

The Role of Non-Covalent Interactions in the Reactions between Palladium Hydrido Complex with Amidoarylphosphine Pincer Ligand and Brønsted Acids

 , , , , ,
, , , , ,  ,
,  and
and
Abstract
:1. Introduction
2. Results and Discussion
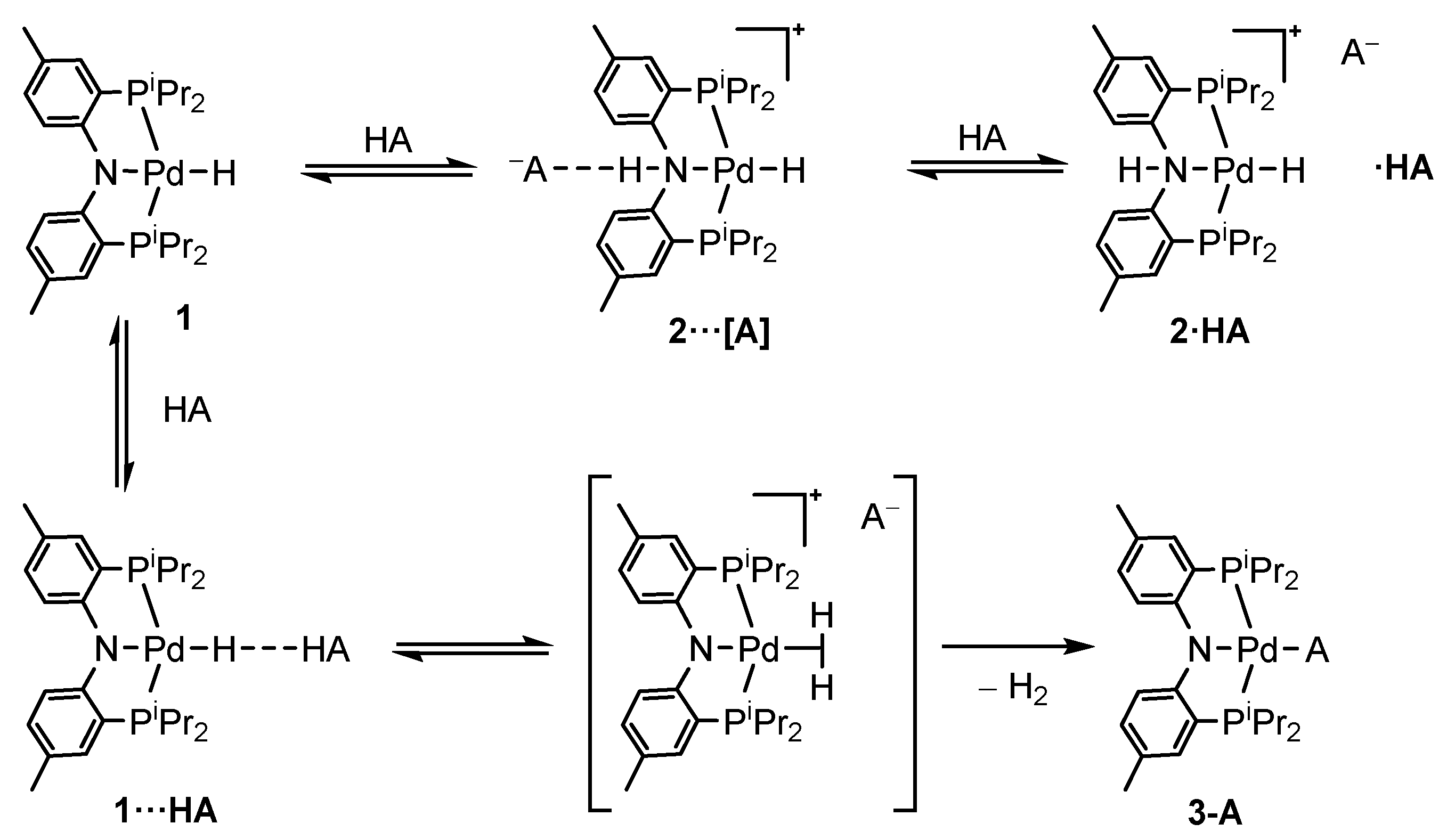
2.1. (PNP)PdH Interaction with Strong Brønsted Acids TfOH and HBF4, Chemical Properties of [(PN(H)P)PdH]+
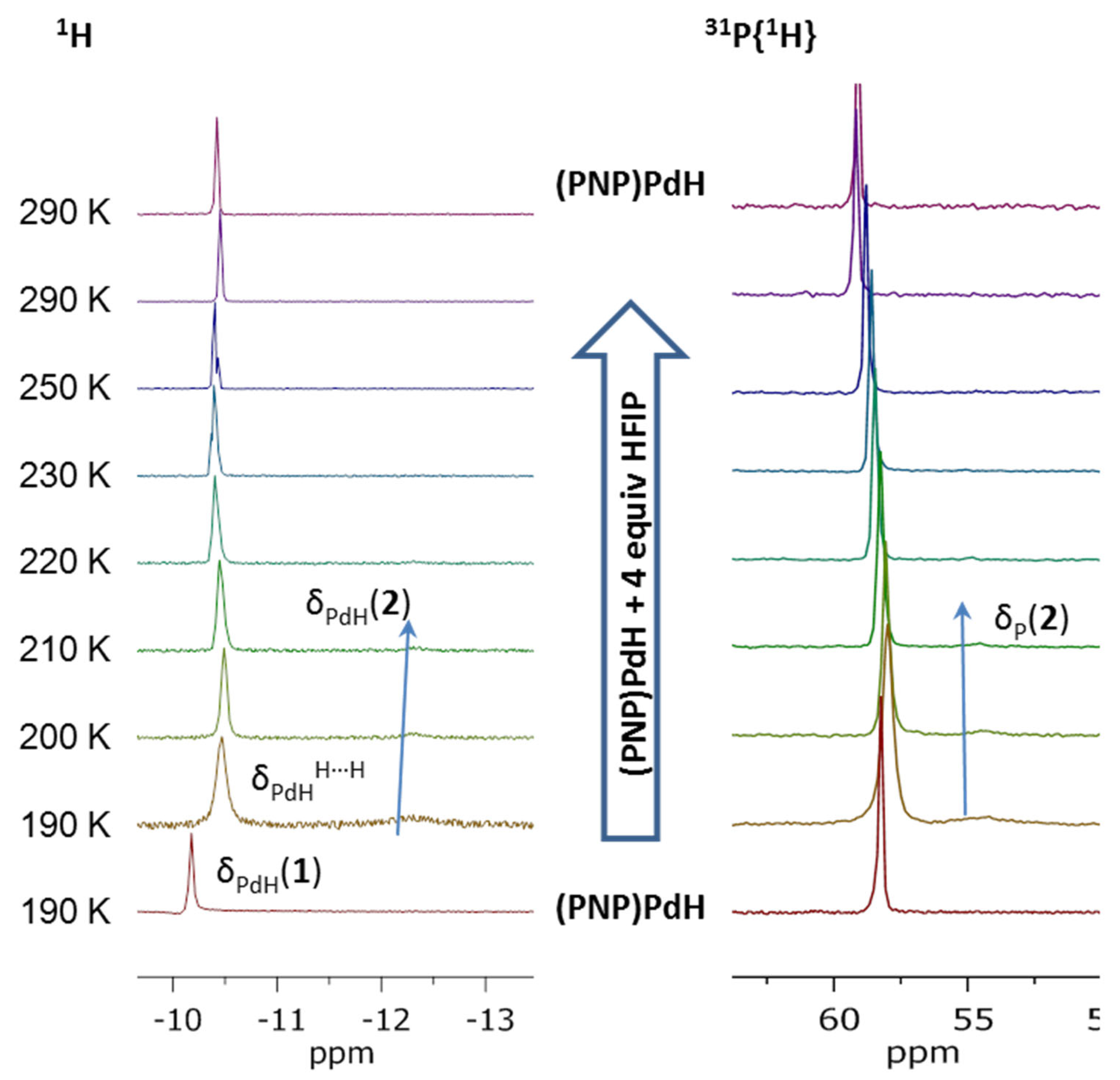
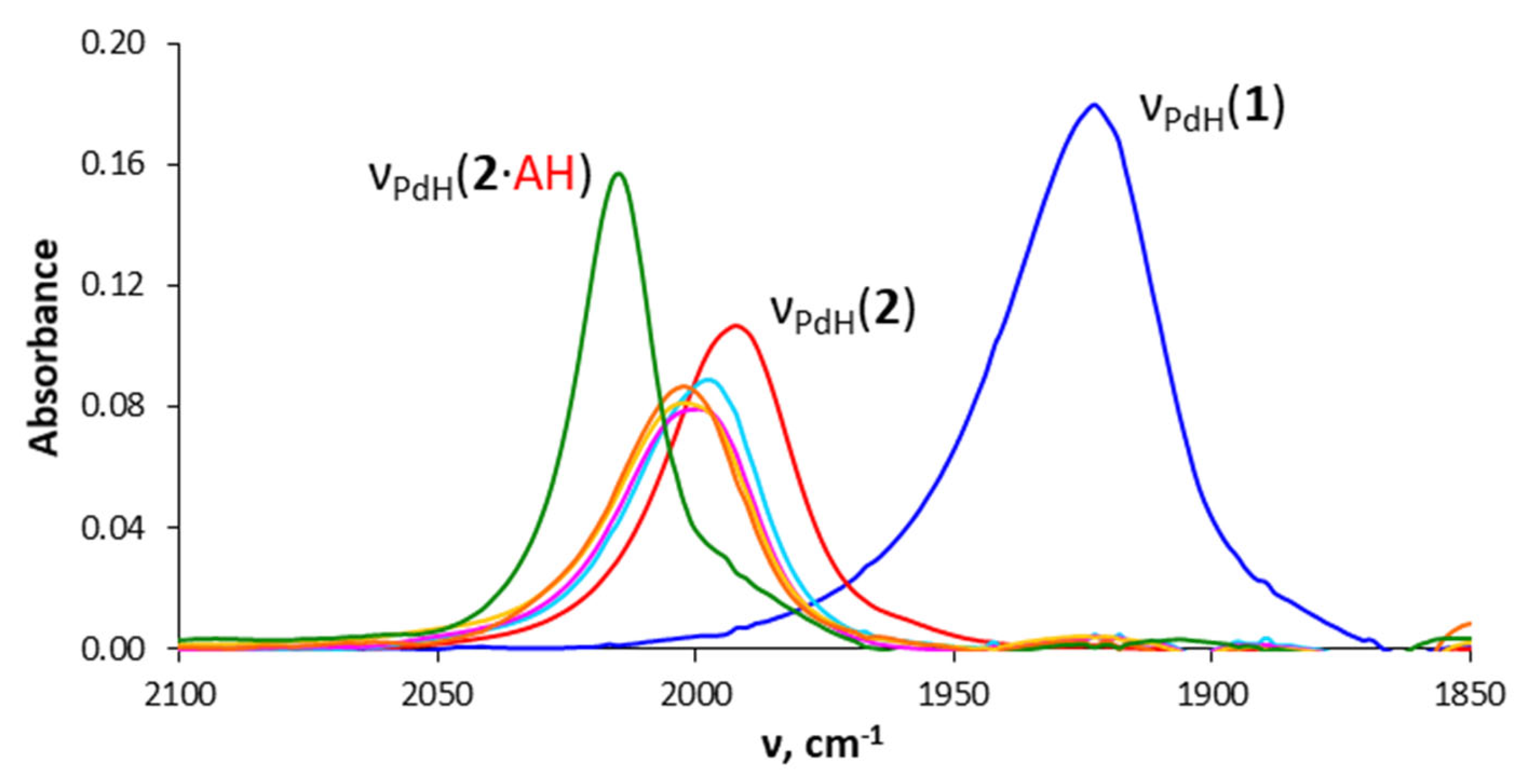
2.2. Interaction with Fluorinated Alcohols
2.3. DFT Calculations
2.4. Interaction with Formic Acid
3. Materials and Methods
- a.
- General procedure for the interaction of 1 with TfOH and HBF4 and of 2 with bases
- b.
- General procedure for the interaction of 1 with fluorinated alcohols
- c.
- General procedure for the studies of interaction of 1 with formic acid
- d.
- Formic acid monomer concentration calculation.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References and Notes
- van Koten, G.; Milstein, D. (Eds.) Organometallic Pincer Chemistry; Topics in Organometallic Chemistry; Springer: Berlin/Heidelberg, Germany, 2013; Volume 40. [Google Scholar] [CrossRef]
- Alig, L.; Fritz, M.; Schneider, S. First-Row Transition Metal (De)Hydrogenation Catalysis Based on Functional Pincer Ligands. Chem. Rev. 2019, 119, 2681–2751. [Google Scholar] [CrossRef]
- Piccirilli, L.; Lobo Justo Pinheiro, D.; Nielsen, M. Recent Progress with Pincer Transition Metal Catalysts for Sustainability. Catalysts 2020, 10, 773. [Google Scholar] [CrossRef]
- Khusnutdinova, J.R.; Milstein, D. Metal–Ligand Cooperation. Angew. Chem. Intern. Ed. 2015, 54, 12236–12273. [Google Scholar] [CrossRef]
- Belkova, N.V.; Filippov, O.A.; Osipova, E.S.; Safronov, S.V.; Epstein, L.M.; Shubina, E.S. Influence of phosphine (pincer) ligands on the transition metal hydrides reactivity. Coord. Chem. Rev. 2021, 438, 213799. [Google Scholar] [CrossRef]
- Kumar, A.; Daw, P.; Milstein, D. Homogeneous Catalysis for Sustainable Energy: Hydrogen and Methanol Economies, Fuels from Biomass, and Related Topics. Chem. Rev. 2022, 122, 385–441. [Google Scholar] [CrossRef]
- Fan, L.; Foxman, B.M.; Ozerov, O.V. N−H Cleavage as a Route to Palladium Complexes of a New PNP Pincer Ligand. Organometallics 2004, 23, 326–328. [Google Scholar] [CrossRef]
- Gregor, L.C.; Chen, C.-H.; Fafard, C.M.; Fan, L.; Guo, C.; Foxman, B.M.; Gusev, D.G.; Ozerov, O.V. Heterolytic splitting of H-X bonds at a cationic (PNP)Pd center. Dalton Trans. 2010, 39, 3195–3202. [Google Scholar] [CrossRef] [PubMed]
- Although the synthesis of (PNP)PdH (1) has been described in reference 7, its structure was determined by X-ray diffraction in this work for the first time. See CCDC 2257866 for crystallographic information.
- Raamat, E.; Kaupmees, K.; Ovsjannikov, G.; Trummal, A.; Kütt, A.; Saame, J.; Koppel, I.; Kaljurand, I.; Lipping, L.; Rodima, T.; et al. Acidities of strong neutral Brønsted acids in different media. J. Phys. Org. Chem. 2013, 26, 162–170. [Google Scholar] [CrossRef]
- Fourmond, V.; Jacques, P.-A.; Fontecave, M.; Artero, V. H2 Evolution and Molecular Electrocatalysts: Determination of Overpotentials and Effect of Homoconjugation. Inorg. Chem. 2010, 49, 10338–10347. [Google Scholar] [CrossRef]
- Fong, T.P.; Forde, C.E.; Lough, A.J.; Morris, R.H.; Rigo, P.; Rocchini, E.; Stephan, T. Synthesis of the acidic dihydrogen complexes trans-[M(H2)(CN)L2]+ and trans-[M(H2)(CNH)L2]2+ where M = Fe, Ru, Os and L = dppm, dppe, dppp, depe, and dihydrogen substitution by the trifluoromethanesulfonate anion to give trans-[Ru(OTf)(CN)L2] or trans-[Ru(OTf)(CNH)L2]OTf. J. Chem. Soc. Dalton Trans. 1999, 4475–4486. [Google Scholar] [CrossRef]
- Tshepelevitsh, S.; Kütt, A.; Lõkov, M.; Kaljurand, I.; Saame, J.; Heering, A.; Plieger, P.G.; Vianello, R.; Leito, I. On the Basicity of Organic Bases in Different Media. Eur. J. Org. Chem. 2019, 2019, 6735–6748. [Google Scholar] [CrossRef]
- Matrosov, E.I.; Tsvetkov, E.N.; Mironova, Z.N.; Malevannaya, R.A.; Kabachnik, M.I. Acid-base properties of phosphine oxides in nitromethane. Bull. Acad. Sci. USSR Chem. Sci. Div. 1975, 24, 1231–1234. [Google Scholar] [CrossRef]
- Belkova, N.V.; Epstein, L.M.; Filippov, O.A.; Shubina, E.S. Hydrogen and Dihydrogen Bonds in the Reactions of Metal Hydrides. Chem. Rev. 2016, 116, 8545–8587. [Google Scholar] [CrossRef] [PubMed]
- Estimated from the pKa value in DMSO using correlation equiation pKa(MeCN) = 12.31 + 0.98pKa(DMSO) from reference:Kuejtt, A.; Leito, I.; Kaljurand, I.; Soovaeli, L.; Vlasov, V.M.; Yagupolskii, L.M.; Koppel, I.A. A Comprehensive Self-Consistent Spectrophotometric Acidity Scale of Neutral Broensted Acids in Acetonitrile. J. Org. Chem. 2006, 71, 2829–2838. [Google Scholar] [CrossRef]
- Belkova, N.V.; Epstein, L.M.; Shubina, E.S. Dihydrogen Bonding, Proton Transfer and Beyond: What We Can Learn from Kinetics and Thermodynamics. Eur. J. Inorg. Chem. 2010, 2010, 3555–3565. [Google Scholar] [CrossRef]
- Osipova, E.S.; Belkova, N.V.; Epstein, L.M.; Filippov, O.A.; Kirkina, V.A.; Titova, E.M.; Rossin, A.; Peruzzini, M.; Shubina, E.S. Dihydrogen Bonding and Proton Transfer from MH vs. OH acids to Group 10 Metal Hydrides (tBuPCP)MH [tBuPCP = κ3-2,6-(tBu2PCH2)2C6H3; M = Ni, Pd]. Eur. J. Inorg. Chem. 2016, 2016, 1415–1424. [Google Scholar] [CrossRef]
- Filippov, O.A.; Kirkina, V.A.; Belkova, N.V.; Stoccoro, S.; Zucca, A.; Babakhina, G.M.; Epstein, L.M.; Shubina, E.S. First Example of Hydrogen Bonding to Platinum Hydride. Z. Phys. Chem. 2013, 227, 869–880. [Google Scholar] [CrossRef]
- Stirling, M.J.; Sweeney, G.; MacRory, K.; Blacker, A.J.; Page, M.I. The kinetics and mechanism of the organo-iridium-catalysed enantioselective reduction of imines. Org. Biomol. Chem. 2016, 14, 3614–3622. [Google Scholar] [CrossRef]
- Osipova, E.; Sedlova, D.; Gutsul, E.; Nelyubina, Y.; Dorovatovskii, P.; Epstein, L.; Filippov, O.; Shubina, E.; Belkova, N. Reactivity of heterobimetallic ion pairs in formic acid dehydrogenation. Organometallics 2023. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. PCCP 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. PCCP 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FA Amount, Equiv. | keff, M−1·s−1 | t of Full FA Conversion |
|---|---|---|
| 0.5 | 3.5·10−7 | 40 min |
| 1 | 3.5·10−7 | 40 min |
| 2 | 4.1·10−7 | 2.5 h |
| 5 | 9.1·10−8 | more than 24 h |
| 10 | Ca. 10% conversion after 3 days |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kirkina, V.A.; Kulikova, V.A.; Gutsul, E.I.; Gafurov, Z.N.; Sakhapov, I.F.; Yakhvarov, D.G.; Nelyubina, Y.V.; Filippov, O.A.; Shubina, E.S.; Belkova, N.V. The Role of Non-Covalent Interactions in the Reactions between Palladium Hydrido Complex with Amidoarylphosphine Pincer Ligand and Brønsted Acids. Inorganics 2023, 11, 212. https://doi.org/10.3390/inorganics11050212
Kirkina VA, Kulikova VA, Gutsul EI, Gafurov ZN, Sakhapov IF, Yakhvarov DG, Nelyubina YV, Filippov OA, Shubina ES, Belkova NV. The Role of Non-Covalent Interactions in the Reactions between Palladium Hydrido Complex with Amidoarylphosphine Pincer Ligand and Brønsted Acids. Inorganics. 2023; 11(5):212. https://doi.org/10.3390/inorganics11050212
Chicago/Turabian StyleKirkina, Vladislava A., Vasilisa A. Kulikova, Evgenii I. Gutsul, Zufar N. Gafurov, Ilias F. Sakhapov, Dmitry G. Yakhvarov, Yulia V. Nelyubina, Oleg A. Filippov, Elena S. Shubina, and Natalia V. Belkova. 2023. "The Role of Non-Covalent Interactions in the Reactions between Palladium Hydrido Complex with Amidoarylphosphine Pincer Ligand and Brønsted Acids" Inorganics 11, no. 5: 212. https://doi.org/10.3390/inorganics11050212
APA StyleKirkina, V. A., Kulikova, V. A., Gutsul, E. I., Gafurov, Z. N., Sakhapov, I. F., Yakhvarov, D. G., Nelyubina, Y. V., Filippov, O. A., Shubina, E. S., & Belkova, N. V. (2023). The Role of Non-Covalent Interactions in the Reactions between Palladium Hydrido Complex with Amidoarylphosphine Pincer Ligand and Brønsted Acids. Inorganics, 11(5), 212. https://doi.org/10.3390/inorganics11050212