) ppm; 13C{1H} NMR (125.76 MHz, CD2Cl2): δ = 138.5 (s, 1C, NCHN), 128.4 (s, 1C, CH), 125.5 (qdt, 1C, CF3, 1J(19F,13C) = 284.2 Hz, 2J(19F,13C) = 25.9 Hz, 3J(19F,13C) = 4.8 Hz), 121.5 (s, 1C, CH), 115.4 (tdq, 1C, CF2H, 1J(19F,13C) = 246.4 Hz, 2J(19F,13C) = 26.7 Hz, 3J(19F,13C) = 2.8 Hz), 95.8 (d, br, 1C, CF, 1J(19F,13C) = 180.4 Hz), 35.5 (s, 1C, CH3) ppm; 13C{11B,1H} NMR (75.48 MHz, CD2Cl2): δ = 138.5 (s, 1C, NCHN), 128.2 (s, 1C, CH), 125.6 (qdt, 1C, CF3, 1J(19F,13C) = 284.2 Hz, 2J(19F,13C) = 25.9 Hz, 3J(19F,13C) = 4.8 Hz), 121.7 (s, 1C, CH), 115.5 (tdq, 1C, CF2H, 1J(19F,13C) = 246.4 Hz, 2J(19F,13C) = 26.7 Hz, 3J(19F,13C) = 2.8 Hz), 95.9 (dqt, 1C, CF, 1J(19F,13C) = 180.4 Hz, 2J(19F,13C) = 28.5 Hz, 2J(19F,13C) = 23.3 Hz), 35.4 (s, 1C, CH3) ppm; 13C{19F} NMR (125.76 MHz, CD2Cl2): δ = 138.5 (d, 1C, NCHN, 1J(13C,1H) = 214.9 Hz), 128.4 (dm, 1C, CH, 1J(13C,1H) = 198.8 Hz), 125.5 (s, 1C, CF3), 121.5 (dm, 1C, CH, 1J(13C,1H) = 197.9 Hz), 115.4 (dt, 1C, CF2H, 1J(13C,1H) = 189.1 Hz, 3J(13C,1H) = 3.3 Hz), 95.8 (m, br, 1C, CF), 35.5 (q, 1C, CH3, 1J(13C,1H) = 142.4 Hz) ppm; 19F NMR (376.82 MHz, CD2Cl2): δ = −74.0 (dtt, 3F, CF3, 3J(19F,19F) = 10.2 Hz, 4J(19F,19F) = 8.0 Hz, 4J(19F,1H) = 2.2 Hz), −129.7 (dm, 2F, CF2H, 2J(19F,1H) = 54.3 Hz), −205.8 (m, 1F, CF) ppm; 19F{1H} NMR (376.82 MHz, CD2Cl2): δ = −74.0 (dt, 3F, CF3, 3J(19F,19F) = 10.2 Hz, 4J(19F,19F) = 8.0 Hz), −129.7 (m, 2F, CF2H), −205.8 (m, 1F, CF) ppm; 19F{11B} NMR (470.59 MHz, CD2Cl2): δ = −74.0 (dtt, 3F, CF3, 3J(19F,19F) = 10.3 Hz, 4J(19F,19F) = 7.9 Hz, 4J(19F,1H) = 2.2 Hz), −129.5 (dm, 2F, CF2H, 2J(19F,1H) = 54.3 Hz), −205.8 (m, 1F, CF) ppm.
IR (ATR): 2395 cm−1 ((B–H)).
Raman: 2407 cm−1 ((B–H)).
Elemental analysis: Calculated (%) for C7H9BF6N2, C 34.18, H 3.69, N 11.39; found, C 34.16, H 3.61, N 11.24.
HMRS ((-)-APCI): m/z (isotopic abundance) calculated for deprotonated 3b C7H8BF6N2−: 245.07 (100.0%), 244.07 (24.8%), 246.07 (7.6%), 245.08 (1.9%); found, 245.07 (100.0%), 244.07 (24.1%), 246.07 (7.6%), 245.08 (1.0%).
Crystals of 3b suitable for an X-ray diffraction study were obtained via the slow evaporation of a toluene solution or from a saturated aqueous solution.
Hexafluoroisopropylborane-1-butylimidazole adduct 3c was prepared using 1-butylimidazole (621 mg, 657 µL, 5.00 mmol). Yield: 1.24 g (4.31 mmol, 86%, calculated for 1) of a colorless liquid.
1H NMR (500.13 MHz, CD2Cl2): δ = 7.80 (s, 1H, NCHN), 7.11 (s, br, 1H, CH), 6.98(m, 1H, CH), 5.88 (tdm, 1H, CF2H, 2J(19F,1H) = 54.5 Hz, 3J(19F,1H) = 9.3 Hz), 4.00 (t, 2H, CH2, 3J(1H,1H) = 7.2), 2.40 (qm, 2H, BH2, 1J(11B,1H) = 97.5), 1.80 (m, 2H, CH2), 1.33 (m, 2H, CH2), 0.95 (t, 3H, CH3, 3J(1H,1H) = 7.4) ppm; 1H{11B} NMR (500.13 MHz, CD2Cl2): δ = 7.80 (s, 1H, NCHN), 7.11 (s, br, 1H, CH), 6.98(m, 1H, CH), 5.88 (tdm, 1H, CF2H, 2J(19F,1H) = 54.5 Hz, 3J(19F,1H) = 9.3 Hz), 4.00 (t, 2H, CH2, 3J(1H,1H) = 7.2), 2.40 (dq, 2H, BH2, 3J(19F,1H) = 27.2, 4J(19F,1H) = 2.7), 1.80 (m, 2H, CH2), 1.33 (m, 2H, CH2), 0.95 (t, 3H, CH3, 3J(1H,1H) = 7.4) ppm; 1H{19F} NMR (400.30 MHz, CD2Cl2): δ = 7.80 (s, 1H, NCHN), 7.11 (s, br, 1H, CH), 6.98(m, 1H, CH), 5.88 (m, 1H, CF2H), 4.00 (t, 2H, CH2, 3J(1H,1H) = 7.2), 2.40 (qm, 2H, BH2, 1J(11B,1H) = 97.5), 1.80 (m, 2H, CH2), 1.33 (m, 2H, CH2), 0.95 (t, 3H, CH3, 3J(1H,1H) = 7.4) ppm; 11B NMR (160.46 MHz, CD2Cl2): δ = −15.6 (t, 1B, BH2, 1J(11B,1H) = 97.5 Hz) ppm; 11B{1H} NMR (160.46 MHz, CD2Cl2): δ = −15.6 (s, 1B, BH2) ppm; 11B{19F} NMR (160.46 MHz, CD2Cl2): δ = −15.6 (t, 1B, BH2, 1J(11B,1H) = 97.5 Hz) ppm; 13C{1H} NMR (125.76 MHz, CD2Cl2): δ = 137.8 (s, 1C, NCHN), 128.3 (s, 1C, CH), 125.6 (qdt, 1C, CF3, 1J(19F,13C) = 283.7 Hz, 2J(19F,13C) = 26.0 Hz, 3J(19F,13C) = 4.8 Hz), 120.2 (s, 1C, CH), 115.4 (tdq, 1C, CF2H, 1J(19F,13C) = 247.0 Hz, 2J(19F,13C) = 27.0 Hz, 3J(19F,13C) = 2.9 Hz), 95.8 (d, br, 1C, CF, 1J(19F,13C) = 181.0 Hz), 49.1 (s, 1C, CH2), 32.7 (s, 1C, CH2), 19.8 (s, 1C, CH2) 13.5 (s, 1C, CH3) ppm; 13C{11B,1H} NMR (75.48 MHz, CD2Cl2): δ = 137.9 (s, 1C, NCHN), 128.2 (s, 1C, CH), 125.7 (qdt, 1C, CF3, 1J(19F,13C) = 283.7 Hz, 2J(19F,13C) = 26.0 Hz, 3J(19F,13C) = 4.8 Hz), 120.4 (s, 1C, CH), 115.5 (tdq, 1C, CF2H, 1J(19F,13C) = 247.0 Hz, 2J(19F,13C) = 27.0 Hz, 3J(19F,13C) = 2.9 Hz), 96.0 (dqt, 1C, CF, 1J(19F,13C) = 181.0 Hz, 2J(19F,13C) = 28.6 Hz, 2J(19F,13C) = 23.6 Hz), 49.1 (s, 1C, CH2), 32.7 (s, 1C, CH2), 19.8 (s, 1C, CH2) 13.5 (s, 1C, CH3) ppm; 13C{19F} NMR (125.76 MHz, CD2Cl2): δ = 137.8 (d, 1C, NCHN, 1J(13C,1H) = 214.3 Hz), 128.3 (dm, 1C, CH, 1J(13C,1H) = 198.7 Hz), 125.6 (s, 1C, CF3), 120.2 (dm, 1C, CH, 1J(13C,1H) = 195.9 Hz), 115.4 (dt, 1C, CF2H, 1J(13C,1H) = 188.8 Hz, 3J(13C,1H) = 3.3 Hz), 95.8 (m, br, 1C, CF), 49.1 (tm, 1C, CH2, 1J(13C,1H) = 141.3 Hz), 32.7 (tm, 1C, CH2, 1J(13C,1H) = 127.7 Hz), 19.8 (tm, 1C, CH2, 1J(13C,1H) = 126.0 Hz) 13.5 (qm, 1C, CH3, 1J(13C,1H) = 125.1 Hz) ppm; 19F NMR (376.82 MHz, CD2Cl2): δ = −74.1 (dtt, 3F, CF3, 3J(19F,19F) = 10.2 Hz, 4J(19F,19F) = 8.0 Hz, 4J(19F,1H) = 2.3 Hz), −129.7 (dm, 2F, CF2H, 2J(19F,1H) = 54.6 Hz), −205.7 (m, 1F, CF) ppm; 19F{1H} NMR (376.82 MHz, CD2Cl2): δ = −74.1 (dt, 3F, CF3, 3J(19F,19F) = 10.2 Hz, 4J(19F,19F) = 8.0 Hz), −129.7 (m, 2F, CF2H), −205.7 (m, 1F, CF) ppm; 19F{11B} NMR (470.59 MHz, CD2Cl2): δ = −74.1 (dtt, 3F, CF3, 3J(19F,19F) = 10.3 Hz, 4J(19F,19F) = 7.9 Hz, 4J(19F,1H) = 2.2 Hz), −129.7 (dm, 2F, CF2H, 2J(19F,1H) = 54.3 Hz), −205.7 (m, 1F, CF) ppm.
IR (ATR): 2412 cm−1 ((B–H)).
Raman: 2417 cm−1 ((B–H)).
Elemental analysis: Calculated (%) for C10H15BF6N2, C 41.70, H 5.25, N 9.73; found, C 41.76, H 5.41, N 10.51.
HMRS ((-)-APCI): m/z (isotopic abundance) calculated for deprotonated 3c C10H14BF6N2−: 287.12 (100.0%), 286.12 (24.8%), 288.12 (10.8%), 287.12 (2.7%); found, 287.12 (100.0%), 286.12 (24.3%), 288.12 (11.0%), 287.12 (0.4%).
Hexafluoroisopropyl-1-vinylimidazole adduct 3d was prepared using 1-vinylimidazole (470 mg, 452 µL, 5.00 mmol). Yield: 1.14 g (4.40 mmol, 88%, calculated for 1) of a colorless liquid.
1H NMR (500.13 MHz, CD2Cl2): δ = 8.02 (s, 1H, NCHN), 7.28 (m, 1H, CH), 7.19 (s, br, 1H, CH), 6.92 (dd, 1H, CHVn, 3J(1H,1H) = 15.7 Hz, 3J(1H,1H) = 8.7 Hz), 5.91 (tdq, 1H, CF2H, 2J(19F,1H) = 54.6 Hz, 3J(19F,1H) = 9.4 Hz, 4J(19F,1H) = 0.6 Hz), 5.56 (dd, 1H, CH2Vn, 3J(1H,1H) = 15.7 Hz, 3J(1H,1H) = 2.6 Hz), 5.26 (dd, 1H, CH2Vn, 3J(1H,1H) = 8.7 Hz, 3J(1H,1H) = 2.6 Hz), 2.43 (qm, 2H, BH2, 1J(11B,1H) = 99.5) ppm; 1H{11B} NMR (500.13 MHz, CD2Cl2): δ = 8.02 (s, 1H, NCHN), 7.28 (m, 1H, CH), 7.19 (s, br, 1H, CH), 6.92 (dd, 1H, CHVn, 3J(1H,1H) = 15.7 Hz, 3J(1H,1H) = 8.7 Hz), 5.91 (tdq, 1H, CF2H, 2J(19F,1H) = 54.6 Hz, 3J(19F,1H) = 9.4 Hz, 4J(19F,1H) = 0.6 Hz), 5.56 (dd, 1H, CH2Vn, 3J(1H,1H) = 15.7 Hz, 3J(1H,1H) = 2.6 Hz), 5.26 (dd, 1H, CH2Vn, 3J(1H,1H) = 8.7 Hz, 3J(1H,1H) = 2.6 Hz), 2.43 (dq, 2H, BH2, 3J(19F,1H) = 26.5, 4J(19F,1H) = 2.8) ppm; 1H{19F} NMR (400.30 MHz, CD2Cl2): δ = 8.02 (s, 1H, NCHN), 7.28 (m, 1H, CH), 7.19 (s, br, 1H, CH), 6.92 (dd, 1H, CHVn, 3J(1H,1H) = 15.7 Hz, 3J(1H,1H) = 8.7 Hz), 5.91 (m, 1H, CF2H), 5.56 (dd, 1H, CH2Vn, 3J(1H,1H) = 15.7 Hz, 3J(1H,1H) = 2.6 Hz), 5.26 (dd, 1H, CH2Vn, 3J(1H,1H) = 8.7 Hz, 3J(1H,1H) = 2.6 Hz), 2.43 (qm, 2H, BH2, 1J(11B,1H) = 99.5) ppm; 11B NMR (160.46 MHz, CD2Cl2): δ = −15.5 (t, 1B, BH2, 1J(11B,1H) = 99.5 Hz) ppm; 11B{1H} NMR (160.46 MHz, CD2Cl2): δ = −15.5 (s, 1B, BH2) ppm; 11B{19F} NMR (160.46 MHz, CD2Cl2): δ = −15.5 (t, 1B, BH2, 1J(11B,1H) = 99.5 Hz) ppm; 13C{1H} NMR (125.76 MHz, CD2Cl2): δ = 136.7 (s, 1C, NCHN), 129.0 (s, 1C, CH), 128.7 (s, 1C, CHVn), 125.5 (qdt, 1C, CF3, 1J(19F,13C) = 283.4 Hz, 2J(19F,13C) = 25.7 Hz, 3J(19F,13C) = 4.9 Hz), 117.4 (s, 1C, CH), 115.3 (tdq, 1C, CF2H, 1J(19F,13C) = 246.5 Hz, 2J(19F,13C) = 27.1 Hz, 3J(19F,13C) = 2.9 Hz), 107.6 (s, 1C, CHVn), 95.7 (d, br, 1C, CF, 1J(19F,13C) = 181.0 Hz) ppm; 13C{11B,1H} NMR (75.48 MHz, CD2Cl2): δ = 136.8 (s, 1C, NCHN), 128.9 (s, 1C, CH), 128.7 (s, 1C, CHVn), 125.5 (qdt, 1C, CF3, 1J(19F,13C) = 283.4 Hz, 2J(19F,13C) = 25.7 Hz, 3J(19F,13C) = 4.9 Hz), 117.5 (s, 1C, CH), 115.4 (tdq, 1C, CF2H, 1J(19F,13C) = 246.5 Hz, 2J(19F,13C) = 27.1 Hz, 3J(19F,13C) = 2.9 Hz), 107.5 (s, 1C, CHVn), 95.7 (dqt, 1C, CF, 1J(19F,13C) = 181.0 Hz, 2J(19F,13C) = 29.0 Hz, 2J(19F,13C) = 23.5 Hz) ppm; 13C{19F} NMR (125.76 MHz, CD2Cl2): δ = 136.7 (d, 1C, NCHN, 1J(13C,1H) = 217.1 Hz), 129.0 (dm, 1C, CH, 1J(13C,1H) = 200.0 Hz), 128.7 (dm, 1C, CHVn, 1J(13C,1H) = 184.1 Hz), 125.5 (s, 1C, CF3), 117.4 (dm, 1C, CH, 1J(13C,1H) = 198.9 Hz), 115.3 (dt, 1C, CF2H, 1J(13C,1H) = 188.8 Hz, 3J(13C,1H) = 3.3 Hz), 107.6 (dd, 1C, CHVn, 1J(13C,1H) = 167.1, 1J(13C,1H) = 159.4 Hz), 95.7 (m, br, 1C, CF) ppm; 19F NMR (376.82 MHz, CD2Cl2): δ = −74.1 (dtt, 3F, CF3, 3J(19F,19F) = 10.2 Hz, 4J(19F,19F) = 8.0 Hz, 4J(19F,1H) = 2.2 Hz), −129.6 (dm, 2F, CF2H, 2J(19F,1H) = 54.5 Hz), −205.8 (m, 1F, CF) ppm; 19F{1H} NMR (376.82 MHz, CD2Cl2): δ = −74.0 (dt, 3F, CF3, 3J(19F,19F) = 10.1 Hz, 4J(19F,19F) = 8.1 Hz), −129.6 (m, 2F, CF2H), −205.8 (m, 1F, CF) ppm; 19F{11B} NMR (470.59 MHz, CD2Cl2): δ = −74.1 (dtt, 3F, CF3, 3J(19F,19F) = 10.2 Hz, 4J(19F,19F) = 8.0 Hz, 4J(19F,1H) = 2.2 Hz, −129.6 (dm, 2F, CF2H, 2J(19F,1H) = 54.5 Hz), −205.8 (m, 1F, CF) ppm.
IR (ATR): 2415 cm−1 ((B–H)).
Raman: 2420 cm−1 ((B–H)).
Elemental analysis: Calculated (%) for C7H9BF6N2, C 37.25, H 3.52, N 10.89; found, C 37.57, H 3.66, N 11.29.
HMRS ((-)-APCI): m/z (isotopic abundance) calculated for deprotonated 3d C8H8BF6N2−: 257.07 (100.0%), 256.07 (24.8%), 258.07 (8.7%), 257.08 (2.1%); found, 257.07 (100.0%), 256.07 (23.3%), 258.07 (8.3%), 257.08 (0.3%).
Hexafluoroisopropyl-benzimidazole adduct 3e was prepared using benzimidazole (590 mg, 5.00 mmol). Yield: 1.41 g (4.44 mmol, 89%, calculated for 1) of a colorless liquid.
1H NMR (500.13 MHz, CD2Cl2): δ = 10.40 (s, br, 1H, NH), 8.39 (s, 1H, NCHN), 7.98 (m. 1H, CH), 7.61 (m, 1H, CH), 7.50 (m, 2H, CH), 6.00 (tdq, 1H, CF2H, 2J(19F,1H) = 54.5 Hz, 3J(19F,1H) = 9.4 Hz, 4J(19F,1H) = 0.6 Hz), 2.76 (m, 2H, BH2) ppm; 1H{11B} NMR (500.13 MHz, CD2Cl2): δ = 10.40 (s, br, 1H, NH), 8.39 (s, 1H, NCHN), 7.98 (m, 1H, CH), 7.61 (m, 1H, CH), 7.50 (m, 2H, CH), 6.00 (td, 1H, CF2H, 2J(19F,1H) = 54.5 Hz, 3J(19F,1H) = 9.4 Hz), 2.76 (dq, 2H, BH2, 3J(19F,1H) = 27.1, 4J(19F,1H) = 2.7) ppm; 1H{19F} NMR (400.30 MHz, CD2Cl2): δ = 10.47 (s, br, 1H, NH), 8.39 (s, 1H, NCHN), 7.98 (m. 1H, CH), 7.61 (m, 1H, CH), 7.50 (m, 2H, CH), 6.00 (m, 1H, CF2H, 2J(19F,1H) = 54.5 Hz, 3J(19F,1H) = 9.4 Hz, 4J(19F,1H) = 0.6 Hz), 2.76 (m, 2H, BH2) ppm; 11B NMR (160.46 MHz, CD2Cl2): δ = −17.1 (t, 1B, BH2, 1J(11B,1H) = 95.7 Hz) ppm; 11B{1H} NMR (160.46 MHz, CD2Cl2): δ = −17.1 (s, 1B, BH2) ppm; 11B{19F} NMR (160.46 MHz, CD2Cl2): δ = −17.1 (t, 1B, BH2, 1J(11B,1H) = 95.7 Hz) ppm; 13C{1H} NMR (125.76 MHz, CD2Cl2): δ = 142.2 (s, 1C, NCHN), 136.7 (s, 1C, CH), 131.1 (s, 1C, CH), 126.2 (s, 1C, CH), 125.7 (s, 1C, CH), 125.6 (qdt, 1C, CF3, 1J(19F,13C) = 283.5 Hz, 2J(19F,13C) = 26.0 Hz, 3J(19F,13C) = 5.0 Hz), 117.6 (m, 1C, CH), 115.4 (tdq, 1C, CF2H, 1J(19F,13C) = 246.5 Hz, 2J(19F,13C) = 26.9 Hz, 3J(19F,13C) = 2.9 Hz), 112.9 (s, 1C, CH), 96.5 (d, br, 1C, CF, 1J(19F,13C) = 180.5 Hz) ppm; 13C{11B,1H} NMR (75.48 MHz, CD2Cl2): δ = 142.2 (s, 1C, NCHN), 136.6 (s, 1C, CH), 131.2 (s, 1C, CH), 126.2 (s, 1C, CH), 125.7 (s, 1C, CH), 125.6 (qdt, 1C, CF3, 1J(19F,13C) = 283.6 Hz, 2J(19F,13C) = 25.9 Hz, 3J(19F,13C) = 4.9 Hz), 117.4 (m, 1C, CH), 115.5 (tdq, 1C, CF2H, 1J(19F,13C) = 246.5 Hz, 2J(19F,13C) = 26.9 Hz, 3J(19F,13C) = 2.9 Hz), 113.0 (s, 1C, CH), 96.6 (dqt, 1C, CF, 1J(19F,13C) = 181.0 Hz, 2J(19F,13C) = 28.9 Hz, 2J(19F,13C) = 23.7 Hz) ppm; 13C{19F} NMR (125.76 MHz, CD2Cl2): δ = 142.2 (dm, 1C, NCHN, 1J(13C,1H) = 214.3 Hz), 136.6 (m, 1C, CH), 131.2 (m, 1C, CH), 126.2 (dm, 1C, CH, 1J(13C,1H) = 162.9 Hz), 125.7 (s, 1C, CH, 1J(13C,1H) = 162.4 Hz), 125.6 (s, 1C, CF3), 117.6 (dm, 1C, CH, 1J(13C,1H) = 168.4 Hz), 115.3 (dt, 1C, CF2H, 1J(13C,1H) = 188.6 Hz, 3J(13C,1H) = 3.3 Hz), 112.9 (dm, 1C, CH, 1J(13C,1H) = 167.3 Hz), 96.5 (m, br, 1C, CF) ppm; 19F NMR (376.82 MHz, CD2Cl2): δ = −74.0 (dtt, 3F, CF3, 3J(19F,19F) = 10.2 Hz, 4J(19F,19F) = 8.0 Hz, 4J(19F,1H) = 2.2 Hz), −129.4 (dm, 2F, CF2H, 2J(19F,1H) = 54.4 Hz), −203.9 (m, 1F, CF) ppm; 19F{1H} NMR (376.82 MHz, CD2Cl2): δ = −74.0 (dt, 3F, CF3, 3J(19F,19F) = 10.2 Hz, 4J(19F,19F) = 8.0 Hz), −129.4 (m, 2F, CF2H), −203.9 (m, 1F, CF) ppm; 19F{11B} NMR (470.59 MHz, CD2Cl2): δ = −74.0 (dtt, 3F, CF3, 3J(19F,19F) = 10.3 Hz, 4J(19F,19F) = 7.9 Hz, 4J(19F,1H) = 2.2 Hz), −129.3 (dm, 2F, CF2H, 2J(19F,1H) = 54.4 Hz), −203.9 (m, 1F, CF) ppm.
IR (ATR): 3434 cm−1 ((N–H)), 2415 cm−1 ((B–H)).
Raman: 2420 cm−1, 2421 cm−1 ((B–H)).
Elemental analysis: Calculated (%) for C10H8BF6N2, C 42.59, H 3.22, N 9.93; found, C 42.73, H 3.39, N 10.28.
HMRS ((-)-APCI): m/z (isotopic abundance) calculated for deprotonated 3e C10H8BF6N2−: 281.07 (100.0%), 280.07 (24.8%), 282.07 (10.8%), 281.08 (2.7%); found, 281.07 (100.0%), 280.07 (24.8%), 282.07 (10.4%).
3.4.4. Hexafluoroisopropylborane-1,3-bis(2,6-diisopropylphenyl)-imidazolin-2-ylidene Adduct (4)
A solution of 1 (0.2 M in toluene, 5.00 mL, 1.00 mmol) was added to IDipp 1,3-bis-(2,6-diisopropylphenyl)-imidazolin-2-ylidene (389 mg, 1.00 mmol). After stirring for 48 h at room temperature, all volatiles were removed under reduced pressure. The solid residue was taken up in Et2O (25 mL) and washed with water (3 × 5 mL). Subsequently, the organic phase was dried over Na2SO4, filtered, and the organic solvent was removed under reduced pressure. After purification by column chromatography (CH2Cl2/hexane 85:15), the product was dried under vacuum. Yield: 321 mg (0.584 mmol, 58%, calculated for 1) of a colorless solid.
1H NMR (500.13 MHz, CD2Cl2): δ = 7.52 (t, 2H, p-CH, C3H6, 3J(1H,1H) = 7.7 Hz), 7.34 (d, 4H, m-CH, C3H6, 3J(1H,1H) = 7.8 Hz), 7.15 (s, 2H, CH, 3J(1H,1H) = 7.8 Hz), 5.95 (td, 1H, CF2H, 2J(19F,1H) = 54.5 Hz, 3J(19F,1H) = 9.5 Hz), 2.59 (sept, 4H, CH(CH3)2, 3J(1H,1H) = 6.9 Hz), 1.33 (dm, 12H, CH(CH)3(CH)3, 3J(1H,1H) = 6.7 Hz), 1.23 (m, 2H, BH2), 1.14 (dm, 12H, CH(CH)3(CH)3, 3J(1H,1H) = 6.7 Hz) ppm; 1H{11B} NMR (500.13 MHz, CD2Cl2): δ = 7.52 (t, 2H, p-CH, C3H6, 3J(1H,1H) = 7.7 Hz), 7.34 (d, 4H, m-CH, C3H6, 3J(1H,1H) = 7.8 Hz), 7.15 (s, 2H, CH, 3J(1H,1H) = 7.8 Hz), 5.35 (td, 1H, CF2H, 2J(19F,1H) = 54.5 Hz, 3J(19F,1H) = 9.5 Hz), 2.59 (sept, 4H, CH(CH3)2, 3J(1H,1H) = 6.9 Hz), 1.33 (dm, 12H, CH(CH)3(CH)3, 3J(1H,1H) = 6.7 Hz), 1.23 (m, 2H, BH2), 1.14 (dm, 12H, CH(CH)3(CH)3, 3J(1H,1H) = 6.7 Hz) ppm; 1H{19F} NMR (400.30 MHz, CD2Cl2): δ = 7.53 (t, 2H, p-CH, C3H6, 3J(1H,1H) = 7.7 Hz), 7.34 (d, 4H, m-CH, C3H6, 3J(1H,1H) = 7.8 Hz), 7.15 (s, 2H, CH, 3J(1H,1H) = 7.8 Hz), 5.35 (d, 1H, CF2H, 3J(19F,1H) = 9.5 Hz), 2.59 (sept, 4H, CH(CH3)2, 3J(1H,1H) = 6.9 Hz), 1.33 (dm, 12H, CH(CH)3(CH)3, 3J(1H,1H) = 6.7 Hz), 1.23 (m, 2H, BH2), 1.14 (dm, 12H, CH(CH)3(CH)3, 3J(1H,1H) = 6.7 Hz) ppm; 11B NMR (160.46 MHz, CD2Cl2): δ = −29.2 (t, 1B, BH2, 1J(11B,1H) = 88.1 Hz) ppm; 11B{1H} NMR (160.46 MHz, CD2Cl2): δ = −29.2 (s, 1B, BH2) ppm; 11B{19F} NMR (160.46 MHz, CD2Cl2): δ = −29.2 (t, 1B, BH2, 1J(11B,1H) = 88.1 Hz) ppm; 13C{1H} NMR (125.76 MHz, CD2Cl2): δ = 170.0 (m. br, 1C, NCN), 146.1 (m, 4C, ArC), 134.7 (m, 2C, ArC), 130.7 (m, 2C, ArCH), 125.6 (qdm, 1C, CF3, 1J(19F,13C) = 283.4 Hz, 2J(19F,13C) = 26.5 Hz), 124.4 (m, 4C, ArCH), 124.0 (m, 2C, NCH), 115.5 (tdq, 1C, CF2H, 1J(19F,13C) = 247.6 Hz, 2J(19F,13C) = 25.3 Hz, 3J(19F,13C) = 2.6 Hz), 96.7 (d, br, 1C, CF, 1J(19F,13C) = 177.6 Hz), 29.0 (s, 4C, iPrCH), 26.1 (s, 4C, iPrCH3), 22.3 (m, 4C, iPrCH3) ppm; 13C{19F} NMR (125.76 MHz, CD2Cl2): δ = 170.0 (m. br, 1C, NCN), 146.1 (m, 4C, ArC), 134.7 (m, 2C, ArC), 130.7 (dm, 2C, ArCH, 1J(13C,1H) = 158.2 Hz), 125.6 (s, 1C, CF3), 124.4 (dm, 4C, ArCH, 1J(13C,1H) = 123.7 Hz), 124.0 (dm, 2C, NCH, 1J(13C,1H) = 198.2 Hz), 115.5 (dm, 1C, CF2H, 1J(13C,1H) = 188.8 Hz), 96.7 (m, br, 1C, CF), 29.0 (dm, 4C, iPrCH, 1J(13C,1H) = 128.9 Hz), 26.1 (qm, 4C, iPrCH3, 1J(13C,1H) = 126.7 Hz), 22.3 (qm, 4C, iPrCH3, 1J(13C,1H) = 125.8 Hz) ppm; 19F NMR (376.82 MHz, CD2Cl2): δ = −74.1 (dt, 3F, CF3, 3J(19F,19F) ≈ 10.3 Hz, 4J(19F,19F) ≈ 7.9 Hz), −129.2 (dm, 2F, CF2H, 2J(19F,1H) = 53.8 Hz), −200.3 (m, 1F, CF) ppm; 19F{1H} NMR (376.82 MHz, CD2Cl2): δ = −74.1 (dt, 3F, CF3, 3J(19F,19F) ≈ 10.3 Hz, 4J(19F,19F) ≈ 7.9 Hz), −129.2 (m, 2F, CF2H), −200.3 (m, 1F, CF) ppm; 19F{11B} NMR (470.59 MHz, CD2Cl2): δ = −74.1 (dt, 3F, CF3, 3J(19F,19F) ≈ 10.3 Hz, 4J(19F,19F) ≈ 7.9 Hz), −129.2 (dm, 2F, CF2H, 2J(19F,1H) = 53.8 Hz), −200.3 (m, 1F, CF) ppm.
IR (ATR): 2430, 2402 cm−1 ((B–H)).
Raman: 2434, 2403 cm−1 ((B–H)).
Elemental analysis: Calculated (%) for C30H39BF6N2, C 65.22, H 7.12, N 5.07; found, C 65.13, H 7.13, N 5.03.
HMRS ((-)-APCI): m/z (isotopic abundance) calculated for deprotonated 3 C30H38BF6N2−: 551.30 (100.0%), 552.31 (32.4%), 550.31 (24.8%), 551.31 (5.6%), 553.31 (3.4%); found, 551.30 (100.0%), 552.31 (34.1%), 550.31 (25.8%), 553.31 (4.4%).
Crystals of 4 suitable for an X-ray diffraction study were obtained via the slow evaporation of a CH2Cl2 solution.
3.4.5. Benzyltriphenylphosphonium Hexafluoroisopropylcyanodihydridoborate ([BnPPh3]5)
Tetrabutylammonium cyanide (805 mg, 4.20 mmol) was added to a solution of 1 (0.2 M in toluene, 15 mL, 3.00 mmol). After stirring for 14 h at room temperature, all volatiles were removed under reduced pressure. Subsequently, a solution of [BnPPh3]Cl (1.17 g, 3.00 mmol) in ethanol (30 mL) and hydrochloric acid (0.5 M, 3 mL) was added to the solid residue. The product was precipitated from the solution by dropwise addition of water (200 mL). After stirring the suspension for 14 h at room temperature the product was filtered off, washed with water (3 × 20 mL) and diethyl ether (2 × 10 mL), and dried under vacuum. Yield: 750 mg (1.38 mmol, 35%, calculated for 1) of a colorless solid.
1H NMR (500.13 MHz, (CD3)2CO): δ = 7.95 (m, 3H, CH), 7.77 (m, 12H, CH), 7.35 (m, 1H, CH), 7.26 (m, 2H, CH), 7.11 (m, 2H, CH), 5.93 (tdm, 1H, CF2H, 2J(19F,1H) = 54.5 Hz, 3J(19F,1H) = 8.6 Hz), 5.10 (d, 2H, CH2, 2J(31P,1H) = 15.0 Hz), 1.14 (qd, 2H, BH2, 1J(11B,1H) = 92.6, 3J(19F,1H) = 29.2) ppm; 1H{11B} NMR (500.13 MHz, (CD3)2CO): δ = 7.95 (m, 3H, CH), 7.77 (m, 12H, CH), 7.35 (m, 1H, CH), 7.26 (m, 2H, CH), 7.11 (m, 2H, CH), 5.93 (tdm, 1H, CF2H, 2J(19F,1H) = 54.5 Hz, 3J(19F,1H) = 8.6 Hz), 5.10 (d, 2H, CH2, 2J(31P,1H) = 15.0 Hz), 1.14 (dm, 2H, BH2, 3J(19F,1H) = 29.2) ppm; 1H{19F} NMR (400.30 MHz, (CD3)2CO): δ = 7.94 (m, 3H, CH), 7.77 (m, 12H, CH), 7.34 (m, 1H, CH), 7.25 (m, 2H, CH), 7.12 (m, 2H, CH), 5.95 (m, 1H, CF2H), 5.10 (d, 2H, CH2, 2J(31P,1H) = 15.0 Hz), 1.20 (qd, 2H, BH2, 1J(11B,1H) = 92.6, 3J(19F,1H) = 29.2) ppm; 11B NMR (160.46 MHz, (CD3)2CO): δ = −33.3 (td, 1B, BH2, 1J(11B,1H) = 92.6 Hz, 2J(19F,11B) = 10.5 Hz) ppm; 11B{1H} NMR (160.46 MHz, (CD3)2CO): δ = −33.3 (d, 1B, BH2, 2J(19F,11B) = 10.5 Hz) ppm; 11B{19F} NMR (160.46 MHz, (CD3)2CO): δ = −33.3 (t, 1B, BH2, 1J(11B,1H) = 92.6 Hz) ppm; 13C{1H} NMR (125.76 MHz, (CD3)2CO): δ = 136.2 (d, 3C, p-Ph-CH, 4J(31P,13C) = 3.0 Hz), 135.1 (d, 12C, o-Ph-CH, 2J(31P,13C) = 9.8 Hz), 131.9 (d, 2C, o-Bn-CH, 3J(31P,13C) = 5.5 Hz), 131.1 (d, 12C, m-Ph-CH, 3J(31P,13C) = 12.6 Hz), 129.8 (d, 2C, m-Bn-CH, 4J(31P,13C) = 3.3 Hz), 129.5 (d, 1C, p-Bn-CH, 5J(31P,13C) = 3.8 Hz), 128.4 (d, 1C, i-Bn-CH, 2J(31P,13C) = 8.5 Hz), 126.8 (qdm, 1C, CF3, 1J(19F,13C) = 282.6 Hz, 2J(19F,13C) = 26.6 Hz), 118.7 (d, 3C, i-Ph-CH, 1J(31P,13C) = 85.8 Hz), 116.7 (tdm, 1C, CF2H, 1J(19F,13C) = 247.6 Hz, 2J(19F,13C) = 27.2 Hz), 97.0 (m, br, 1C, CF), 30.4 (d, 1C, PCH2, 1J(31P,13C) = 29.3 Hz) ppm (CN not detected); 13C{11B,1H} NMR (75.48 MHz, (CD3)2CO): δ = 136.0 (d, 3C, p-Ph-CH, 4J(31P,13C) = 3.0 Hz), 135.0 (d, 12C, o-Ph-CH, 2J(31P,13C) = 9.8 Hz), 131.8 (d, 2C, o-Bn-CH, 3J(31P,13C) = 5.5 Hz), 131.0 (d, 12C, m-Ph-CH, 3J(31P,13C) = 12.6 Hz), 129.7 (d, 2C, m-Bn-CH, 4J(31P,13C) = 3.3 Hz), 129.4 (d, 1C, p-Bn-CH, 5J(31P,13C) = 3.8 Hz), 128.3 (d, 1C, i-Bn-CH, 2J(31P,13C) = 8.5 Hz), 124.7 (qdm, 1C, CF3, 1J(19F,13C) = 282.6 Hz, 2J(19F,13C) = 26.6 Hz), 118.5 (d, 3C, i-Ph-CH, 1J(31P,13C) = 85.8 Hz), 116.5 (tdm, 1C, CF2H, 1J(19F,13C) = 247.6 Hz, 2J(19F,13C) = 27.2 Hz), 96.9 (dqt, 1C, CF, 1J(19F,13C) = 180.5 Hz, 2J(19F,13C) = 29.1 Hz, 2J(19F,13C) = 21.6 Hz), 30.3 (d, 1C, PCH2, 1J(31P,13C) = 48.1 Hz) ppm (CN not detected); 13C{19F} NMR (125.76 MHz, (CD3)2CO): δ = 136.2 (m, 3C, p-Ph-CH), 135.1 (m, 12C, o-Ph-CH), 131.9 (m, 2C, o-Bn-CH), 131.1 (m, 12C, m-Ph-CH), 129.8 (m, 2C, m-Bn-CH), 129.5 (m, 1C, p-Bn-CH), 128.4 (m, 1C, i-Bn-CH), 126.8 (m, 1C, CF3), 118.7 (m, 3C, i-Ph-CH), 116.7 (dt, 1C, CF2H, 1J(13C,1H) = 188.8 Hz, 3J(13C,1H) = 4.2 Hz), 97.0 (m, br, 1C, CF), 30.4 (m, 1C, PCH2) ppm (CN not detected); 19F NMR (376.82 MHz, (CD3)2CO): δ = −73.4 (m, 3F, CF3), −129.5 (dm, 2F, CF2H, 2J(19F,1H) = 54.2 Hz), −200.0 (m, 1F, CF) ppm; 19F{1H} NMR (376.82 MHz, (CD3)2CO): δ = −73.4 (m, 3F, CF3), −129.5 (m, 2F, CF2H), −200.0 (m, 1F, CF) ppm; 19F{11B} NMR (470.59 MHz, (CD3)2CO): δ = −73.5 (m, 3F, CF3), −129.7 (dm, 2F, CF2H, 2J(19F,1H) = 54.2 Hz), −200.3 (m, 1F, CF) ppm; 31P{1H} NMR (202.46 MHz, (CD3)2CO): δ = 23.0 (s, 1P, BnPh3P) ppm.
IR (ATR): 2390 cm−1 ((B–H)), 2183 cm−1 ((C≡N)).
Raman: 2395 cm−1 ((B–H)), 2187 cm−1 ((C≡N)).
Elemental analysis: Calculated (%) for C29H25BF6NP, C 64.11, H 4.64, N 2.58; found, C 64.30, H 4.84, N 2.54.
HMRS ((-)-ESI): m/z (isotopic abundance) calculated for C4H3BF6N−: 190.03 (100.0%), 189.03 (24.8%), 191.03 (4.3%); found, 190.03 (100%), 189.03 (24.7%), 191.03 (3.8%).
Crystals of [BnPPh3]5 suitable for an X-ray diffraction study were obtained from a saturated ethanol solution.
3.4.6. Tetramethylammonium Hexafluoroisopropylfluorodihydridoborate ([Me4N]6) and Benzyltriphenylphosphonium Hexafluoroisopropylfluorodihydridoborate ([BnPPh3]6)
Tetrametylammonium fluoride (931 mg, 10.0 mmol) was added to a solution of 1 (0.2 M in toluene, 50 mL, 10.0 mmol). After stirring for 72 h at room temperature, the suspension was filtered. The solid product was washed with toluene (2 × 50 mL) and dried under vacuum. Yield: 2.36 g (9.17 mmol, 92%, calculated for 1) of a colorless solid.
1H NMR (400.30 MHz, (CD3)2CO): δ = 5.94 (tdm, 1H, CF2H, 2J(19F,1H) = 54.6 Hz, 3J(19F,1H) = 8.8 Hz), 3.41 (s, 12H, CH3), 2.75 (m, br, 2H, BH2) ppm; 1H{11B} NMR (400.30 MHz, (CD3)2CO): δ = 5.94 (tdm, 1H, CF2H, 2J(19F,1H) = 54.6 Hz, 3J(19F,1H) = 8.8 Hz), 3.41 (s, 12H, CH3), 2.75 (m, 2H, BH2) ppm; 1H{19F} NMR (400.30 MHz, (CD3)2CO): δ = 5.95 (m, 1H, CF2H), 3.41 (s, 12H, CH3), 2.75 (m, br, 2H, BH2) ppm; 11B NMR (128.43 MHz, (CD3)2CO): δ = −3.3 (td, 1B, BH2, 1J(11B,1H) = 100.5 Hz, 1J(19F,11B) = 79.6 Hz) ppm; 11B{1H} NMR (128.43 MHz, (CD3)2CO): δ = −3.3 (d, 1B, BH2, 1J(19F,11B) = 79.6 Hz) ppm; 19F NMR (376.82 MHz, (CD3)2CO): δ = −72.3 (m, 3F, CF3), −131.2 (dm, 2F, CF2H, 2J(19F,1H) = 53.1 Hz), −209.4 (m, 1F, CF), −245.8 (m, 1F, BF) ppm; 19F{1H} NMR (376.82 MHz, (CD3)2CO): δ = −72.3 (m, 3F, CF3), −131.2 (m, 2F, CF2H), −209.4 (m, 1F, CF), −245.8 (m, 1F, BF) ppm.
[Me4N]6 (386 mg, 1.50 mmol) was dissolved in ethanol (20 mL) and [BnPPh3]Cl (583 mg, 1.50 mmol) was added. The product was precipitated from the solution by dropwise addition of water (150 mL). The product was filtered off, washed with water (3 × 20 mL), and dried under vacuum. Yield: 707 mg (1.32 mmol, 88%, calculated for [Me4N]6) of a colorless solid.
1H NMR (500.13 MHz, (CD3)2CO): δ = 7.94 (m, 3H, CH), 7.77 (m, 12H, CH), 7.34 (m, 1H, CH), 7.25 (m, 2H, CH), 7.11 (m, 2H, CH), 5.94 (tdm, 1H, CF2H, 2J(19F,1H) = 54.7 Hz, 3J(19F,1H) = 8.6 Hz), 5.07 (d, 2H, CH2, 2J(31P,1H) = 14.0 Hz), 2.85 (qm, 2H, BH2, 1J(11B,1H) = 100.5) ppm; 1H{11B} NMR (500.13 MHz, (CD3)2CO): δ = 7.94 (m, 3H, CH), 7.77 (m, 12H, CH), 7.34 (m, 1H, CH), 7.25 (m, 2H, CH), 7.11 (m, 2H, CH), 5.94 (tdm, 1H, CF2H, 2J(19F,1H) = 54.7 Hz, 3J(19F,1H) = 8.6 Hz), 5.07 (d, 2H, CH2, 2J(31P,1H) = 14.0 Hz), 2.85 (m, 2H, BH2) ppm; 1H{19F} NMR (400.30 MHz, (CD3)2CO): δ = 7.94 (m, 3H, CH), 7.77 (m, 12H, CH), 7.34 (m, 1H, CH), 7.25 (m, 2H, CH), 7.11 (m, 2H, CH), 5.94 (m, 1H, CF2H), 5.07 (d, 2H, CH2, 2J(31P,1H) = 14.0 Hz), 2.85 (qm, 2H, BH2, 1J(11B,1H) = 100.5) ppm; 11B NMR (160.46 MHz, (CD3)2CO): δ = −3.2 (td, 1B, BH2, 1J(11B,1H) = 100.5 Hz, 1J(19F,11B) = 79.6 Hz) ppm; 11B{1H} NMR (160.46 MHz, (CD3)2CO): δ = −3.2 (d, 1B, BH2, 2J(19F,11B) = 79.6 Hz) ppm; 11B{19F} NMR (160.46 MHz, (CD3)2CO): δ = −3.2 (t, 1B, BH2, 1J(11B,1H) = 100.5 Hz) ppm; 13C{1H} NMR (125.76 MHz, (CD3)2CO): δ = 136.1 (d, 3C, p-Ph-CH, 4J(31P,13C) = 3.0 Hz), 135.1 (d, 12C, o-Ph-CH, 2J(31P,13C) = 9.8 Hz), 132.0 (d, 2C, o-Bn-CH, 3J(31P,13C) = 5.6 Hz), 131.0 (d, 12C, m-Ph-CH, 3J(31P,13C) = 12.6 Hz), 129.8 (d, 2C, m-Bn-CH, 4J(31P,13C) = 3.3 Hz), 129.5 (d, 1C, p-Bn-CH, 5J(31P,13C) = 3.8 Hz), 128.5 (d, 1C, i-Bn-CH, 2J(31P,13C) = 8.5 Hz), 127.2 (qdm, 1C, CF3, 1J(19F,13C) = 283.2 Hz, 2J(19F,13C) = 26.7 Hz), 118.7 (d, 3C, i-Ph-CH, 1J(31P,13C) = 86.1 Hz), 117.0 (tdm, 1C, CF2H, 1J(19F,13C) = 248.5 Hz, 2J(19F,13C) = 26.7 Hz), 98.1 (m, br, 1C, CF), 30.2 (d, 1C, PCH2, 1J(31P,13C) = 48.1 Hz) ppm; 13C{11B,1H} NMR (75.48 MHz, (CD3)2CO): δ = 136.0 (d, 3C, p-Ph-CH, 4J(31P,13C) = 3.0 Hz), 135.0 (d, 12C, o-Ph-CH, 2J(31P,13C) = 9.8 Hz), 131.9 (d, 2C, o-Bn-CH, 3J(31P,13C) = 5.6 Hz), 131.0 (d, 12C, m-Ph-CH, 3J(31P,13C) = 12.6 Hz), 129.7 (d, 2C, m-Bn-CH, 4J(31P,13C) = 3.3 Hz), 129.4 (d, 1C, p-Bn-CH, 5J(31P,13C) = 3.8 Hz), 128.4 (d, 1C, i-Bn-CH, 2J(31P,13C) = 8.5 Hz), 127.2 (qdm, 1C, CF3, 1J(19F,13C) = 283.2 Hz, 2J(19F,13C) = 26.7 Hz), 118.6 (d, 3C, i-Ph-CH, 1J(31P,13C) = 86.1 Hz), 116.9 (tdm, 1C, CF2H, 1J(19F,13C) = 248.5 Hz, 2J(19F,13C) = 26.7 Hz), 98.1 (dm, 1C, CF, 1J(19F,13C) = 179.2 Hz), 30.1 (d, 1C, PCH2, 1J(31P,13C) = 48.1 Hz) ppm; 13C{19F} NMR (125.76 MHz, (CD3)2CO): δ = 136.1 (m, 3C, p-Ph-CH), 135.1 (m, 12C, o-Ph-CH), 132.0 (m, 2C, o-Bn-CH), 131.0 (m, 12C, m-Ph-CH), 129.8 (m, 2C, m-Bn-CH), 129.5 (m, 1C, p-Bn-CH), 128.5 (m, 1C, i-Bn-CH), 127.2 (m, 1C, CF3), 118.7 (m, 3C, i-Ph-CH), 117.0 (dt, 1C, CF2H, 1J(13C,1H) = 188.8 Hz, 3J(13C,1H) = 4.2 Hz), 98.1 (m, br, 1C, CF), 30.2 (m, 1C, PCH2) ppm; 19F NMR (376.82 MHz, (CD3)2CO): δ = −72.0 (m, 3F, CF3), −131.1 (dm, 2F, CF2H, 2J(19F,1H) = 53.8 Hz), −209.0 (m, 1F, CF), −245.3 (m, 1F, BF) ppm; 19F{1H} NMR (376.82 MHz, (CD3)2CO): δ = −72.0 (m, 3F, CF3), −131.1 (m, 2F, CF2H), −209.0 (m, 1F, CF), −245.3 (m, 1F, BF) ppm; 19F{11B} NMR (470.59 MHz, (CD3)2CO): δ = −72.0 (m, 3F, CF3), −131.1 (dm, 2F, CF2H, 2J(19F,1H) = 54.2 Hz), −208.9 (m, 1F, CF), −245.3 (tm, 1F, BF, 2J(19F,1H) = 43.5 Hz) ppm; 31P{1H} NMR (202.46 MHz, (CD3)2CO): δ = 23.0 (s, 1P, BnPh3P) ppm.
IR (ATR): 2311, 2241 cm−1 ((B–H)).
Raman: 2330, 2245 cm−1 ((B–H)).
Elemental analysis: Calculated (%) for C28H25BF7P, C 62.71, H 4.70; found, C 62.72, H 4.95.
HMRS ((-)-ESI): m/z (isotopic abundance) calculated for C3H3BF7−: 183.02 (100.0%), 182.03 (24.8%), 184.02 (3.2%); found, 183.02 (100.0%), 182.03 (24.8%), 184.02 (3.3%).
Crystals of [BnPPh3]6 suitable for an X-ray diffraction study were obtained from a saturated ethanol solution.
3.4.7. Benzyltriphenylphosphonium Hexafluoroisopropyltrifluoroborate ([BnPPh3]7)
[Me4N]6 (1.03 g, 4.00 mmol) was added to a solution of KHF2 (3.12 g, 40.0 mmol) in trifluoroacetic acid (5 mL) under gas evolution. After stirring for 30 min at room temperature, all volatiles were removed under reduced pressure. Subsequently, a solution of [BnPPh3]Cl (1.56 g, 4.00 mmol) in ethanol (20 mL) was added to the solid residue. The product was precipitated from the solution by dropwise addition of dilute hydrochloric acid (0.083 M, 300 mL). The product was filtered off, washed with diethyl ether (4 × 5 mL), and dried under vacuum. The solid product was crystallized by slow evaporation of an acetone solution. Yield: 1.86 g (3.25 mmol, 81%, calculated for [Me4N]6) of a colorless solid.
1H NMR (500.13 MHz, (CD3)2CO): δ = 7.94 (m, 3H, CH), 7.77 (m, 12H, CH), 7.34 (m, 1H, CH), 7.25 (m, 2H, CH), 7.11 (m, 2H, CH), 5.94 (tdm, 1H, CF2H, 2J(19F,1H) = 53.9 Hz, 3J(19F,1H) = 8.6 Hz), 5.08 (d, 2H, CH2, 2J(31P,1H) = 14.9 Hz) ppm; 1H{11B} NMR (500.13 MHz, (CD3)2CO): δ = 7.95 (m, 3H, CH), 7.77 (m, 12H, CH), 7.35 (m, 1H, CH), 7.26 (m, 2H, CH), 7.11 (m, 2H, CH), 5.94 (tdm, 1H, CF2H, 2J(19F,1H) = 53.9 Hz, 3J(19F,1H) = 8.6 Hz), 5.08 (d, 2H, CH2, 2J(31P,1H) = 14.9 Hz) ppm; 1H{19F} NMR (400.30 MHz, (CD3)2CO): δ = 7.95 (m, 3H, CH), 7.77 (m, 12H, CH), 7.35 (m, 1H, CH), 7.26 (m, 2H, CH), 7.11 (m, 2H, CH), 6.00 (m, 1H, CF2H), 5.07 (d, 2H, CH2, 2J(31P,1H) = 14.9 Hz) ppm; 11B NMR (160.46 MHz, (CD3)2CO): δ = 0.6 (qd, 1B, BF3, 1J(19F,11B) = 43.6 Hz, 2J(19F,11B) = 12.2 Hz) ppm; 11B{1H} NMR (160.46 MHz, (CD3)2CO): δ = 0.6 (qd, 1B, BF3, 1J(19F,11B) = 43.6 Hz, 2J(19F,11B) = 12.2 Hz) ppm; 11B{19F} NMR (160.46 MHz, (CD3)2CO): δ = 0.6 (s, 1B, BF3) ppm; 13C{1H} NMR (125.76 MHz, (CD3)2CO): δ = 136.2 (d, 3C, p-Ph-CH, 4J(31P,13C) = 3.1 Hz), 135.1 (d, 12C, o-Ph-CH, 2J(31P,13C) = 9.8 Hz), 132.0 (d, 2C, o-Bn-CH, 3J(31P,13C) = 5.5 Hz), 131.1 (d, 12C, m-Ph-CH, 3J(31P,13C) = 12.6 Hz), 129.9 (d, 2C, m-Bn-CH, 4J(31P,13C) = 3.3 Hz), 129.5 (d, 1C, p-Bn-CH, 5J(31P,13C) = 3.8 Hz), 128.5 (d, 1C, i-Bn-CH, 2J(31P,13C) = 8.5 Hz), 125.9 (qdm, 1C, CF3, 1J(19F,13C) = 283.0 Hz, 2J(19F,13C) = 25.3 Hz), 118.7 (d, 3C, i-Ph-CH, 1J(31P,13C) = 86.0 Hz), 115.9 (tdm, 1C, CF2H, 1J(19F,13C) = 247.0 Hz, 2J(19F,13C) = 26.3 Hz), 91.6 (m, br, 1C, CF), 30.3 (d, 1C, PCH2, 1J(31P,13C) = 48.1 Hz) ppm; 13C{11B,1H} NMR (75.48 MHz, (CD3)2CO): δ = 136.1 (m, br, 3C, p-Ph-CH), 135.0 (d, 12C, o-Ph-CH, 2J(31P,13C) = 9.8 Hz), 131.9 (d, 2C, o-Bn-CH, 3J(31P,13C) = 5.6 Hz), 131.0 (d, 12C, m-Ph-CH, 3J(31P,13C) = 12.6 Hz), 129.6 (d, 2C, m-Bn-CH, 4J(31P,13C) = 3.3 Hz), 129.4 (d, 1C, p-Bn-CH, 5J(31P,13C) = 3.8 Hz), 128.4 (d, 1C, i-Bn-CH, 2J(31P,13C) = 8.5 Hz), 125.8 (qdm, 1C, CF3, 1J(19F,13C) = 283.2 Hz, 2J(19F,13C) = 26.7 Hz), 118.5 (d, 3C, i-Ph-CH, 1J(31P,13C) = 86.1 Hz), 115.8 (tdm, 1C, CF2H, 1J(19F,13C) = 248.5 Hz, 2J(19F,13C) = 26.7 Hz), 91.5 (dm, 1C, CF, 1J(19F,13C) = 178.1 Hz), 30.1 (d, 1C, PCH2, 1J(31P,13C) = 48.1 Hz) ppm; 13C{19F} NMR (125.76 MHz, (CD3)2CO): δ = 136.2 (m, 3C, p-Ph-CH), 135.1 (m, 12C, o-Ph-CH), 132.0 (m, 2C, o-Bn-CH), 131.1 (m, 12C, m-Ph-CH), 129.9 (m, 2C, m-Bn-CH), 129.5 (m, 1C, p-Bn-CH), 128.5 (m, 1C, i-Bn-CH), 125.9 (m, 1C, CF3), 118.7 (m, 3C, i-Ph-CH), 115.9 (d, 1C, CF2H, 1J(13C,1H) = 189.9 Hz), 91.6 (qm, 1C, CF, 1J(13C,11B) = 67.7 Hz), 30.3 (m, 1C, PCH2) ppm; 19F NMR (376.82 MHz, (CD3)2CO): δ = −72.4 (m, 3F, CF3), −131.9 (dm, 2F, CF2H, 2J(19F,1H) = 53.0 Hz), −148.7 (qm, 1F, BF3, qm, 3F, BF3, 1J(19F,11B) = 43.6 Hz), −217.1 (m, 1F, CF) ppm; 19F{1H} NMR (376.82 MHz, (CD3)2CO): δ = −72.4 (m, 3F, CF3), −131.9 (m, 2F, CF2H), −148.7 (qm, 3F, BF3, 1J(19F,11B) = 43.6 Hz), −217.1 (m, 1F, CF) ppm; 19F{11B} NMR (470.59 MHz, (CD3)2CO): δ = −72.1 (m, 3F, CF3), −131.8 (dm, 2F, CF2H, 2J(19F,1H) = 53.0 Hz), −149.3 (qm, 1F, BF3, m, 3F, BF3), −217.4 (m, 1F, CF) ppm; 31P{1H} NMR (202.46 MHz, (CD3)2CO): δ = 22.9 (s, 1P, BnPh3P) ppm.
Elemental analysis: Calculated (%) for C28H23BF9P, C 58.77, H 4.05; found, C 58.71, H 4.27.
HMRS ((-)-ESI): m/z (isotopic abundance) calculated for C3HBF9−: 219.00 (100.0%), 218.01 (24.8%), 220.01 (3.2%); found, 219.00 (100.0%), 218.01 (26.0%), 220.01 (3.0%).
Crystals of [BnPPh3]7 suitable for an X-ray diffraction study were obtained from a saturated ethanol solution.
4. Conclusions
A large-scale synthesis of hexafluoroisopropylborane (CF3)(CF2H)CFBH2·SMe2 (1) was developed, which is the prerequisite for the follow-up synthesis of selected borane adducts as well as borates with the (CF3)(CF2H)CFB moiety presented in this study. All borane adducts and borate salts were fully characterized by NMR and vibrational spectroscopy, elemental analysis, single-crystal X-ray diffraction, DSC, and cyclic voltammetry. Whereas the borane imidazole adducts 3a–e are of particular interest for the synthesis of novel anionic NHCs, borane NHC adduct 4 might be a promising additive in electrochemical devices for overvoltage protection. Furthermore, the [BnPPh3]+ borates reveal interesting properties and [BnPPh3]7 particularly shows an astonishingly high electrochemical stability. These results highlight the potential of borate anions with a hexafluoroisopropyl substituent for applications in materials science in general and in electrochemical devices especially. Such applications seem promising due to the easy and scalable synthesis of 1, which relies on the hydroboration of an industrial product, whereas the preparation of related fluoroalkyl boron compounds requires potentially explosive lithium organyls, Grignard reagents, or reactions that require special techniques (e.g., handling of chlorine monofluoride) [1,2,3,4].
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/inorganics11120471/s1. Spectral data, information on quantum chemical calculations, single-crystal X-ray diffraction, and electrochemical measurements (Figures S1–S8, Tables S1 and S2). References [59,60,61,62,63,64,65,66,67,68,69,70,71] are cited in the Supplementary Materials.
Author Contributions
Conceptualization, synthesis, analysis, quantum chemical calculations, L.Z.; writing—original draft preparation, L.Z.; writing—review and editing, M.F..; supervision, M.F. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Julius-Maximilians-Universität Würzburg (Germany).
Data Availability Statement
CCDC 2304957 (2), 2304958 ((S)-3b), 2304959 ((R)-3b), 2304960 (4), 2304961 ([BnPPh3]5), 2304962 ([BnPPh3]6), and 2304963 ([BnPPh3]7) contain the supplementary crystallographic data for this paper. These data are provided free of charge by The Cambridge Crystallographic Data Centre.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pawelke, G.; Bürger, H. Trifluoromethyl-substituted Aminoboranes and Amine Boranes Revealing Alkene and Alkane Chemistry. Appl. Organomet. Chem. 1996, 10, 147–174. [Google Scholar] [CrossRef]
- Pawelke, G.; Bürger, H. Perfluoroalkyl Borates: Congeners of Perfluoroalkanes. Coord. Chem. Rev. 2001, 215, 243–266. [Google Scholar] [CrossRef]
- Finze, M.; Bernhardt, E.; Willner, H. Trifluoromethylboranes and -Borates: New Synthetic Strategies and Applications. Angew. Chem. Int. Ed. 2007, 46, 9180–9196. [Google Scholar] [CrossRef] [PubMed]
- Adonin, N.Y.; Bardin, V.V. Polyfluorinated organic compounds of boron. Russ. Chem. Rev. 2010, 79, 757–785. [Google Scholar] [CrossRef]
- Krossing, I.; Raabe, I. Noncoordinating anions—Fact or fiction? A survey of likely candidates. Angew. Chem. Int. Ed. Engl. 2004, 43, 2066–2090. [Google Scholar] [CrossRef]
- Riddlestone, I.M.; Kraft, A.; Schaefer, J.; Krossing, I. Taming the Cationic Beast: Novel Developments in the Synthesis and Application of Weakly Coordinating Anions. Angew. Chem. Int. Ed. 2018, 57, 13982–14024. [Google Scholar] [CrossRef]
- Krossing, I.; Raabe, I. Relative Stabilities of Weakly Coordinating Anions: A Computational Study. Chem. Eur. J. 2004, 10, 5017–5030. [Google Scholar] [CrossRef]
- Engesser, T.A.; Lichtenthaler, M.R.; Schleep, M.; Krossing, I. Reactive p-block cations stabilized by weakly coordinating anions. Chem. Soc. Rev. 2016, 45, 789–899. [Google Scholar] [CrossRef]
- Bernhardt, E.; Henkel, G.; Willner, H.; Pawelke, G.; Bürger, H. Synthesis and properties of the tetrakis(trifluoromethyl)borate anion, [B(CF3)4]–: Structure determination of Cs[B(CF3)4] by single-crystal X-ray diffraction. Chem. Eur. J. 2001, 7, 4696–4705. [Google Scholar] [CrossRef]
- Bernhardt, E.; Finze, M.; Willner, H. Mechanistic Study on the Fluorination of K[B(CN)4] with ClF Enabling the High Yield and Large Scale Synthesis of K[B(CF3)4] and K[(CF3)3BCN]. Inorg. Chem. 2011, 50, 10268–10273. [Google Scholar] [CrossRef]
- Wilson, W.W.; Vij, A.; Vij, V.; Bernhardt, E.; Christe, K.O. Polynitrogen Chemistry: Preparation and Characterization of (N5)2SnF6, N5SnF5, and N5B(CF3)4. Chem. Eur. J. 2003, 9, 2840–2844. [Google Scholar] [CrossRef] [PubMed]
- Bernhardt, E.; Finze, M.; Willner, H. A new synthesis for nitrosyl salts with weakly coordinating anions exemplified by NO[B(CF3)4]. Z. Anorg. Allg. Chem. 2006, 632, 248–250. [Google Scholar] [CrossRef]
- Finze, M.; Bernhardt, E.; Berkei, M.; Willner, H.; Hung, J.; Waymouth, R.M. [H(OEt2)2]+ and [Ph3C]+ Salts of the Borate Anions [B(CF3)4]–, [(CF3)3BCN]–, and [B(CN)4]–. Organometallics 2005, 24, 5103–5109. [Google Scholar] [CrossRef]
- Bernhardt, E.; Finze, M.; Willner, H.; Lehmann, C.W.; Aubke, F. Salts of the Cobalt(I) Complexes [Co(CO)5]+ and[Co(CO)2(NO)2]+ and the Lewis Acid–Base Adduct [Co2(CO)7CO–B(CF3)3]. Chem. Eur. J. 2006, 12, 8276–8283. [Google Scholar] [CrossRef] [PubMed]
- Brauer, D.J.; Bürger, H.; Chebude, Y.; Pawelke, G. Halogenation of (CF3)3B·NH3: N,N-Dihaloamino- and Halo-tris(trifluoromethyl)borates [(CF3)3B-NX2]– and [(CF3)3B-X]–, X = F, Cl, Br. Inorg. Chem. 1999, 38, 3972–3977. [Google Scholar] [CrossRef]
- Bernhardt, E.; Finze, M.; Willner, H.; Lehmann, C.W.; Aubke, F. [Co(CO)5][(CF3)3BF]: A stable salt of a homoleptic trigonal-bipyramidal metal-carbonyl cation. Angew. Chem. Int. Ed. Engl. 2003, 42, 2077–2079. [Google Scholar] [CrossRef]
- Geier, J.; Willner, H.; Lehmann, C.W.; Aubke, F. Formation of Hexacarbonylmanganese(I) Salts, [Mn(CO)6]+X−, in Anhydrous HF. Inorg. Chem. 2007, 46, 7210–7214. [Google Scholar] [CrossRef]
- Finze, M.; Bernhardt, E.; Zähres, M.; Willner, H. Rearrangement reactions of the transient Lewis acids (CF3)3B and (CF3)3BCF2: An experimental and theoretical study. Inorg. Chem. 2004, 43, 490–505. [Google Scholar] [CrossRef]
- Terheiden, A.; Bernhardt, E.; Willner, H.; Aubke, F. Carbonyltris(trifluormethyl)borane, (CF3)3BCO, An Unusual Boron Carbonyl. Angew. Chem. Int. Ed. 2002, 41, 799–801. [Google Scholar] [CrossRef]
- Finze, M.; Bernhardt, E.; Terheiden, A.; Berkei, M.; Willner, H.; Christen, D.; Oberhammer, H.; Aubke, F. Tris(trifluoromethyl)borane carbonyl, (CF3)3BCO—Synthesis, physical, chemical and spectroscopic properties, gas phase, and solid state structure. J. Am. Chem. Soc. 2002, 124, 15385–15398. [Google Scholar] [CrossRef]
- Finze, M.; Bernhardt, E.; Willner, H.; Lehmann, C.W. [(CF3)3BCP]– and [(CF3)3BCAs]–: Thermally stable phosphaethynyl and arsaethynyl complexes. Angew. Chem. Int. Ed. 2004, 43, 4160–4163. [Google Scholar] [CrossRef] [PubMed]
- Finze, M.; Bernhardt, E.; Willner, H.; Lehmann, C.W. Cyano- and Isocyanotris(trifluoromethyl)borates: Syntheses, Spectroscopic Properties, and Solid State Structures of K[(CF3)3BCN] and K[(CF3)3BNC]. J. Am. Chem. Soc. 2005, 127, 10712–10722. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.-B.; Matsumoto, H.; Tatsumi, K. Low-Melting, Low-Viscous, Hydrophobic Ionic Liquids: 1-Alkyl(AlkylEther)-3-methylimidazolium Perfluoroalkyltrifluoroborate. Chem. Eur. J. 2004, 10, 6581–6591. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.-B.; Matsumoto, H.; Tatsumi, K. A New Class of Hydrophobic Ionic Liquids: Trialkyl(2-methoxyethyl)ammonium Perfluoroethyltrifluoroborate. Chem. Lett. 2004, 33, 886–887. [Google Scholar] [CrossRef]
- Zhou, Z.-B.; Matsumoto, H.; Tatsumi, K. Low-melting, Low-viscous, Hydrophobic Ionic Liquids: N-Alkyl(alkyl ether)-N-methylpyrrolidinium Perfluoroethyltrifluoroborate. Chem. Lett. 2004, 33, 1636–1637. [Google Scholar] [CrossRef]
- Zhou, Z.-B.; Matsumoto, H.; Tatsumi, K. Low-Viscous, Low-Melting, Hydrophobic Ionic Liquids: 1-Alkyl-3-methylimidazolium Trifluoromethyltrifluoroborate. Chem. Lett. 2004, 33, 680–681. [Google Scholar] [CrossRef]
- Zhou, Z.-B.; Takeda, M.; Ue, M. New hydrophobic ionic liquids based on perfluoroalkyltrifluoroborate anions. J. Fluorine Chem. 2004, 125, 461–476. [Google Scholar] [CrossRef]
- Zhou, Z.B.; Matsumoto, H.; Tatsumi, K. Low-Melting, Low-Viscous, Hydrophobic Ionic Liquids: Aliphatic Quaternary Ammonium Salts with Perfluoroalkyltrifluoroborates. Chem. Eur. J. 2005, 11, 752–766. [Google Scholar] [CrossRef]
- Zhou, Z.-B.; Matsumoto, H.; Tatsumi, K. Cyclic Quaternary Ammonium Ionic Liquids with Perfluoroalkyltrifluoroborates: Synthesis, Characterization, and Properties. Chem. Eur. J. 2006, 12, 2196–2212. [Google Scholar] [CrossRef]
- Landmann, J.; Sprenger, J.A.P.; Hennig, P.T.; Bertermann, R.; Grüne, M.; Würthner, F.; Ignat’ev, N.V.; Finze, M. Perfluoroalkyltricyanoborate and Perfluoroalkylcyanofluoroborate Anions: Building Blocks for Low-Viscosity Ionic Liquids. Chem. Eur. J. 2018, 24, 608–623. [Google Scholar] [CrossRef]
- Ignat’ev, N.V.; Finze, M. Cyanoborates. Eur. J. Inorg. Chem. 2019, 2019, 3539–3560. [Google Scholar] [CrossRef]
- Ribbeck, T.; Zottnick, S.H.; Kerpen, C.; Landmann, J.; Ignat’ev, N.V.; Müller-Buschbaum, K.; Finze, M. Anhydrous, Homoleptic Lanthanide Frameworks with the Pentafluoroethyltricyanoborate Anion. Inorg. Chem. 2017, 56, 2278–2286. [Google Scholar] [CrossRef] [PubMed]
- Hennig, P.T.; Sprenger, J.A.P.; Schneider, L.N.; Ignat’ev, N.V.; Finze, M. The pentafluoroethyltrihydridoborate anion: From shock sensitive salts to stable room temperature ionic liquids. Chem. Commun. 2019, 55, 6110–6113. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Wang, S.; Dewhurst, R.D.; Ignat’ev, N.V.; Finze, M.; Braunschweig, H. Boron: Its Role in Energy-Related Processes and Applications. Angew. Chem. Int. Ed. 2020, 59, 8800–8816. [Google Scholar] [CrossRef] [PubMed]
- Rüther, T.; Bhatt, A.I.; Best, A.S.; Harris, K.R.; Hollenkamp, A.F. Electrolytes for Lithium (Sodium) Batteries Based on Ionic Liquids: Highlighting the Key Role Played by the Anion. Batter. Supercaps 2020, 3, 793–827. [Google Scholar] [CrossRef]
- Zhou, Z.-B.; Takeda, M.; Ue, M. Novel electrolyte salts based on perfluoroalkyltrifluoroborate anions 1. Synthesis and characterization. J. Fluorine Chem. 2003, 123, 127–131. [Google Scholar] [CrossRef]
- Zhou, Z.-B.; Takeda, M.; Fujii, T.; Ue, M. Li[C2F5BF3] as an electrolyte salt for 4 V class lithium-ion cells. J. Electrochem. Soc. 2005, 152, A351–A356. [Google Scholar] [CrossRef]
- Ue, M.; Fujii, T.; Zhou, Z.-B.; Takeda, M.; Kinoshita, S. Electrochemical properties of Li[CnF2n+1BF3] as electrolyte salts for lithium-ion cells. Solid State Ionics 2006, 177, 323–331. [Google Scholar] [CrossRef]
- Schmidt, M.; Kühner, A.; Willner, H.; Bernhardt, E. Tetrakisfluoroalkylborat-Salze und deren Verwendung als Leitsalze. Merck Patent GmbH. EP120 5480(A2), 2002.
- Clarke-Hannaford, J.; Breedon, M.; Rüther, T.; Spencer, M.J.S. Fluorinated Boron-Based Anions for Higher Voltage Li Metal Battery Electrolytes. Nanomaterials 2021, 11, 2391. [Google Scholar] [CrossRef]
- Gerken, M.; Pawelke, G.; Bernhardt, E.; Willner, H. Syntheses and Characterization of (C2F5)3BCO and (C3F7)3BCO. Chem. Eur. J. 2010, 16, 7527–7536. [Google Scholar] [CrossRef] [PubMed]
- Phillips, N.A.; White, A.J.P.; Crimmin, M.R. Selective Hydrodefluorination of Hexafluoropropene to Industrially Relevant Hydrofluoroolefins. Adv. Synth. Catal. 2019, 361, 3351–3358. [Google Scholar] [CrossRef]
- Riefer, J.; Zapf, L.; Wirthensohn, R.; Hennig, P.T.; Ribbeck, T.; Sprenger, J.A.P.; Ignat’ev, N.V.; Finze, M. Pyridine Adducts of Tricyano- and Dicyanoboranes. Eur. J. Org. Chem. 2023, 26, e202300031. [Google Scholar] [CrossRef]
- Zapf, L.; Radius, U.; Finze, M. 1,3-Bis(tricyanoborane)imidazoline-2-ylidenate Anion—A Ditopic Dianionic N-Heterocyclic Carbene Ligand. Angew. Chem. Int. Ed. 2021, 60, 17974–17980. [Google Scholar] [CrossRef] [PubMed]
- Zapf, L.; Peters, S.; Bertermann, R.; Radius, U.; Finze, M. Tricyanoborane-Functionalized Anionic N-Heterocyclic Carbenes: Adjustment of Charge and Stereo-Electronic Properties. Chem. Eur. J. 2022, 28, e202200275. [Google Scholar] [CrossRef]
- Zapf, L.; Peters, S.; Radius, M.; Finze, M. Boranes Paving the Way to Anionic Cyclic (Alkyl)(amino)carbenes (Ani-cAACs). Angew. Chem. Int. Ed. 2023, 62, e202300056. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Quillian, B.; Wei, P.; Wannere, C.S.; Xie, Y.; King, B.R.; Schaefer III, H.F.; Schleyer, P.v.R.; Robinson, G.H. A stable neutral diborene containing a B=B double bond. J. Am. Chem. Soc. 2007, 129, 12412–12413. [Google Scholar] [CrossRef]
- Vaid, T.P.; Cook, M.E.; Scott, J.D.; Carazo, M.B.; Ruchti, J.; Minteer, S.D.; Sigman, M.S.; McNeil, A.J.; Sanford, M.S. Theoretical and Experimental Investigation of Functionalized Cyanopyridines Yield an Anolyte with an Extremely Low Reduction Potential for Nonaqueous Redox Flow Batteries. Chem. Eur. J. 2022, 28, e202202147. [Google Scholar] [CrossRef]
- von Aspern, N.; Röschenthaler, G.-V.; Winter, M.; Cekic-Laskovic, I. Fluorine and Lithium: Ideal Partners for High-Perfor- mance Rechargeable Battery Electrolytes. Angew. Chem. Int. Ed. 2019, 58, 15978–16000. [Google Scholar] [CrossRef]
- Böttcher, T.; Röschenthaler, G.-V. Highly reactive carbenes as ligands for main group element fluorides. Syntheses and applications. J. Fluorine Chem. 2015, 171, 4–11. [Google Scholar] [CrossRef]
- Janssen, P.; Streipert, B.; Krafft, R.; Murmann, P.; Wagner, R.; Lewis-Alleyne, L.; Röschenthaler, G.-V.; Winter, M.; Cekic-Laskovic, I. Shutdown potential adjustment of modified carbene adducts as additives for lithium ion battery electrolytes. J. Power Sources 2017, 367, 72–79. [Google Scholar] [CrossRef]
- Laoire, C.O.; Plichta, E.; Hendrickson, M.; Mukerjee, S.; Abraham, K.M. Electrochemical studies of ferrocene in a lithium ion conducting organic carbonate electrolyte. Electrochim. Acta 2009, 54, 6560–6564. [Google Scholar] [CrossRef]
- Zapf, L.; Riethmann, M.; Föhrenbacher, S.A.; Finze, M.; Radius, U. An easy-to-perform evaluation of steric properties of Lewis acids. Chem. Sci. 2023, 14, 2275–2288. [Google Scholar] [CrossRef] [PubMed]
- Riefer, J.; Zapf, L.; Sprenger, J.A.P.; Wirthensohn, R.; Endres, S.; Pöppler, A.-C.; Gutmann, M.; Meinel, L.; Ignat’ev, N.V.; Finze, M. Cyano(fluoro)borate and cyano(hydrido)borate ionic liquids: Low-viscosity ionic media for electrochemical applications. Phys. Chem. Chem. Phys. (PCCP) 2023, 25, 5037–5048. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.K.; Becker, E.D.; Cabral de Menezes, S.M.; Goodfellow, R.; Granger, P. NMR Nomenclature. Nuclear Spin Properties and Conventions for Chemical Shifts. Pure Appl. Chem. 2001, 73, 1795–1818. [Google Scholar] [CrossRef]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef]
- Kobler, H.; Munz, R.; Gasser, G.A.; Simchen, G. Eine einfache Synthese von Tetraalkylammoniumsalzen mit funktionellen Anionen. Justus Liebigs Ann. Chem. 1978, 1937–1945. [Google Scholar] [CrossRef]
- Armarego, W.L.F.; Chai, C.L.L. Purification of Laboratory Chemicals, 5th ed.; Butterworth-Heinemann (Elsevier): Amsterdam, The Netherlands, 2003. [Google Scholar]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. A 1965, 140, 1133–1138. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. I. The effect of the exchange-only gradient correction. J. Chem. Phys. 1992, 96, 2155–2160. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter. 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. Iii. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Rienstra-Kiracofe, J.C.; Tschumper, G.S.; Schaefer III, H.F.; Nandi, S.; Ellison, G.B. Atomic and molecular electron affinities: Photoelectron experiments and theoretical computations. Chem. Rev. 2002, 102, 231–282. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A Short History of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT, Program for Crystal Structure Solution; Universität Göttingen: Göttingen, Germany, 2014. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt Graphical User Interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, K. Diamond 4.6.8; Crystal Impact GbR: Bonn, Germany, 1997–2022. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).