
Adding Diversity to Diiron Aminocarbyne Complexes with Amine Ligands

 , , , , and
, , , , and 
Abstract
1. Introduction
2. Results and Discussion
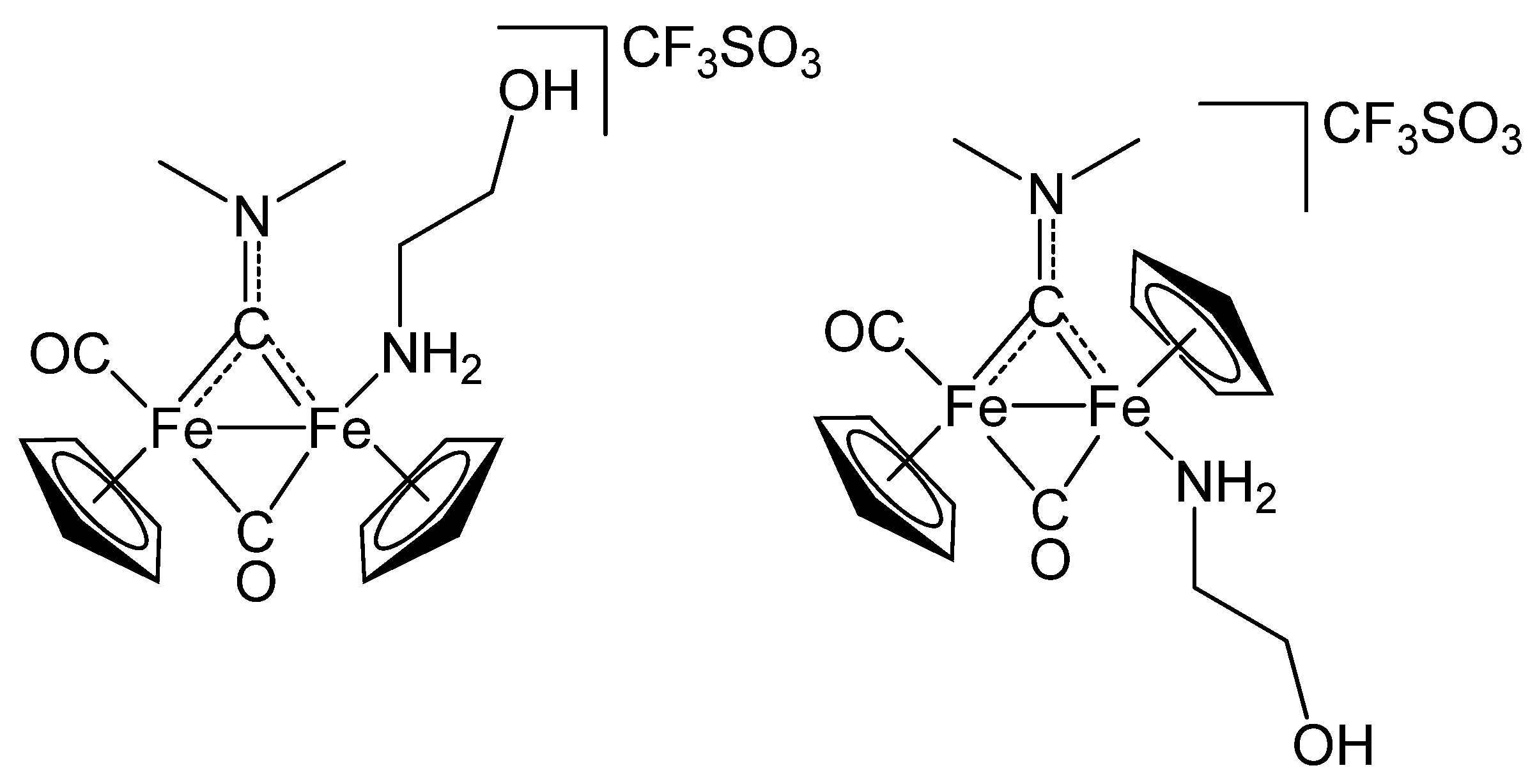
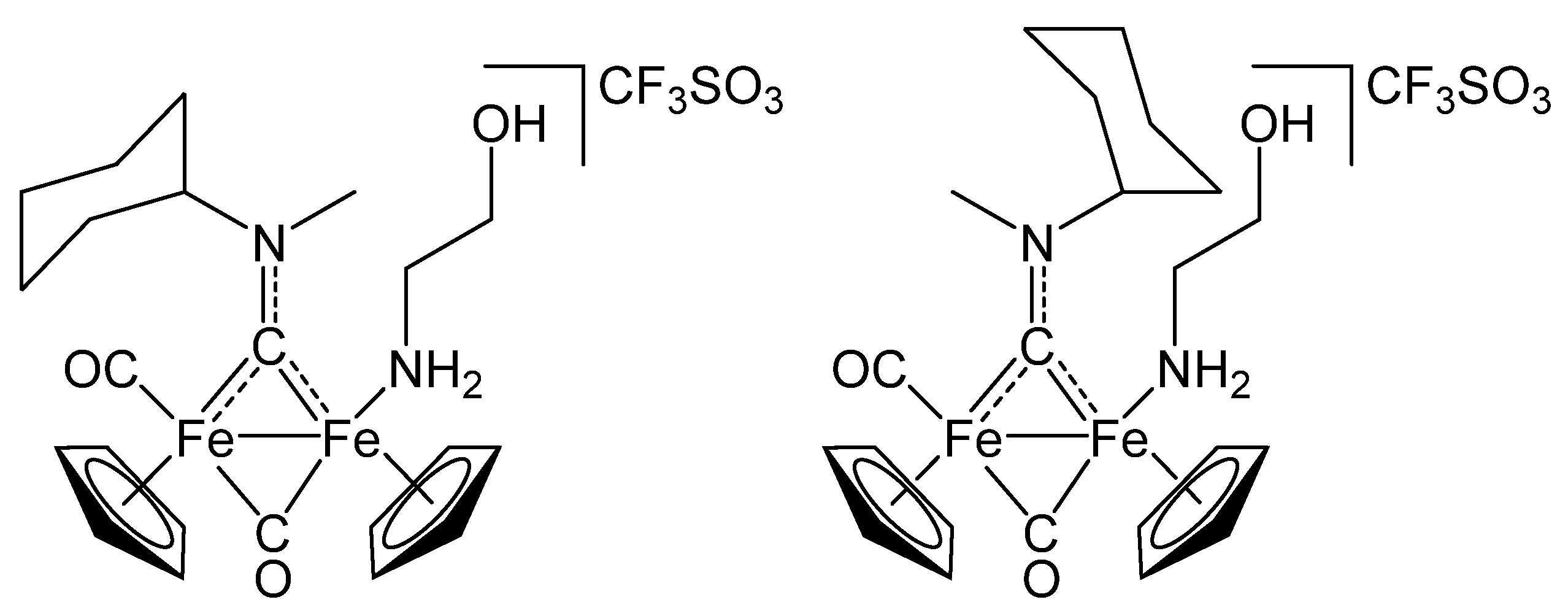
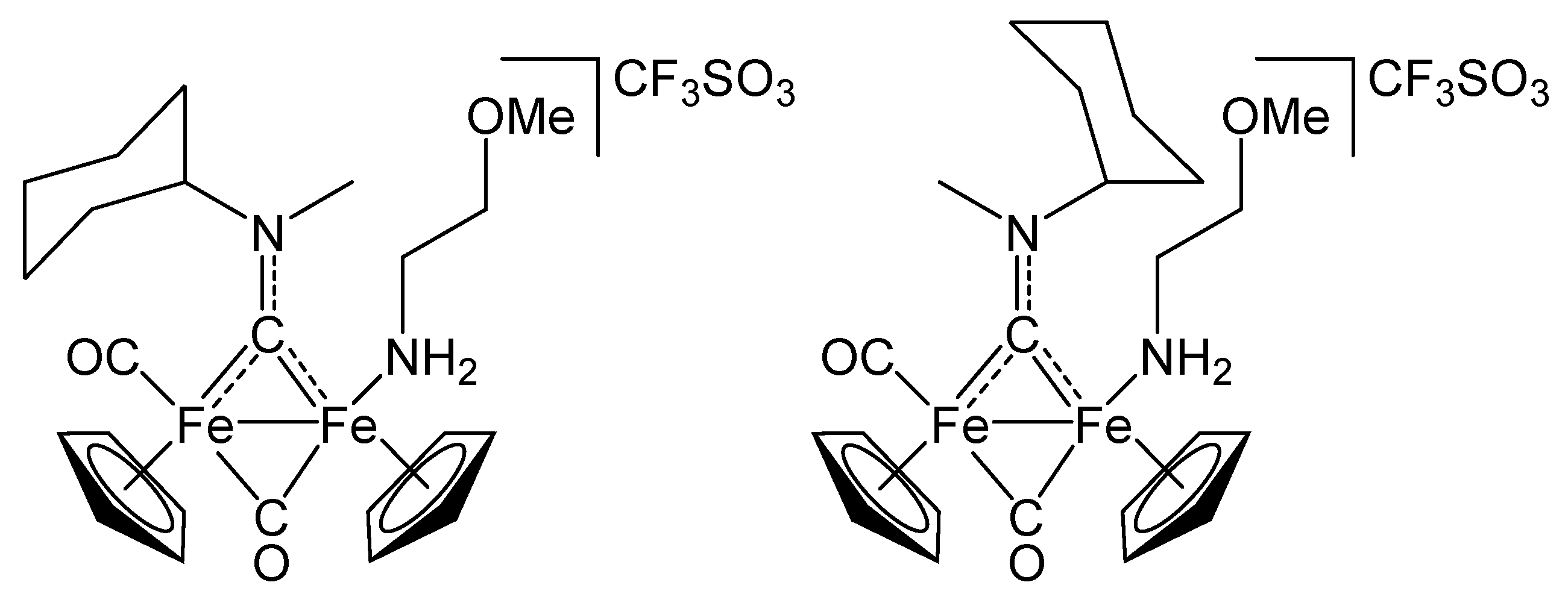
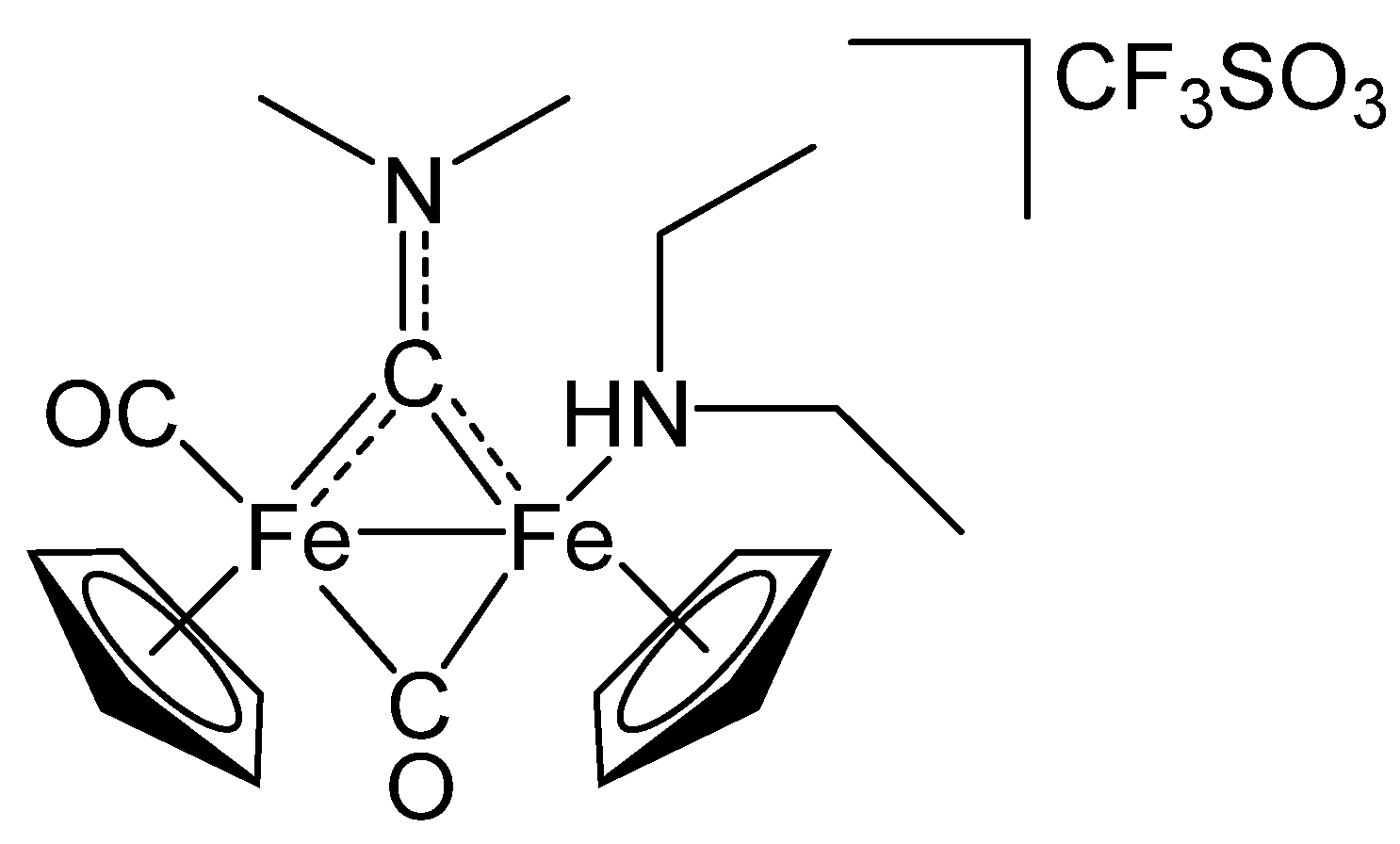
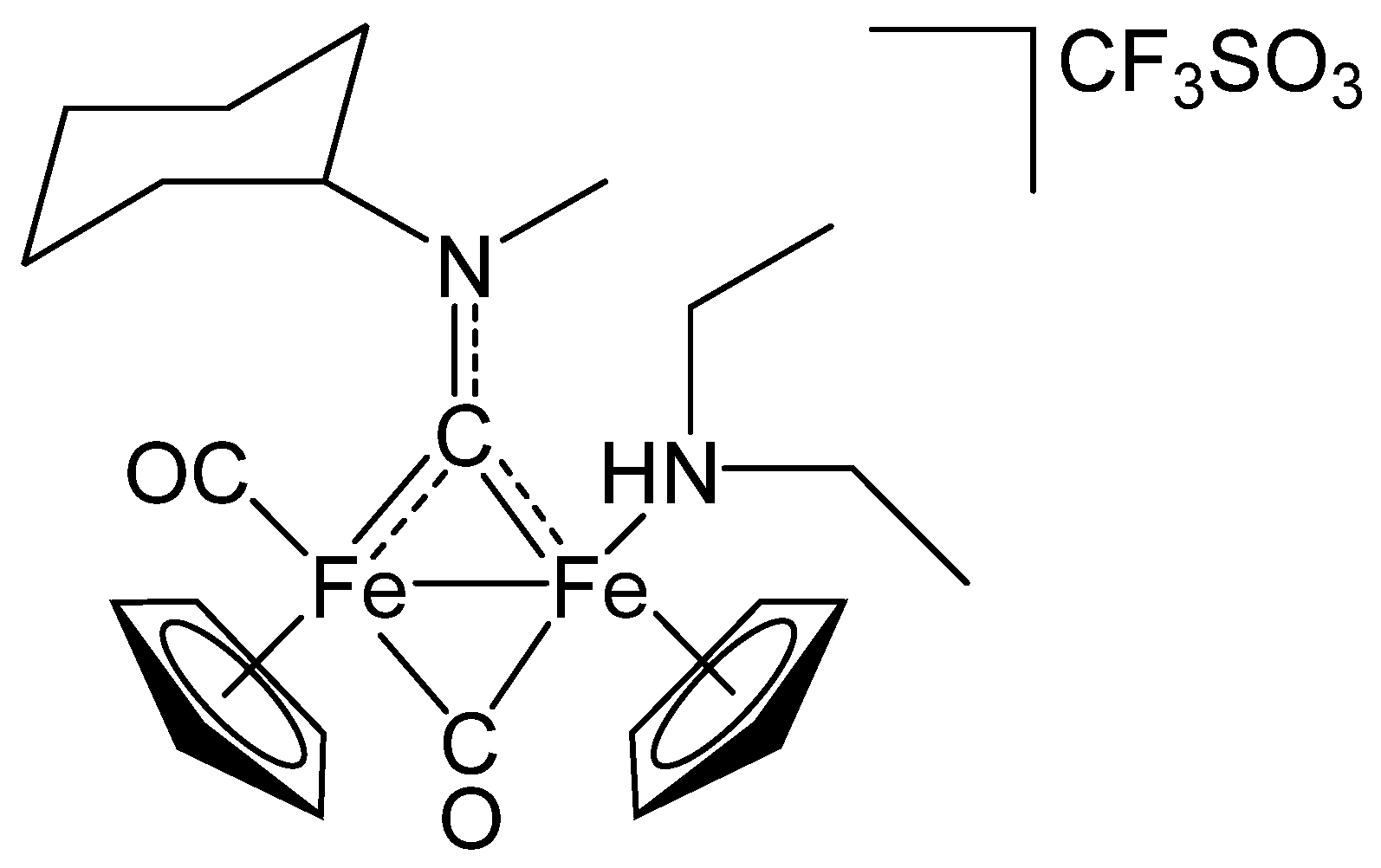
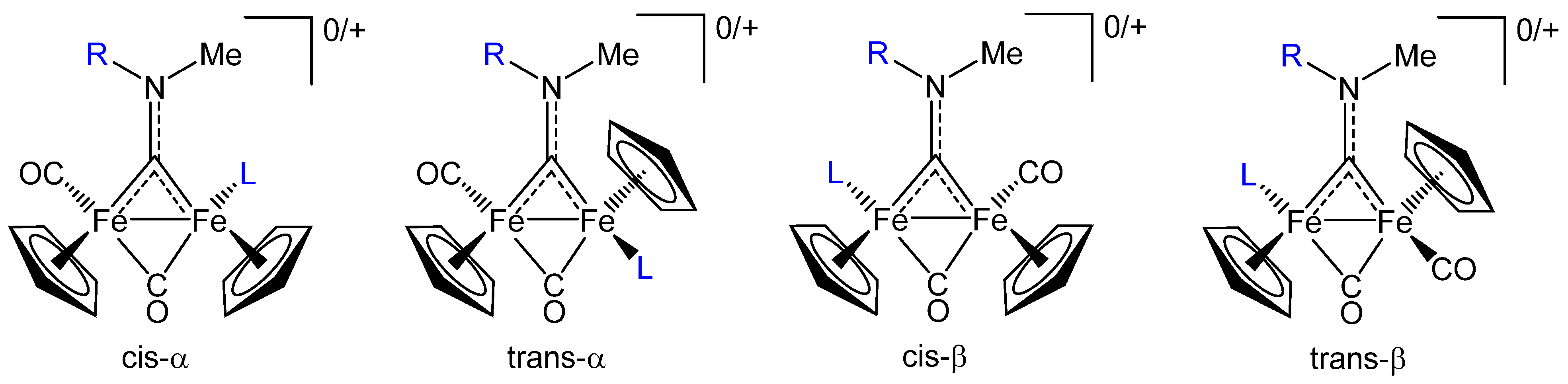
2.1. Synthesis and Spectroscopic Characterization
2.2. Behavior in Aqueous Solutions
3. Experimental Section
3.1. Materials and Methods
3.2. Synthesis and Characterization of Diiron Aminocarbyne Complexes with Primary Amines
3.3. Synthesis of Diiron Aminocarbyne Complex with Oxazolidinone–Amine
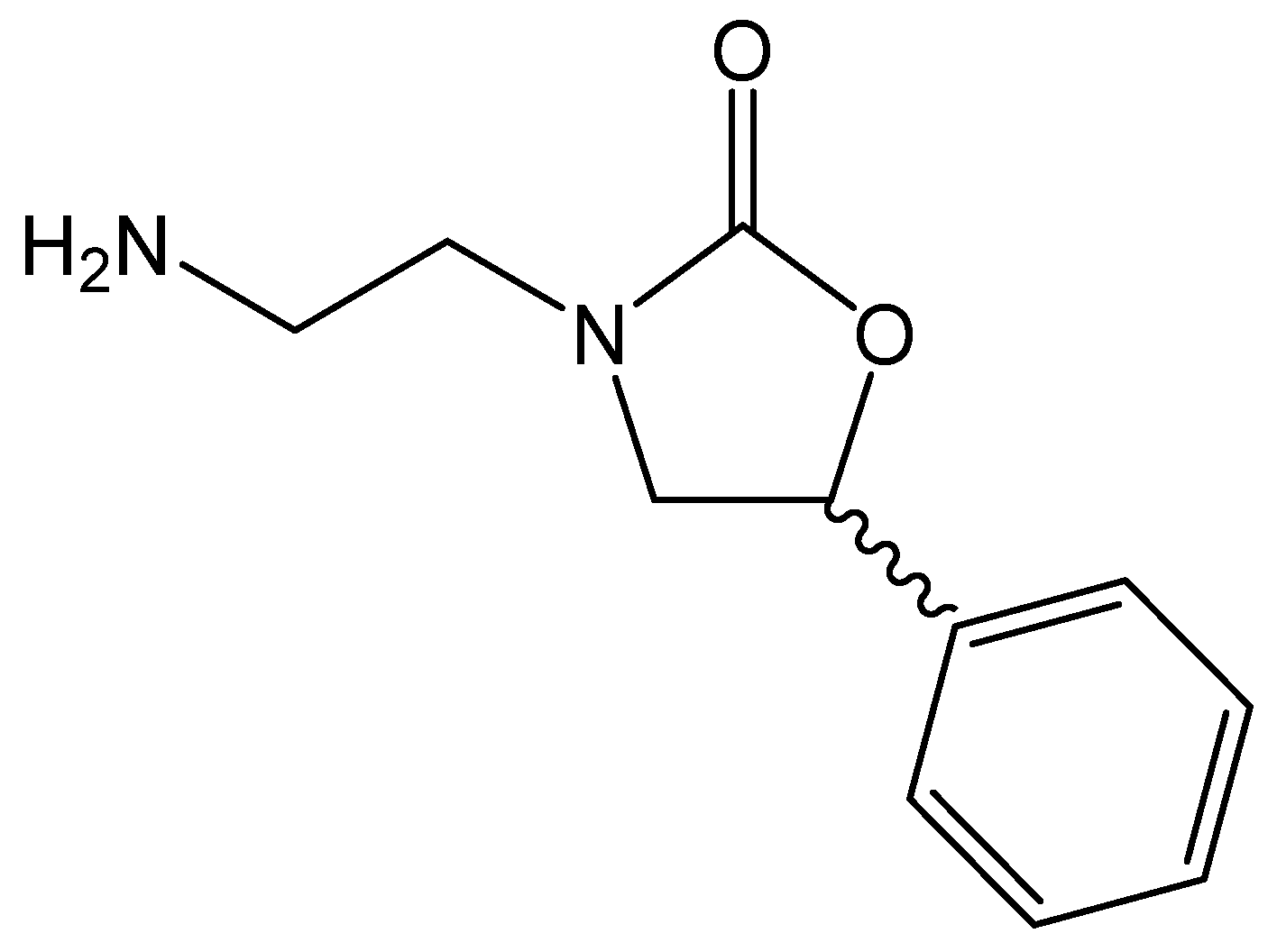
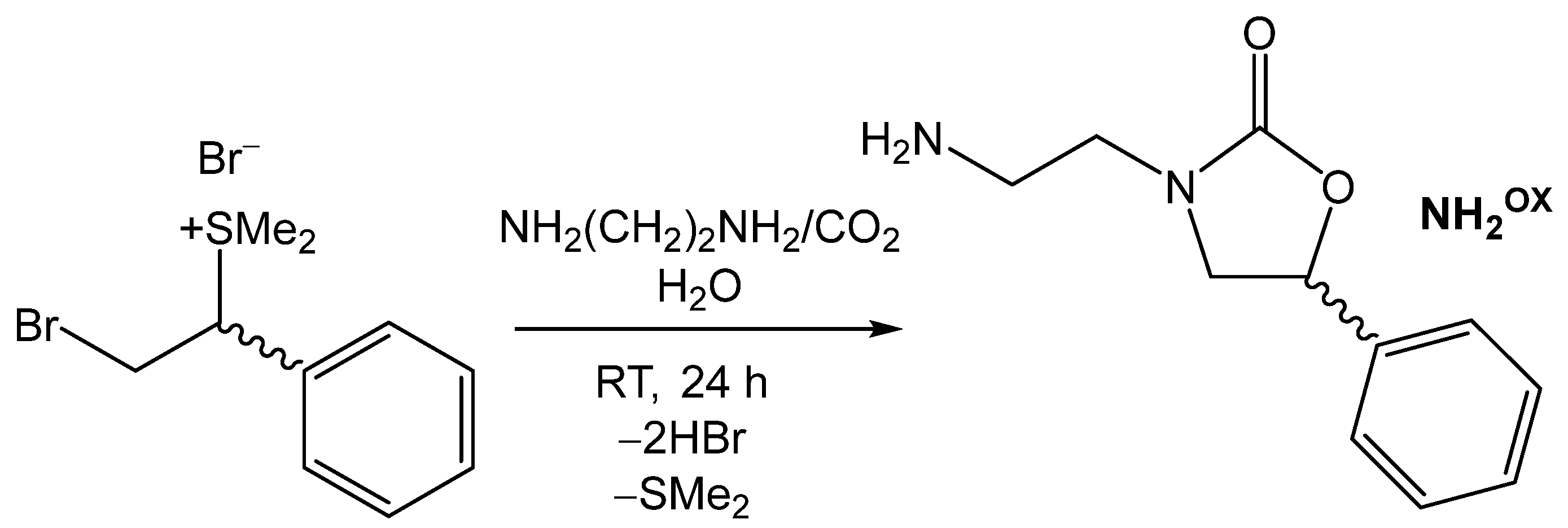
3.3.1. Synthesis and Characterization of 3-(2-Aminoethyl)-5-phenyloxazolidin-2-one (NH2OX, Figure 4) [61]

3.3.2. Synthesis and Characterization of [Fe2Cp2{κN-NH2OX}(CO)(μ-CO){μ-CN(Me)2}]CF3SO3, 3 (Figure 5)

3.4. Synthesis of Diiron Aminocarbyne Complexes with Diethylamine
3.5. Behavior of the Diiron Complexes in Aqueous Media
3.5.1. Solubility in D2O
3.5.2. Stability in Aqueous Solutions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wenger, O.S. Is Iron the New Ruthenium? Chem. Eur. J. 2019, 25, 6043–6052. [Google Scholar] [CrossRef] [PubMed]
- Bresciani, G.; Zacchini, S.; Marchetti, F.; Pampaloni, G. Non-precious metal carbamates as catalysts for the aziridine/CO2 coupling reaction under mild conditions. Dalton Trans. 2021, 50, 5351–5359. [Google Scholar] [CrossRef]
- Bisz, E.; Szostak, M. Iron-Catalyzed C–O Bond Activation: Opportunity for Sustainable Catalysis. ChemSusChem 2017, 10, 3964–3981. [Google Scholar] [CrossRef] [PubMed]
- Enthaler, S.; Junge, K.; Beller, M. Sustainable Metal Catalysis with Iron: From Rust to a Rising Star? Angew. Chem. Int. Ed. 2008, 47, 3317–3321. [Google Scholar] [CrossRef] [PubMed]
- Coufourier, S.; Gaignard Gaillard, Q.; Lohier, J.-F.; Poater, A.; Gaillard, S.; Renaud, J.-L. Hydrogenation of CO2, Hydrogenocarbonate, and Carbonate to Formate in Water using Phosphine Free Bifunctional Iron Complexes. ACS Catal. 2020, 10, 2108–2116. [Google Scholar] [CrossRef]
- van Beek, C.B.; van Leest, N.P.; Lutz, M.; de Vos, S.D.; Klein Gebbink, R.J.M.; de Bruin, B.; Broere, D.L.J. Combining metal-metal cooperativity, metal-ligand cooperativity and chemical non-innocence in diiron carbonyl complexes. Chem. Sci. 2022, 13, 2094–2104. [Google Scholar] [CrossRef]
- Govindarajan, R.; Deolka, S.; Khusnutdinova, J.R. Heterometallic bond activation enabled by unsymmetrical ligand scaffolds: Bridging the opposites. Chem. Sci. 2022, 13, 14008–14031. [Google Scholar] [CrossRef]
- Ritleng, V.; Chetcuti, M.J. Hydrocarbyl Ligand Transformations on Heterobimetallic Complexes. Chem. Rev. 2007, 107, 797−858. [Google Scholar] [CrossRef]
- Desnoyer, A.N.; Nicolay, A.; Rios, P.; Ziegler, M.S.; Don Tilley, T. Bimetallics in a Nutshell: Complexes Supported by Chelating Naphthyridine-Based Ligands. Acc. Chem. Res. 2020, 53, 1944–1956. [Google Scholar] [CrossRef]
- Tsurugi, H.; Laskar, P.; Yamamoto, K.; Mashima, K. Bonding and structural features of metal-metal bonded homo—And hetero-dinuclear complexes supported by unsaturated hydrocarbon ligands. J. Organomet. Chem. 2018, 869, 251–263. [Google Scholar] [CrossRef]
- Busetto, L.; Maitlis, P.M.; Zanotti, V. Bridging vinylalkylidene transition metal complexes. Coord. Chem. Rev. 2010, 254, 470–486. [Google Scholar] [CrossRef]
- Swanson, K.D.; Ortillo, D.O.; Broderick, J.B.; Peters, J.W. [FeFe]-Hydrogenase. In Encyclopedia of Inorganic and Bioinorganic Chemistry, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011. [Google Scholar]
- Lubitz, W.; Ogata, H.; Rüdiger, O.; Reijerse, E. Hydrogenases. Chem. Rev. 2014, 114, 4081–4148. [Google Scholar] [CrossRef] [PubMed]
- Kleinhaus, J.T.; Wittkamp, F.; Yadav, S.; Siegmund, D.; Apfel, U.-P. [FeFe]-Hydrogenases: Maturation and reactivity of enzymatic systems and overview of biomimetic models. Chem. Soc. Rev. 2021, 50, 1668–1784. [Google Scholar] [CrossRef] [PubMed]
- Gee, L.B.; Pelmenschikov, V.; Wang, H.; Mishra, N.; Liu, Y.-C.; Yoda, Y.; Tamasaku, K.; Chiang, M.-H.; Cramer, S.P. Vibrational characterization of a diiron bridging hydride complex—A model for hydrogen catalysis. Chem. Sci. 2020, 11, 5487–5493. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Liu, Y.; Shao, Y.; Jiang, D.; Duan, Q. Iron carbonyl compounds with aromatic dithiolate bridges as organometallic mimics of [FeFe] hydrogenases. Coord. Chem. Rev. 2020, 402, 213081. [Google Scholar] [CrossRef]
- Li, Y.; Rauchfuss, T.B. Synthesis of Diiron(I) Dithiolato Carbonyl Complexes. Chem. Rev. 2016, 116, 7043–7077. [Google Scholar] [CrossRef]
- Mazzoni, R.; Salmi, M.; Zanotti, V. C-C Bond Formation in Diiron Complexes. Chem. Eur. J. 2012, 18, 10174–10194. [Google Scholar] [CrossRef]
- Busetto, L.; Zanotti, V. Carbene ligands in diiron complexes. J. Organomet. Chem. 2005, 690, 5430–5440. [Google Scholar] [CrossRef]
- Chen, J.; Wang, R. Remarkable reactions of cationic carbyne complexes of manganese, rhenium, and diiron with carbonylmetal anions. Coord. Chem. Rev. 2002, 231, 109–149. [Google Scholar] [CrossRef]
- Casey, C.P.; Austin, E.A. Photochemical Reactions of Diiron p-Alkenylidene Complexes with Hydrogen, Trialkylsilanes, and Diazo Compounds: Cleavage to Alkenes, Vinylsilanes, and Allenes. J. Am. Chem. Soc. 1988, 110, 7106–7113. [Google Scholar] [CrossRef]
- Murshid, N.; El-Temtamy, A.; Wang, X. Synthesis and solution behaviour of metal-carbonyl amphiphiles with an Fp (CpFe(CO)2) junction. J. Organomet. Chem. 2017, 851, 40–45. [Google Scholar] [CrossRef]
- García, M.E.; García-Vivó, D.; Ramos, A.; Ruiz, M.A. Phosphinidene-bridged binuclear complexes. Coord. Chem. Rev. 2017, 330, 1–36. [Google Scholar] [CrossRef]
- Biancalana, L.; Marchetti, F. Aminocarbyne ligands in organometallic chemistry. Coord. Chem. Rev. 2021, 449, 214203. [Google Scholar] [CrossRef]
- Marchetti, F. Constructing Organometallic Architectures from Aminoalkylidyne Diiron Complexes. Eur. J. Inorg. Chem. 2018, 2018, 3987–4003. [Google Scholar] [CrossRef]
- Agonigi, G.; Bortoluzzi, M.; Marchetti, F.; Pampaloni, G.; Zacchini, S.; Zanotti, V. Regioselective Nucleophilic Additions to Diiron Carbonyl Complexes Containing a Bridging Aminocarbyne Ligand: A Synthetic, Crystallographic and DFT Study. Eur. J. Inorg. Chem. 2018, 2018, 960–971. [Google Scholar] [CrossRef]
- Biancalana, L.; De Franco, M.; Ciancaleoni, G.; Zacchini, S.; Pampaloni, G.; Gandin, V.; Marchetti, F. Easily Available and Amphiphilic Diiron Cyclopentadienyl Complexes Exhibit In Vitro Anticancer Activity in 2D and 3D Human Cancer Cells via Redox Modulation Triggered by CO Release. Chem. Eur. J. 2021, 27, 10169–10185. [Google Scholar] [CrossRef] [PubMed]
- Rocco, D.; Batchelor, L.K.; Agonigi, G.; Braccini, S.; Chiellini, F.; Schoch, S.; Biver, T.; Funaioli, T.; Zacchini, S.; Biancalana, L.; et al. Anticancer Potential of Diiron Vinyliminium Complexes. Chem. Eur. J. 2019, 25, 14801–14816. [Google Scholar] [CrossRef]
- Biancalana, L.; Kubeil, M.; Schoch, S.; Zacchini, S.; Marchetti, F. Switching on Cytotoxicity of Water-Soluble Diiron Organometallics by UV Irradiation. Inorg. Chem. 2022, 61, 7897–7909. [Google Scholar] [CrossRef]
- Arrigoni, F.; Bertini, L.; De Gioia, L.; Cingolani, A.; Mazzoni, R.; Zanotti, V.; Zampella, G. Mechanistic Insight into Electrocatalytic H2 Production by [Fe2(CN){μ-CN(Me)2}(μ-CO)(CO)(Cp)2]: Effects of Dithiolate Replacement in [FeFe] Hydrogenase Models. Inorg. Chem. 2017, 56, 13852–13864. [Google Scholar] [CrossRef]
- Mazzoni, R.; Gabiccini, A.; Cesari, C.; Zanotti, V.; Gualandi, I.; Tonelli, D. Diiron Complexes Bearing Bridging Hydrocarbyl Ligands as Electrocatalysts for Proton Reduction. Organometallics 2015, 34, 3228−3235. [Google Scholar] [CrossRef]
- Bresciani, G.; Biancalana, L.; Zacchini, S.; Pampaloni, G.; Ciancaleoni, G.; Marchetti, F. Diiron Bis-Cyclopentadienyl Complexes as Transfer Hydrogenation Catalysts: The Key Role of the Bridging Aminocarbyne Ligand. Appl. Organomet. Chem. 2023, 37, e6990. [Google Scholar] [CrossRef]
- Bresciani, G.; Schoch, S.; Biancalana, L.; Zacchini, S.; Bortoluzzi, M.; Pampaloni, G.; Marchetti, F. Cyanide-alkene competition in a diiron complex and isolation of a multisite (cyano)alkylidene—Alkene species. Dalton Trans. 2022, 51, 1936–1945. [Google Scholar] [CrossRef] [PubMed]
- Agonigi, G.; Biancalana, L.; Lupo, M.G.; Montopoli, M.; Ferri, N.; Zacchini, S.; Binacchi, F.; Biver, T.; Campanella, B.; Pampaloni, G.; et al. Exploring the Anticancer Potential of Diiron Bis-cyclopentadienyl Complexes with Bridging Hydrocarbyl Ligands: Behavior in Aqueous Media and In Vitro Cytotoxicity. Organometallics 2020, 39, 645–657. [Google Scholar] [CrossRef]
- Boss, K.; Dowling, C.; Manning, A.R. Preparation, spectra and structure of [Fe2(η5-C5H5)2(L)(CN)(μ-CO){μ-CN(R’)R}], [Fe2(η5-C5H5)2(CO)(CN){μ-CNMe2}2]+ and [Fe2(η5-C5H5)2(CN)2{μ-CNMe2}2] zwitterions (L = CO or organoisocyanide) and their reactions with alkyl and protic electrophiles. J. Organomet. Chem. 1996, 509, 197–207. [Google Scholar] [CrossRef]
- Marchetti, F.; Zacchini, S.; Zanotti, V. Coupling of Isocyanide and μ-Aminocarbyne Ligands in Diiron Complexes Promoted by Hydride Addition. Organometallics 2014, 33, 3990–3997. [Google Scholar] [CrossRef]
- Busetto, L.; Marchetti, F.; Zacchini, S.; Zanotti, V. Diiron and diruthenium aminocarbyne complexes containing pseudohalides: Stereochemistry and reactivity. Inorg. Chim. Acta 2005, 358, 1204–1216. [Google Scholar] [CrossRef]
- Howell, J.A.S.; Rowan, A.J. Synthesis and fluxional character of complexes of the type [M2(cp)2(CO)3(CNR)](M = Fe or Ru, Cp =η-C5H5). J. Chem. Soc. Dalton Trans. 1980, 503–510. [Google Scholar] [CrossRef]
- Adams, R.D.; Cotton, F.A. On the Pathway of Bridge-Terminal Ligand Exchange in Some Binuclear Metal Carbonyls. J. Am. Chem. Soc. 1973, 95, 6589–6594. [Google Scholar] [CrossRef]
- Biancalana, L.; Ciancaleoni, G.; Zacchini, S.; Pampaloni, G.; Marchetti, F. Carbonyl-isocyanide mono-substitution in [Fe2Cp2(CO)4]: A re-visitation. Inorg. Chim. Acta 2020, 517, 120181. [Google Scholar] [CrossRef]
- Luh, T.-Y. Trimethylamine N-Oxide-A Versatile Reagent for Organometallic Chemistry. Coord. Chem. Rev. 1984, 60, 255–276. [Google Scholar] [CrossRef]
- Albano, V.G.; Busetto, L.; Monari, M.; Zanotti, V. Reactions of acetonitrile di-iron μ-aminocarbyne complexes; synthesis and structure of [Fe2(μ-CNMe2)(μ-H)(CO)2(Cp)2]. J. Organomet. Chem. 2000, 606, 163–168. [Google Scholar] [CrossRef]
- Bresciani, G.; Boni, S.; Zacchini, S.; Pampaloni, G.; Bortoluzzi, M.; Marchetti, F. Alkyne–alkenyl coupling at a diruthenium complex. Dalton Trans. 2022, 51, 15703–15715. [Google Scholar] [CrossRef]
- Bresciani, G.; Zacchini, S.; Pampaloni, G.; Marchetti, F. Carbon-Carbon Bond Coupling of Vinyl Molecules with an Allenyl Ligand at a Diruthenium Complex. Organometallics 2022, 41, 1006–1014. [Google Scholar] [CrossRef]
- Bresciani, G.; Zacchini, S.; Pampaloni, G.; Bortoluzzi, M.; Marchetti, F. η6-Coordinated ruthenabenzenes from three-component assembly on a diruthenium μ-allenyl scaffold. Dalton Trans. 2022, 51, 8390–8400. [Google Scholar] [CrossRef] [PubMed]
- Bresciani, G.; Biancalana, L.; Pampaloni, G.; Zacchini, S.; Ciancaleoni, G.; Marchetti, F. A Comprehensive Analysis of the Metal–Nitrile Bonding in an Organo-Diiron System. Molecules 2021, 26, 7088. [Google Scholar] [CrossRef] [PubMed]
- Kufelnicki, A.; Tomyn, S.V.; Nedelkov, R.V.; Haukka, M.; Jaciubek-Rosinska, J.; Pajak, M.; Jaszczak, J.; Swiatek, M.; Fritsky, I.O. Synthesis of cobalt(III) complexes with novel open chain oxime ligands and metal–ligand coordination in aqueous solution. Inorg. Chim. Acta 2010, 363, 2996–3003. [Google Scholar] [CrossRef]
- Dahlenburg, L.; Treffert, H.; Farr, C.; Heinemann, F.W.; Zahl, A. Rhodium(I) Complexes Containing β-Amino Alcohol and 1,2-Diamine Ligands: Syntheses, Structures, and Catalytic Applications. Eur. J. Inorg. Chem. 2007, 1738–1751. [Google Scholar] [CrossRef]
- Bortoluzzi, M.; Bresciani, G.; Marchetti, F.; Pampaloni, G.; Zacchini, S. MoCl5 as an effective chlorinating agent towards α-amino acids: Synthesis of α-ammonium-acylchloride salts and α-amino-acylchloride complexes. Dalton Trans. 2015, 44, 10030–10037. [Google Scholar] [CrossRef] [PubMed]
- Niemi, T.; Repo, T. Antibiotics from Carbon Dioxide: Sustainable Pathways to Pharmaceutically Relevant Cyclic Carbamates. Eur. J. Org. Chem. 2019, 1180–1188. [Google Scholar] [CrossRef]
- Jadhavar, P.S.; Vaja, M.D.; Dhameliya, T.M.; Chakraborti, A.K. Oxazolidinones as Anti-tubercular Agents: Discovery, Development and Future Perspectives. Curr. Med. Chem. 2015, 22, 4379–4397. [Google Scholar] [CrossRef]
- Zhao, Q.; Xin, L.; Liu, Y.; Liang, C.; Li, J.; Jian, Y.; Li, H.; Shi, Z.; Liu, H.; Cao, W. Current Landscape and Future Perspective of Oxazolidinone Scaffolds Containing Antibacterial Drugs. J. Med. Chem. 2021, 64, 10557–10580. [Google Scholar] [CrossRef]
- Xie, Y.; Lu, C.; Zhao, B.; Wang, Q.; Yao, Y. Cycloaddition of Aziridine with CO2/CS2 Catalyzed by Amidato Divalent Lanthanide Complexes. J. Org. Chem. 2019, 84, 1951−1958. [Google Scholar] [CrossRef] [PubMed]
- Sengoden, M.; North, A.C. Whitwood. ChemSusChem 2019, 12, 3296–3303. [Google Scholar] [CrossRef] [PubMed]
- Bresciani, G.; Bortoluzzi, M.; Pampaloni, G.; Marchetti, F. Diethylammonium iodide as catalyst for the metal-free synthesis of 5-aryl-2-oxazolidinones from aziridines and carbon dioxide. Org. Biomol. Chem. 2021, 19, 4152–4161. [Google Scholar] [CrossRef] [PubMed]
- Sonzini, P.; Damiano, C.; Intrieri, D.; Manca, G.; Gallo, E. A Metal-Free Synthesis of N-Aryl Oxazolidin-2-Ones by the One-Pot Reaction of Carbon Dioxide with N-Aryl Aziridines. Adv. Synth. Catal. 2020, 362, 2961–2969. [Google Scholar] [CrossRef]
- Bresciani, G.; Bortoluzzi, M.; Ghelarducci, C.; Marchetti, F.; Pampaloni, G. Synthesis of a-alkylidene cyclic carbonates via CO2 fixation under ambient conditions promoted by an easily available silver carbamate. N. J. Chem. 2021, 45, 4340–4346. [Google Scholar] [CrossRef]
- Bresciani, G.; Busto, N.; Ceccherini, V.; Bortoluzzi, M.; Pampaloni, G.; Garcia, B.; Marchetti, F. Screening the biological properties of transition metal carbamates reveals gold(I) and silver(I) complexes as potent cytotoxic and antimicrobial agents. J. Inorg. Biochem. 2022, 227, 111667. [Google Scholar] [CrossRef]
- Biancalana, L.; Bresciani, G.; Chiappe, C.; Marchetti, F.; Pampaloni, G.; Pomelli, C.S. Modifying bis(triflimide) ionic liquids by dissolving early transition metal carbamates. Phys. Chem. Chem. Phys. 2018, 20, 5057—5066. [Google Scholar] [CrossRef]
- Bortoluzzi, M.; Bresciani, G.; Marchetti, F.; Pampaloni, G.; Zacchini, S. Synthesis and structural characterization of mixed halide–N,N-diethylcarbamates of group 4 metals, including a case of unusual tetrahydrofuran activation. New J. Chem. 2017, 41, 1781–1789. [Google Scholar] [CrossRef]
- Bresciani, G.; Antico, E.; Ciancaleoni, G.; Zacchini, S.; Pampaloni, G.; Marchetti, F. Bypassing the Inertness of Aziridine/CO2 Systems to Access 5-Aryl-2-Oxazolidinones: Catalyst-Free Synthesis Under Ambient Conditions. ChemSusChem 2020, 13, 5586–5594. [Google Scholar] [CrossRef]
- Bresciani, G.; Zacchini, S.; Famlonga, L.; Pampaloni, G.; Marchetti, F. Trapping carbamates of α-Amino acids: One-Pot and catalyst-free synthesis of 5-Aryl-2-Oxazolidinonyl derivatives. J. CO2 Util. 2021, 47, 101495. [Google Scholar] [CrossRef]
- Busetto, L.; Marchetti, F.; Zacchini, S.; Zanotti, V.; Zoli, E. Diiron-aminocarbyne complexes with amine or imine ligands: C–N coupling between imine and aminocarbyne ligands promoted by tolylacetylide addition to [Fe2{μ-CN(Me)R}(μ-CO)(CO)(NH=CPh2)(Cp)2][SO3CF3]. J. Organomet. Chem. 2005, 690, 348–357. [Google Scholar] [CrossRef]
- Labinger, J.A. Does cyclopentadienyl iron dicarbonyl dimer have a metal–metal bond? Who’s asking? Inorg. Chim. Acta 2015, 424, 14–19. [Google Scholar] [CrossRef]
- Frenking, G.; Frohlich, N. The Nature of the Bonding in Transition-Metal Compounds. Chem. Rev. 2000, 100, 717–774. [Google Scholar] [CrossRef] [PubMed]
- Schoch, S.; Iacopini, D.; Dalla Pozza, M.; Di Pietro, S.; Degano, I.; Gasser, G.; Di Bussolo, V.; Marchetti, F. Tethering Carbohydrates to the Vinyliminium Ligand of Antiproliferative Organometallic Diiron Complexes. Organometallics 2022, 41, 514–526. [Google Scholar] [CrossRef]
- Marchetti, F. Alkylidyne and Alkylidene Complexes of Iron. In Comprehensive Organometallic Chemistry IV; Parkin, G., Meyer, K., O’Hare, D., Eds.; Elsevier: Kidlington, UK, 2022; Volume 7, pp. 210–257. [Google Scholar] [CrossRef]
- Zhou, X.; Barton, B.E.; Chambers, G.M.; Rauchfuss, T.B. Preparation and Protonation of Fe2(pdt)(CNR)6, Electron-Rich Analogues of Fe2(pdt)(CO)6. Inorg. Chem. 2016, 55, 3401−3412. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Matlock, J.V.; Fritz, S.P.; Harrison, S.A.; Coe, D.M.; McGarrigle, E.M.; Aggarwal, V.K. Synthesis of α-substituted vinylsulfonium salts and their application as annulation reagents in the formation of epoxide-and cyclopropane-fused heterocycles. J. Org. Chem. 2014, 79, 10226–10239. [Google Scholar] [CrossRef]
- Menges, F. Spectragryph-Optical Spectroscopy Software, Version 1.2.5, 2016–2017. Available online: http://www.effemm2.de/spectragryph (accessed on 29 January 2023).
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef]
- Willker, W.; Leibfritz, D.; Kerssebaum, R.; Bermel, W. Gradient selection in inverse heteronuclear correlation spectroscopy. Magn. Reson. Chem. 1993, 31, 287–292. [Google Scholar] [CrossRef]
- Rundlöf, T.; Mathiasson, M.; Bekiroglu, S.; Hakkarainen, B.; Bowden, T.; Arvidsson, T. Survey and qualification of internal standards for quantification by 1H NMR spectroscopy. J. Pharm. Biomed. Anal. 2010, 52, 645–651. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
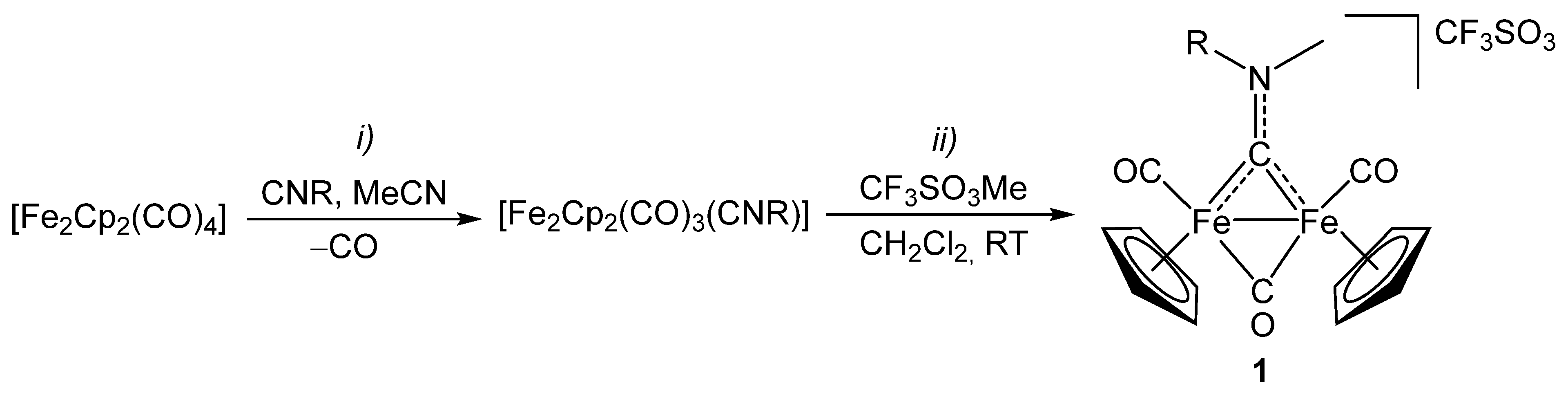{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | Solubility/mol·L−1 | % Stability D2O/CD3OD | % Stability DMEM-d/CD3OD | Solvent/CD3OD Ratio |
|---|---|---|---|---|
| 2a | 2.0·10−3 | 45 | 45 | 2 |
| 49 | 29 | ∞ | ||
| 2b | 0.2·10−3 | 29 | ≈10 | 2 |
| 2c | 9.6·10−3 | 19 | 17 | 2 |
| 3 | 4.7·10−4 | ≈0 | ≈0 | 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saviozzi, C.; Stocchetti, S.; Bresciani, G.; Biancalana, L.; Pampaloni, G.; Marchetti, F. Adding Diversity to Diiron Aminocarbyne Complexes with Amine Ligands. Inorganics 2023, 11, 91. https://doi.org/10.3390/inorganics11030091
Saviozzi C, Stocchetti S, Bresciani G, Biancalana L, Pampaloni G, Marchetti F. Adding Diversity to Diiron Aminocarbyne Complexes with Amine Ligands. Inorganics. 2023; 11(3):91. https://doi.org/10.3390/inorganics11030091
Chicago/Turabian StyleSaviozzi, Chiara, Sara Stocchetti, Giulio Bresciani, Lorenzo Biancalana, Guido Pampaloni, and Fabio Marchetti. 2023. "Adding Diversity to Diiron Aminocarbyne Complexes with Amine Ligands" Inorganics 11, no. 3: 91. https://doi.org/10.3390/inorganics11030091
APA StyleSaviozzi, C., Stocchetti, S., Bresciani, G., Biancalana, L., Pampaloni, G., & Marchetti, F. (2023). Adding Diversity to Diiron Aminocarbyne Complexes with Amine Ligands. Inorganics, 11(3), 91. https://doi.org/10.3390/inorganics11030091