Abstract
We report a straightforward alkane elimination strategy to prepare well-defined heterobimetallic Al/Mo species. Notably, the reaction of the monohydride complex of molybdenum, Cp*MoH(CO)3, with triisobutyl aluminum affords a new heterobimetallic [MoAl]2 tetranuclear compound, [Cp*Mo(CO)(µ-CO)2Al(iBu)2]2, (1), featuring a 12-membered C4O4Mo2Al2 ring in which isocarbonyls bridge the Mo and Al centers. The addition of pyridine to this complex successfully results in the dissociation of the dimer into a new discrete binuclear complex, [Cp*Mo(CO)2(µ-CO)Al(Py)(iBu)2], (2). Switching the nature of the Lewis base from pyridine to tetrahydrofuran does not lead to the THF analogue of adduct 2, but rather to a complex reaction where one of the identified products corresponds to a tetranuclear species, [Cp*Mo(CO)3(μ-CH2CH2CH2CH2O)Al(iBu)2]2, (3), featuring two bridging alkoxybutyl fragments originating from the C-O ring opening of THF. Compound 3 adds to the unusual occurrences of THF ring opening by heterobimetallic complexes, which is evocative of masked metal-only frustrated Lewis pair behavior and highlights the high reactivity of these Al/Mo assemblies.
1. Introduction
Aluminum-based heterobimetallic complexes are burgeoning as captivating entities in coordination chemistry [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15], presenting unique structures and reactivities. The distinct properties of aluminum, such as its hard Lewis acidity or its large palette of coordination modes, coupled with the diverse reactivity of transition metals, pave the way for innovative catalytic and synthetic applications [15,16,17,18,19,20]. Furthermore, aluminum is the most earth-abundant metal, which is appealing in view of developing more sustainable chemical processes.
As investigations into their applications continue, the exploration of novel Al/M combinations as well as the tuning of the steric and electronic features in these complexes are indispensable. This customization is essential for achieving tailored reactivity towards efficient and selective catalytic transformations. Yet one big limitation of this field is the limited number of synthetic routes available to access Al/M compounds. The prevailing synthetic approach commonly employed involves salt metathesis between either Al(III) halide reagents and transition metal –ate complexes, or via aluminyl Al(I) anions which emerged recently in the literature. However, these methodologies exhibit inherent limitations. Challenges include the intricate control of ligand redistribution phenomena, the difficulty to control the final species’ nuclearity, and the complications associated with isolating the desired compounds from salt co-products formed during the process. Additionally, the preparation of low-valent aluminum species is very challenging, the latter being stable only under a limited spectrum of ligand frameworks and experimental conditions [21,22,23].
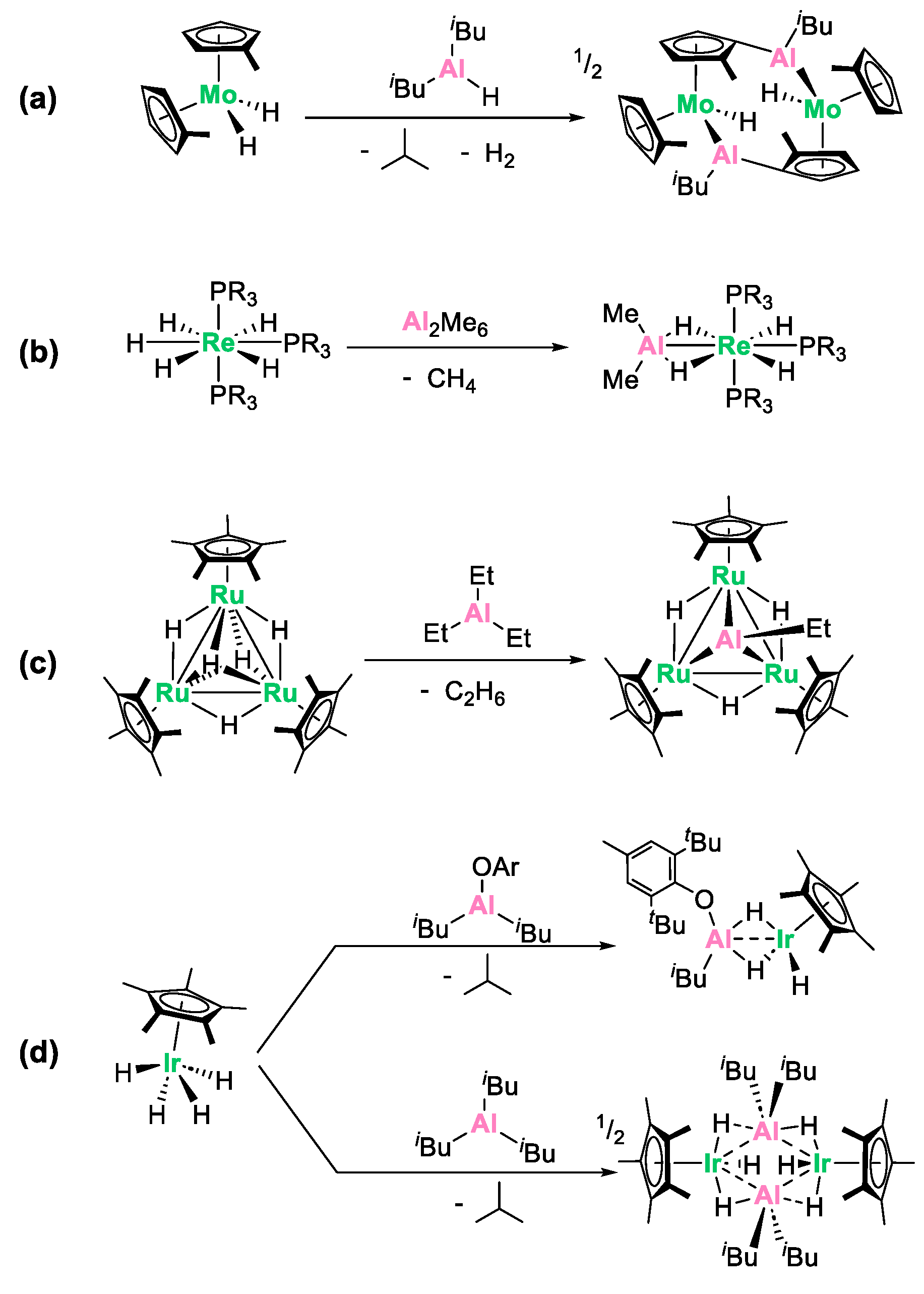
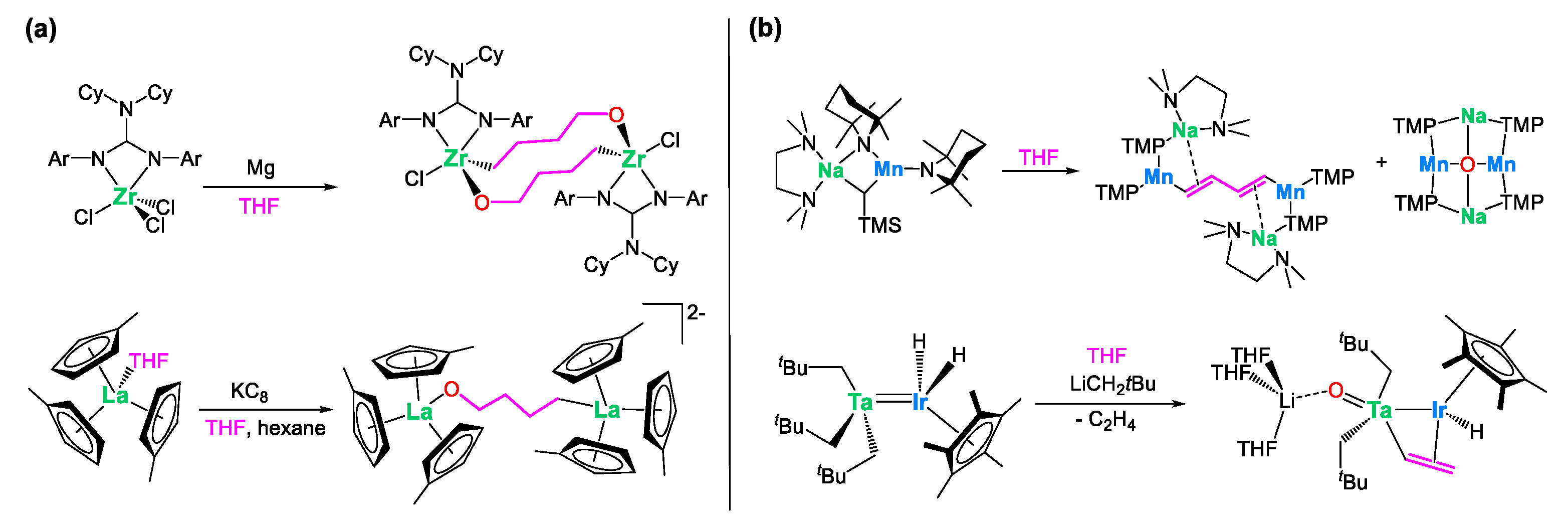
An alternative proficient strategy to synthesize heterobimetallic complexes involves the alkane elimination reaction [24,25,26,27,28] between a transition metal hydride derivative and alkyl aluminum reagents [9,10,29,30,31,32,33]. This reaction can be conceptualized as an electrophilic hydride abstraction process. Although less frequently used, this method has proved to be a powerful synthetic tool for preparing original Al/M species, often featuring metal–metal bonds. An inherent advantage of this synthetic route lies in the release of alkane volatile coproducts, which is a thermodynamic driving force, generally resulting in clean, easy to work-up reactions. Representative literature precedents are shown in Scheme 1. The group of Hey-Hawkins investigated the reaction between Cp2′MoH2 (Cp’ = C5H4Me) and HAlR2 (R = iBu or Et) [29]. This resulted in a complex reaction process involving H2 and alkane elimination, alongside C-H activation of the cyclopentadienyl ligand, leading to the formation of a pseudo “tuck-over” tetranuclear compound featuring a direct aluminum–molybdenum bond (Scheme 1a). Caulton and coworkers reported the reaction of d0 rhenium pentahydrides with Al2Me6 (Scheme 1b), quickly releasing methane at 25 °C and leading to the formation of the heterobimetallic complexes ReH4AlMe2P3 (P = PMe2Ph and PMePh2), in which the Al and Re atoms are hold at close distance (2.508(2) Å) by bridging hydrides [30]. Another typical example is the reaction of a trinuclear ruthenium pentahydride cluster which, upon treatment with trimethylaluminum, cleanly leads to the incorporation of a triply bridging ethylaluminum ligand into the Ru3 core (Scheme 1c) [31].
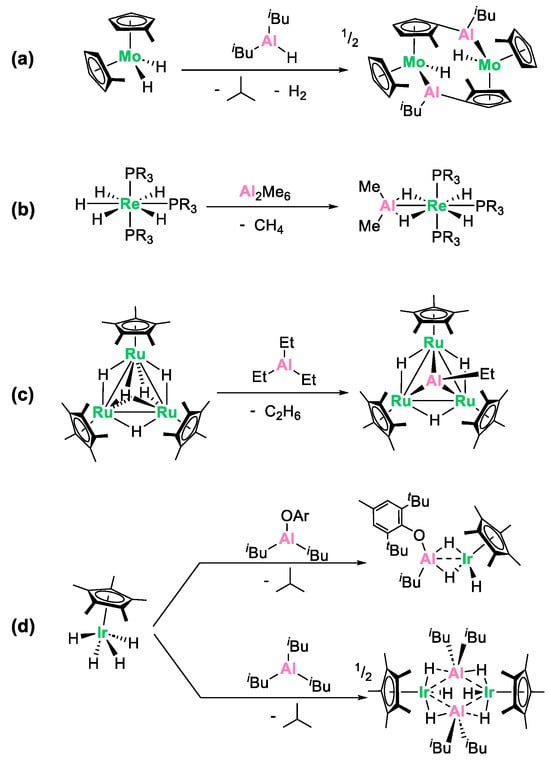
Scheme 1.
Literature precedents for the synthesis of aluminum/transition metal complexes through alkane elimination from (a) molybdenum [29], (b) rhenium [30], (c) ruthenium [31] and (d) iridium [9,10,32]) polyhydrides.
As part of our ongoing research focus on designing novel heterobimetallic architectures [26,28,34,35,36,37], we thus chose to explore the reactivity of alkylaluminum reagents with transition metal hydrides, hopping to trigger alkane elimination reactions. Prior work within our group has demonstrated the capability of an iridium polyhydride complex, Cp*IrH4, to react with isobutyl aluminum derivatives, yielding heterobimetallic assemblies. Our research reveals that the steric environment surrounding the aluminum alkyl moiety plays a crucial role in dictating the nuclearity of the resultant species. A binuclear complex is exclusively formed when employing a bulky aryloxide ligand within the coordination sphere of the aluminum metal (Scheme 1d) [9]; conversely, the absence of such bulky ligands leads to the formation of clusters with higher nuclearities [10]. Since the Cp*Mo(CO)3H complex is known to have an acidic hydride [38,39], we thought it would be an ideal candidate for our study, as part of our quest to explore more sustainable metal alternatives to iridium. The results of these investigations are detailed in the following discussion.
2. Results and Discussion
The reaction of triisobutylaluminum with complex Cp*Mo(CO)3H in n-pentane at 35 °C furnishes the tetranuclear compound [Cp*Mo(CO)(µ-CO)2Al(iBu)2]2, (1), as a brown-yellow crystalline solid in 74% isolated yield (Scheme 2).
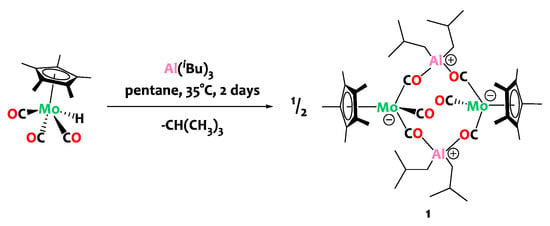
Scheme 2.
Synthesis of the tetranuclear heterobimetallic complex 1.
Compound 1 is diamagnetic. The simplicity of the 1H Nuclear Magnetic Resonance (NMR) spectrum of 1 in deuterated benzene (C6D6) indicates a symmetrical structure in solution on the NMR timescale. Specifically, the spectrum exhibits a singlet at δ = 1.93 ppm for the Cp* ligands and one set of signals for the iBu moieties with a doublet at 0.43 ppm for the methylene group, a doublet at 1.22 ppm for the methyl groups and a multiplet at 2.18 ppm corresponding to the iBu β-H. In the 13C NMR spectrum of 1, a single set of signals for the Cp* ligands on molybdenum is observed (δ = 106.0 and 11.3 ppm) and a characteristic broad signal is found for the carbonyl groups at δ = 253.4 ppm. This value is in agreement with that found in compounds featuring both terminal and bridging isocarbonyls [25,40], which most likely are in fast dynamic exchange on the NMR timescale. The Diffuse Reflectance Infrared Fourier Transform (DRIFT) spectrum of compound 1 reveals ν(CO) bands at 1961, 1917, 1663 and 1615 cm–1. The stretching modes of bridging carbonyls are assigned to the last two bands, in agreement with the literature [25,40,41,42]. The terminal ν(CO) bands are observed, on average, at slightly higher energy (2015 and 1936 cm−1) in the molybdenum precursor complex, Cp*Mo(CO)3H. This indicates a weakening of the terminal carbonyl C-O bonds in compound 1. This weakening suggests an increased backdonation from the molybdenum center in compound 1, implying an augmentation in the electron density at the formally Mo(0) site as a result of hydride loss and aluminum ligation to isocarbonyls. Note that the Mo-H vibration, found at 1796 cm−1 in Cp*Mo(CO)3H, is noticeably absent in the DRIFT spectrum of compound 1, as expected.
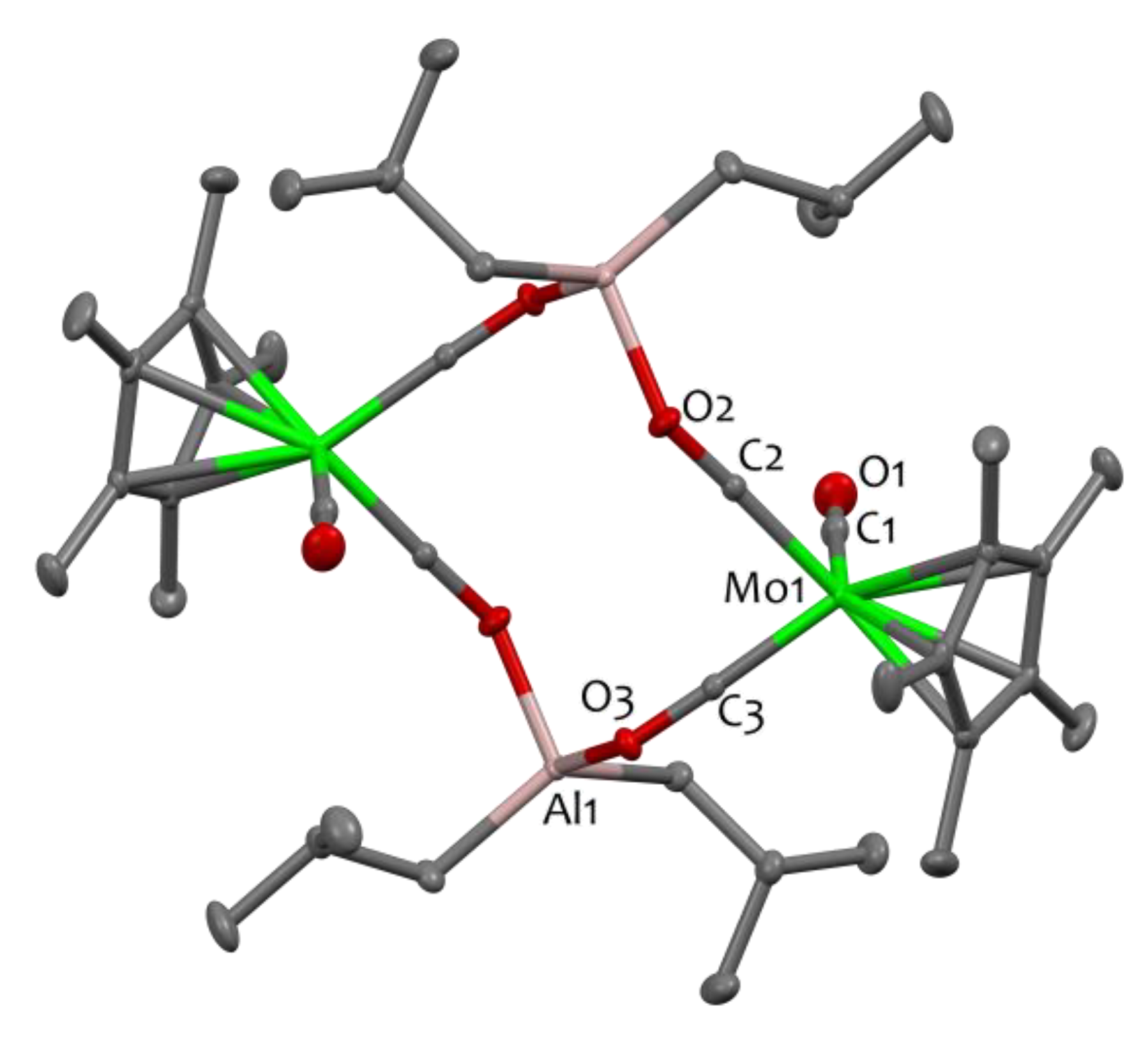
The dimeric structure of 1 is clearly revealed by single crystal X-Ray Diffraction (XRD), in which two sets of the MoAl moiety are connected through isocarbonyl coordination (Mo−CO→Al), forming a [Mo−C−O−Al−O−C−]2 twelve-membered linkage (Figure 1). Compound 1 crystallizes in the triclinic space group P-1. The molecule is placed on the inversion center, located at the center of the metallacycle. The isocarbonyl ligands are characterised by elongated C-O distances (1.216(2) Å and 1.222(2) Å) with respect to the terminal C1-O1 isocarbonyl bond (1.141(3) Å), in agreement with the literature [23,40]. The Al–O bond distances (Al1–O2 1.8562(15) Å and Al1–O3 1.8701(15) Å) are rather long, as expected, and in the range of other isocarbonyl compounds [13,23,43,44], for eg. 1.852(2) Å in [(OC)3Co(μ-H)2Al{(NDippCMe)2CH}(μ-CO)Co(CO)3] [44]. The two isocarbonyl linkages differ strongly in their Al–O–C angles: the Al1–O3–C3 angle is more acute than the Al1–O2–C2 angle (128.4(1) vs. 142.8(1), respectively). Note that deviation from linearity is a typical occurrence in such systems [25,44]. The metrical parameters for the Al(iBu) moieties are as expected [10,45] and the molybdenum atoms exhibit an approximate tetrahedral geometry.
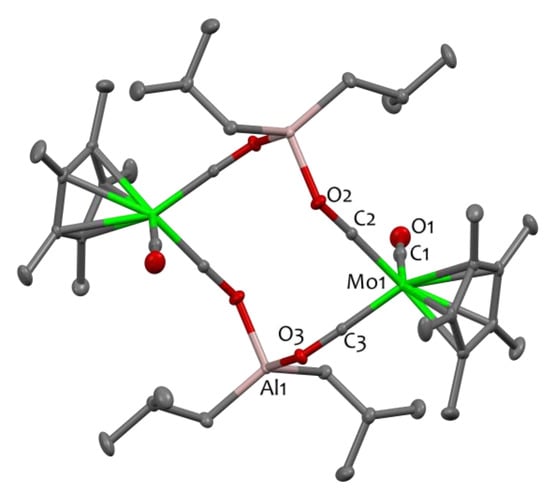
Figure 1.
Solid-state molecular structure of compound 1. Ellipsoids are represented with 30% probability. The color scheme utilized is as follows: carbon atoms are depicted in grey, oxygen atoms in red, aluminum atoms in pink, and molybdenum atoms in green. Hydrogen atoms have been omitted for clarity. Selected bond distances (Å) and angles (°) for 1: Mo1-C2 1.8785(19), Mo1-C3 1.8809(19), Mo1-C1 1.992(2), Al1-O3 1.8701(15), Al1-O2 1.8562(15), O1-C1 1.141(3), O2-C2 1.216(2), O3-C3 1.222(2), C2-Mo1-C3 85.50(8), C2-Mo1-C1 91.42(8), C3-Mo1-C1 88.99(8).
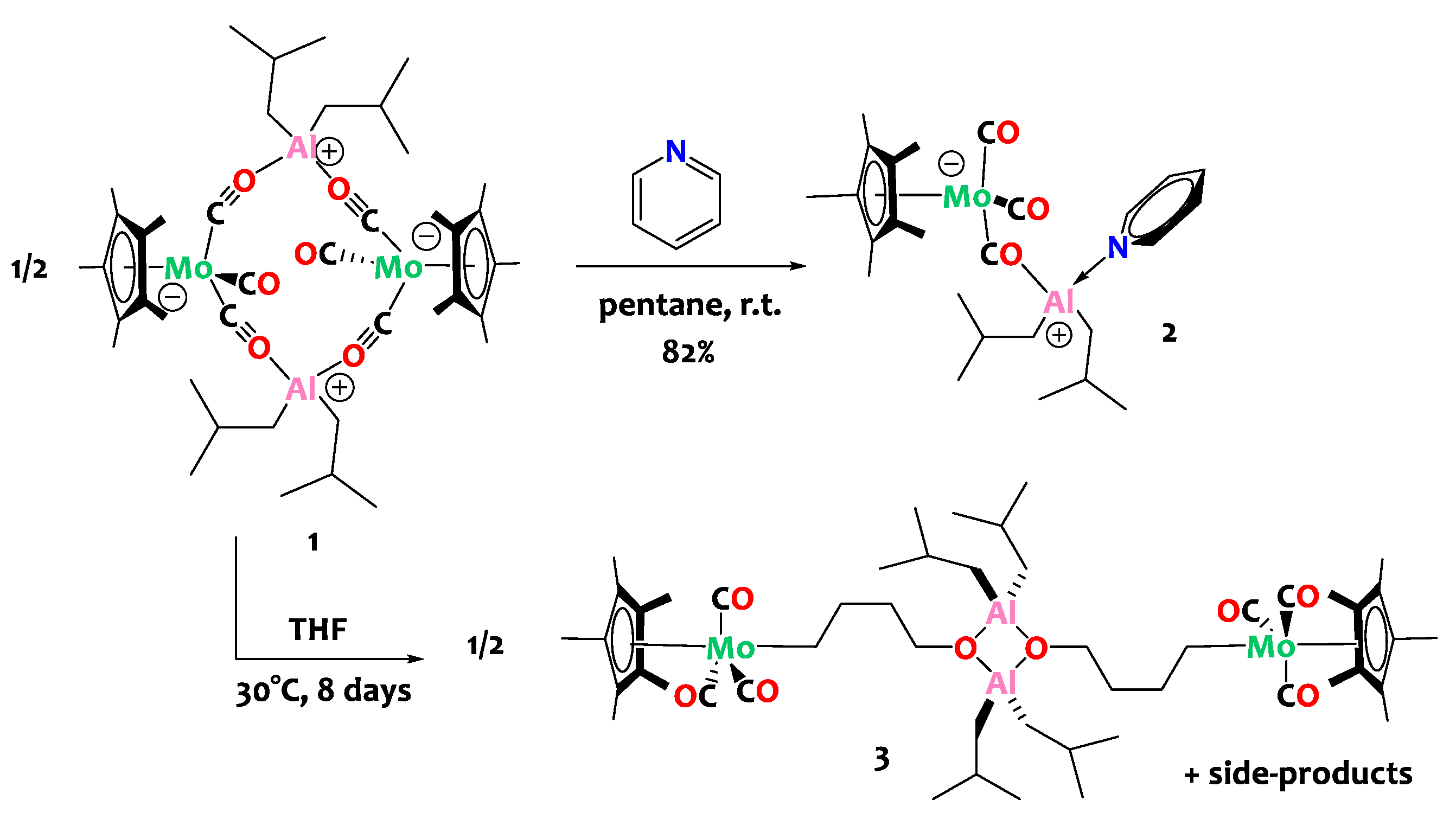
The reactivity of compound 1 towards Lewis bases is then investigated in an attempt to dissociate the tetranuclear assembly and obtain discrete binuclear complexes. Treatment of 1 with stoichiometric amounts of pyridine (1 equiv. of pyridine per Al center) leads to the clean formation of adduct [Cp*Mo(CO)2(µ-CO)Al(Py)(iBu)2], 2, in which a pyridine molecule is N-bound to the Al(III) center (Scheme 3). Note that conducting the reaction with only 0.5 equivalents of pyridine per aluminum center yields an equimolar mixture of compounds 1 and 2. Conversely, when an excess of pyridine is employed (4 equiv. or more), a mixture of novel compounds is obtained; however, their identification and isolation proved unfruitful in our hands.
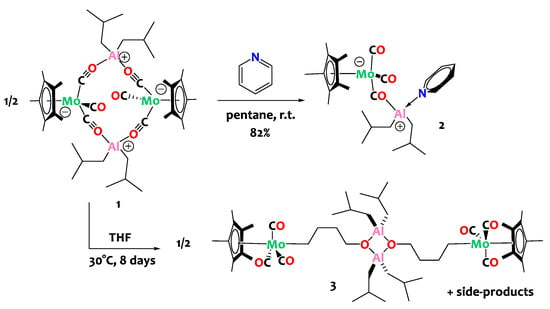
Scheme 3.
Reactivity of complex 1 with Lewis bases: pyridine (top) and tetrahydrofuran (bottom).
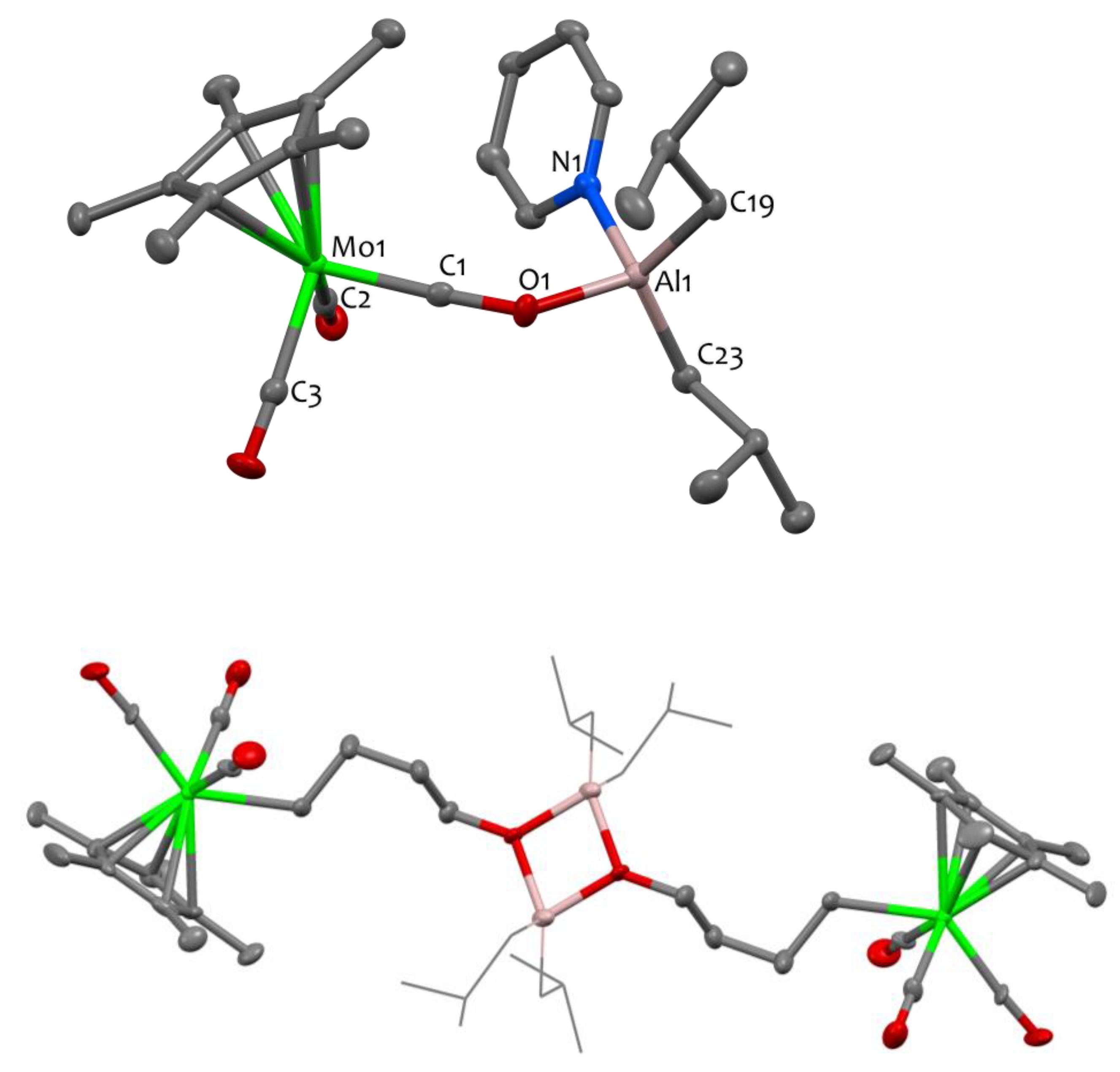
NMR and DRIFT data for compound 2 closely resemble those of compound 1, showcasing similar features, besides additional signals originating from the pyridine ligand, such as νC-H bands at 3079 cm−1, characteristic of Csp2-H bonds. The XRD structure of complex 2 is shown in Figure 2, confirming its dinuclear nature. The Al center is four-coordinated to two carbons from the isobutyl ligands, one nitrogen from the pyridine ligand and one oxygen from an isocarbonyl ligand bridging the Al and Mo sites. As in 1, the isocarbonyl C1-O1 distance (1.251(6)°Å) is elongated compared to the terminal carbonyl C2-O2 and C3-O3 bond lengths (1.163(7)°Å and 1.160(7)°Å, respectively). These carbonyl (terminal and bridging) C-O bond lengths are slightly elongated in monomer 2 vs. dimer 1. The C1-O1-Al1 angle is bent (138.8(4) °), and the Al1-O1 distance (1.836(4) Å) is rather long, in agreement with literature data for Al isocarbonyl complexes [13,23,43,44]. It should be noted that the Al1 center and the Mo1 center are separated by more than 4.6 Å, precluding any possibility of metal–metal interaction.
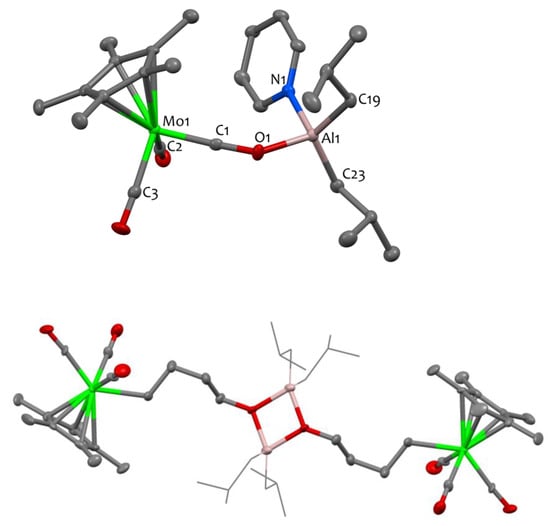
Figure 2.
Solid-state molecular structures of compounds 2 (top) and 3 (bottom). Ellipsoids are represented with 30% probability. The color scheme utilized is as follows: carbon atoms are depicted in grey, oxygen atoms in red, aluminum atoms in pink, nitrogen atoms in blue and molybdenum atoms in green. Hydrogen atoms have been omitted for clarity. In compound 3, the isobutyl moieties attached to the aluminum sites are represented in wireframe format to enhance clarity. Selected bond distances (Å) and angles (°) for 2: Mo1-C1 1.850(6), Mo1-C2 1.962(6), Mo1-C3 1.957(6), Al1-O1 1.836(4), Al1-N1 1.973(5), Al1-C19 1.958(6), Al1-C23 1.948(6), C1-Mo1-C2 87.5(2), C1-Mo1-C3 93.4(2), C3-Mo1-C2 88.2(3), O1-Al1-N1 98.64(19), O1-Al1-C23 109.6(2), C19-Al1-N1 106.8(2), C23-Al1-C19 121.9(3), C1-O1-Al1 138.8(4).
In agreement with Pearson’s Hard-Soft Acid-Base concept, the hard aluminum center prefers to bind to the hard carbonyl oxygens rather than the molybdenum center through unsupported metal–metal bonding (as in Scheme 1a). This suggests that the Mo site in [Cp*Mo(CO)3]- is a weak nucleophile. Unfortunately, attempts to photochemically labilize the CO ligands in 1 or 2 to generate metal–metal bonded species—a strategy that proved effective in some studies [46]—did not succeeded in our hands, leading to intractable mixtures of unidentified species.
The analogous THF adduct could not be isolated since compound 1 is found to be unstable in THF at 30 °C. When conducting the reaction in pentane using stoichiometric quantities of tetrahydrofuran (1 equiv. of THF per aluminum center), a reaction intermediate, presumably corresponding to the THF adduct A, is detected by 1H NMR spectroscopy. However, this intermediate is unstable in these experimental conditions and cannot be isolated. It is only when the reaction is carried out in neat THF (i.e., with a large excess) that the main decomposition product can be isolated in satisfactory yields (80%). The latter is successfully identified by X-ray crystallography as a tetranuclear Al/Mo compound, namely [Cp*Mo(CO)3(μ-CH2CH2CH2CH2O)Al(iBu)2]2, 3 (Scheme 3, bottom), that contains an alkoxy-alkyl bridge derived from the ring opening of THF. Although the quality of the single crystal XRD data for 3 is insufficient to discuss the metrical parameters in great detail (Figure 2—bottom), it is satisfactory enough to prove the atom connectivity and confirm the ring opening of THF. Note that the formation of the bridging C4H8O moiety is also unequivocally validated through 1D and 2D NMR spectroscopy. Specifically, the 1H NMR spectrum displays a triplet at δ = 3.89 ppm (CH2-O), two quintets at δ = 1.98 and 1.87 ppm corresponding to the methylene groups situated at the center of the carbon chain, and a triplet at δ = 1.04 ppm for the CH2-Mo unit.
The cleavage of tetrahydrofuran by monometallic organometallic reagents, resulting in analogous alkoxy-bridged structures, has been documented in a few instances in the literature [47,48]. Such a transformation typically occurs under stringent reducing conditions, as outlined in Scheme 4a. Furthermore, the Mulvey group [49] and our own group [27] have independently demonstrated that the collaborative action of two distinct metals can, in some cases, facilitate a more pronounced degradation of THF. This process results in the removal of the ether oxygen, accompanied by the generation of vinyl or butadiene anions, as depicted in Scheme 4b. The observed cleavage of tetrahydrofuran promoted by compound 1, which occurs under mild conditions without the requirement of external strong reducing agents, is thus noteworthy.
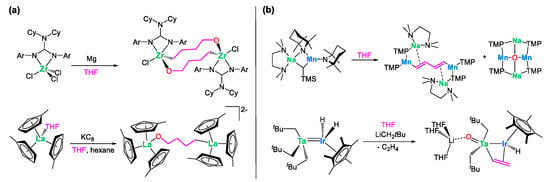
Scheme 4.
Relevant literature precedents for the cleavage of THF by (a) monometallic complexes (M = Zr [47]; La [48]) under stringent reducing conditions and (b) heterobimetallic (M/M’ = Na/Mn [49]; Ta/Ir [27]) complexes.
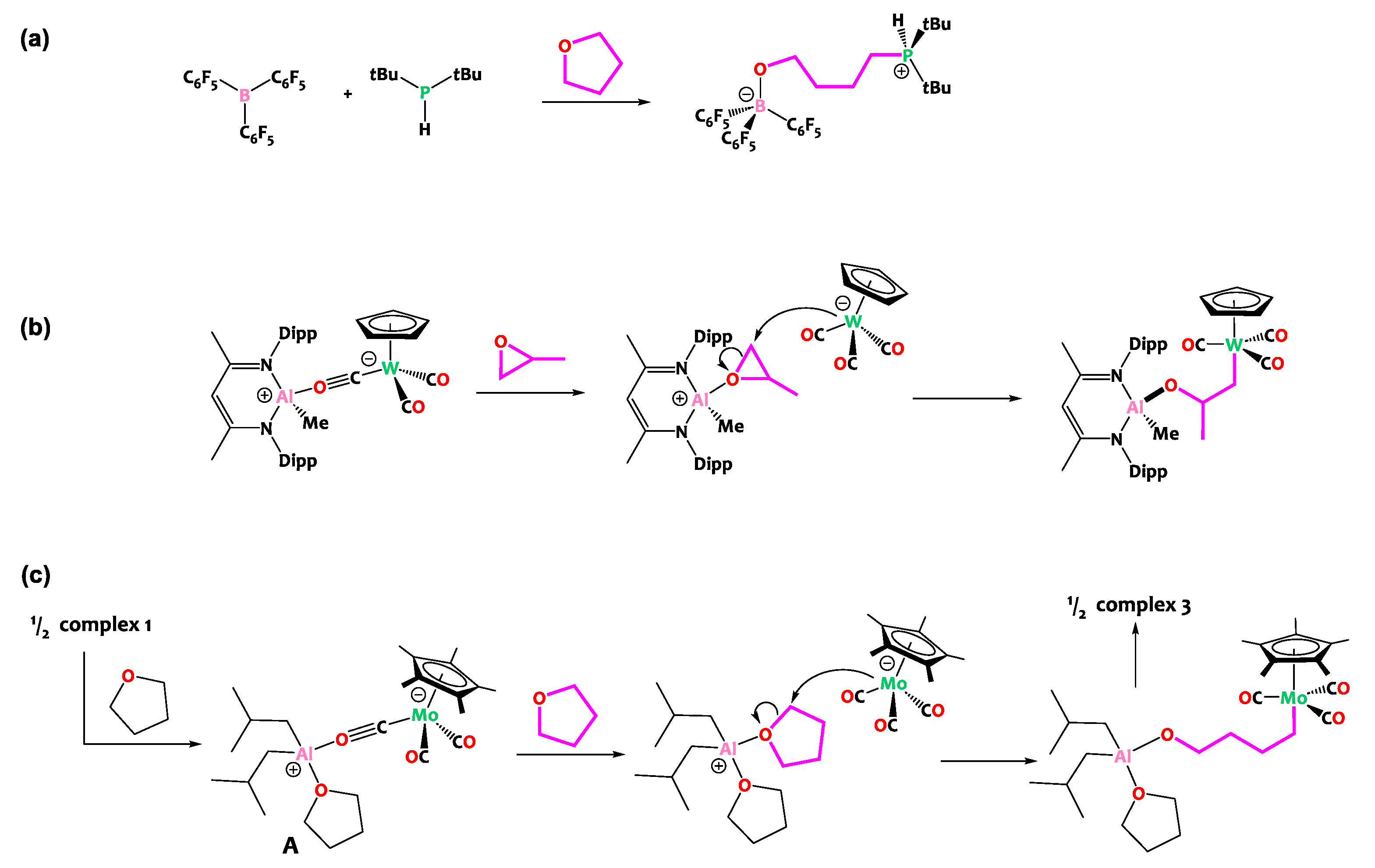
In fact, the reactivity observed here is more evocative of the ring opening of cyclic ethers by Frustrated Lewis Pairs (FLPs, Scheme 5a), wherein ether activation by a Lewis acid is followed by nucleophilic attack. In these examples, the nucleophile is typically a phosphine, amine, carbene or halide and the Lewis acidic partner is classically a main group element (typically boron) [50,51,52,53,54,55,56,57] though examples involving hard metal centers such as titanium [58], yttrium [59], zirconium [60], tantalum [61], thorium [62] or uranium [63] have been described as well. These THF ring-opening reactions share conceptual similarities with the reactivity observed herein. Interestingly, during the preparation of this manuscript, Mankad and coworkers reported a tungsten aluminum isocarbonyl species closely related to our investigation (Scheme 5) [64]. Their study revealed that this [Al](μ-CO)[W] complex behaves as a masked Metal-Only FLP [65,66,67] and is able to ring open propylene oxide with a regioselectivity which confirms that it occurs via the nucleophilic attack of the liberated [CpW(CO)3]− anion. Note that the same group reported an Al/Fe sister compound active in the ring opening of THF, yet in this case THF cleavage is proposed to occur via a radical-pair mechanism as a result of the homolysis of the Al-Fe bond [68] By analogy with these literature precedents, we propose a similar mechanism for the cleavage of THF promoted by compound 1, as depicted in Scheme 5c.
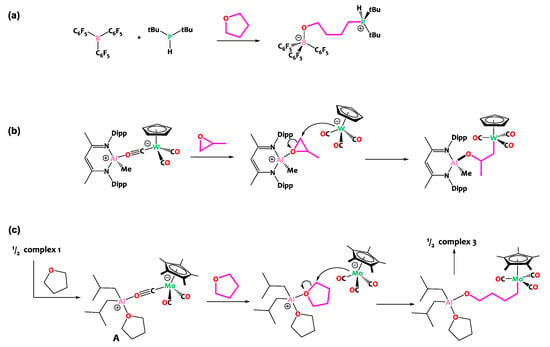
Scheme 5.
(a) THF ring opening by classical Frustrated Lewis Pairs [54,56]. (b) Recently reported ring opening of propylene oxide by a related aluminum tungsten isocarbonyl complex [64], behaving as a masked Metal-Only Frustrated Lewis Pair. (c) Proposed mechanism for THF ring opening promoted by complex 1, by analogy.
3. Materials and Methods
3.1. General Considerations
Unless otherwise stated, all reactions were conducted under an atmosphere of purified argon (<1 ppm O2/H2O) using either an MBraun inert atmosphere glovebox, or standard Schlenk line techniques. Glassware and cannulas were preconditionned in an oven at approximately 100 °C for a minimum of 12 h before utilization. THF and n-pentane were purified by a solvent purification system consisting in passing through an activated alumina column, dried over Na/benzophenone, vacuum-transferred to a storage vessel and freeze-pump-thaw degassed before usage. Hexane and C6D6 were dried over Na/benzophenone, vacuum-transferred to a storage vessel and freeze-pump-thaw degassed before usage. Pyridine was dried over CaH2, vacuum-transferred to a storage vessel and freeze-pump-thaw degassed prior to use. Compound Cp*Mo(CO)3H was synthesized according to the literature procedure [69]. All other reagents were acquired from commercial sources and used as received.
3.2. Analytical Methods
Diffuse Reflectance Infrared Fourier Transform (DRIFT) spectroscopy: prior to analysis, the samples were meticulously prepared under an argon atmosphere in a glovebox and were diluted in potassium bromide (KBr) salt to enhance measurement accuracy. Subsequently, the power was introduced and sealed under argon in a DRIFT cell equipped with KBr windows. The analyses were conducted using a Nicolet 670 FT-IR spectrometer (Thermo Fisher Scientific, Waltham, USA), and the obtained spectra were processed using the OMNIC software (V 7.2).
Elemental analyses were conducted under inert atmosphere at Mikroanalytisches Labor Pascher, Remagen, Germany to determine the elemental composition of the samples.
Solution NMR spectra were acquired using Bruker AV-300, AVQ-400 and AV-500 spectrometers (Bruker Biospin, Billerica, USA). Chemical shifts were determined by referencing the peaks of residual solvents to an external tetramethylsilane (TMS) standard set at 0.00 ppm. To confirm 1H and 13C NMR spectra assignments, the samples were subjected to further analysis through 1H−1H correlation spectroscopy (COSY) and 1H−13C heteronuclear single quantum coherence (HSQC) experiments. The NMR data is reported in the following format: chemical shift (δ) [multiplicity, relative integral, assignment, coupling constant(s) J (Hz) if any]. Multiplicity was denoted as follows: s = singlet, d = doublet, t = triplet, m = multiplet, or combinations thereof.
X-ray structural determinations: while a suitable single crystal for complex 1 and 2 could be selected, only multi-crystals were found for complex 3 which explains the relatively bad statistics. However, its structure could be solved and was confirmed by other experimental techniques. The data collection for these complexes was carried out on Xcalibur Gemini kappa-geometry diffractometer equipped with an Atlas CCD detector and a Copper X-ray source (λ = 1.54184 Å). Intensities were collected at 150 K by means of the CrysalisPro software (Version 1.171.40.67a) [70]. Reflection indexing, refinement of unit-cell parameters, Lorentz-polarization correction, peak integration and background determination were carried out with the CrysalisPro software [70]. An analytical absorption correction was applied using the modeled faces of the crystal [71]. The resulting set of hkl was used for structure solution and refinement. The structures were solved with the ShelXT [72] structure solution program using the intrinsic phasing solution method, with Olex2 [73] serving as the graphical interface. Model refinement was conducted using ShelXL version 2018/3 [74] via least-squares minimization. CCDC 2,330,941 to 2,330,943 contain the supplementary crystallographic data for this paper. These data are provided free of charge by the Cambridge Crystallographic Data Centre. Tables reporting crystallographic parameters for compounds 1, 2 and 3 are provided in the Supplementary Materials.
3.3. Syntheses
Synthesis of [Cp*Mo(CO)(µ-CO)2Al(iBu)2]2, 1. Triisobutylaluminum (329.0 mg, 1.66 mmol) was dissolved in 15 mL of n-pentane. To this colorless solution, 10 mL of a reddish-brown n-pentane solution of Cp*Mo(CO)3H (526.9 mg, 1.67 mmol) were added. The resulting deep red-brown solution was stirred and heated at 35 °C for 2 days, resulting in a brown-yellow color. Then, the volatiles were removed under vacuum yielding a yellow-brown powder. This solid was dissolved in 20 mL of a n-pentane:hexanes (17:3) mixture, filtered and stored at −40 °C in a freezer inside the argon glovebox for 3 days. This yielded 560 mg of single yellow block crystals that were recovered by filtration and dried under vacuum (74% isolated yield). Single crystals suitable for X-ray diffraction were grown similarly. 1H-NMR (500 MHz, C6D6, 293 K) δ 2.18 (m, 4H, CHiBu), 1.93 (s, 30H, Cp*), 1.22 (d, 24H, CH3-iBu, 3JHH = 6.4 Hz), 0.43 (d, 4H, CH2-iBu, 3JHH = 7.0 Hz). 13C{1H}-NMR (125 MHz, C6D6, 293 K) δ 253.4 (µ-CO), 106.0 (CCp*), 28.33 (CH3-iBu), 26.0 (CHiBu), 21.4 (CH2-iBu), 11.3 (CH3-Cp*). DRIFTS (KBr cell) σ 2947 (s, νCH), 2916 (s, νCH), 2860 (s, νCH), 1961 (s, COterminal stretching), 1917 (s, COterminal stretching), 1663 (s, µ-CO stretching), 1615 (s, µ-CO stretching). Elemental analysis calcd (%) for C42H66Al2Mo2O6: C 55.26, H 7.29. Found: C 55.37, H 7.39.
Synthesis of [Cp*Mo(CO)2(µ-CO)Al(Py)(iBu)2], 2. A 1 mL colorless pentane solution of pyridine (23.3 mg, 0.295 mmol, 2 eq.) was added dropwise into a 9 mL yellow pentane solution of 1 (135.5 mg, 0.148 mmol, 1 eq.), at room temperature. Instantly, the solution turned to a deep orange color. The reactional medium was stirred at room temperature for 15 min. Then, pentane volatiles were removed under vacuum yielding a yellow powder. This solid was dissolved in the minimum amount of pentane (3 mL), filtered, and stored at −40 °C for 18 h. This yielded compound 2 as yellow needle-shaped crystals that were recovered by filtration and dried under vacuum (130 mg, 82% yield). Single crystals suitable for X-ray diffraction were grown similarly. 1H NMR (500 MHz, C6D6, 293 K) δ 8.13 (dt, 2H, CHortho-Py, 3JHH = 4.9 Hz, 5JHH = 1.6 Hz), 6.73 (tt, 1H, CHpara-Py, 3JHH = 7.9 Hz, 5JHH = 1.6 Hz), 6.51 (m, 2H, CHmeta-Py, 3JHH = 6.4 Hz), 2.09 (m, 2H, CHiBu), 2.09 (s, 15H, CH3Cp*), 1.17 (d, 12H, CH3-iBu,3JHH = 6.6 Hz), 0.38 (d, 4H, CH2-iBu, 3JHH = 7.0 Hz). 13C{1H} NMR (125 MHz, C6D6, 293 K) δ 146.7 (CHortho-Py), 141.5 (CHpara-Py), 125.8 (CHmeta-Py), 103.3 (CCp*), 28.4 (CH3-iBu), 26.3 (CHiBu), 21.3 (CH2-iBu), 11.8 (CH3-Cp*). DRIFTS (KBr cell) σ 3079 (s, νCH), 2967 (s, νCH), 2788 (s, νCH), 1950 (s, COterminal stretching), 1816 (s, COterminal stretching), 1615 (s, µ-CO stretching). Elemental analysis calcd (%) for C26H38AlMoNO3: C 58.31, H 7.15, N 2.62. Found: C 57.97, H 7.10, N 2.54.
Formation of [Cp*Mo(CO)3(μ-CH2CH2CH2CH2O)Al(iBu)2]2, 3. Compound 1 (176.0 mg, 0.19 mmol) was dissolved in 7 mL of THF. The resulting reddish solution was stirred for 8 days at 30 °C yielding a deep brown solution. THF volatiles were removed under vacuum yielding a brown solid. This solid was dissolved in the minimum amount of pentane (3 mL), filtered, and stored at −40 °C for 16 h, yielding orange crystals that were recovered by filtration and dried under vacuum (160 mg, 0.15 mmol, 80% yield). Single crystals suitable for X-ray diffraction were grown similarly. Compound 3 was unambiguously identified and characterized by XRD and NMR spectroscopy. Nevertheless, despite numerous efforts and attempts, its separation from reaction co-products proved difficult, which hindered the acquisition of a satisfactory elemental analysis. 1H NMR (500 MHz, C6D6, 293 K) δ 3.89 (t, 4H, CH2-O, 3JHH = 7.8 Hz), 2.19 (m, 4H, CHiBu), 1.98 (quintet, 4H, 3JHH = 7.3 Hz, CH2), 1.87 (quintet, 4H, 3JHH = 8.2 Hz, CH2), 1.49 (s, 30H, CH3Cp*), 1.26 (d, 24H, CH3-iBu, 3JHH = 6.5 Hz), 1.04 (t, 4H, CH2-Mo, 3JHH = 8.8 Hz), 0.40 (d, 8H, CH2-iBu, 3JHH = 7.1 Hz). 13C{1H} NMR (125 MHz, C6D6, 293 K) δ 146.66 (CHortho-Py), 141.47 (CHpara-Py), 125.84 (CHmeta-Py), 103.33 (CCp*), 28.37 (CH3-iBu), 26.28 (CHiBu), 21.33 (CH2-iBu), 11.78 (CH3-Cp*). 13C{1H}-NMR (125 MHz, C6D6, 293 K): δ 231.9 (CO), 104.0 (CCp*), 64.0 (CH2-O), 39.6 (CH2), 32.1 (CH2), 28.8 (CH3-iBu), 26.3 (CHiBu), 24.9 (CH2-iBu), 12.4 (CH2-Mo), 10.0 (CH3-Cp*).
4. Conclusions
We have shown that molybdenum-aluminum heterobimetallic complexes can be prepared in high yields from an alkane elimination reaction between the acidic molybdenum hydride complex Cp*Mo(CO)3H and triisobutylaluminum. The new complex [{Al(iBu)2[Mo(Cp*)(CO)3]}2, (1), is tetranuclear in the solid state and exhibits two Al(iBu)2 units which are linked by two bridging isocarbonyl groups to two [Mo(Cp*)(CO)3] anions. This results in the formation of a centrosymmetric, 12-membered C4O4Mo2Al2 ring. Interestingly enough, we have demonstrated the possibility to dissociate this tetranuclear dimer 1 into a discrete and well-defined binuclear complex, [Cp*Mo(CO)2(µ-CO)Al(Py)(iBu)2], (2), using pyridine as a Lewis base. Switching to an oxygenated Lewis base such as THF under mild conditions drastically changed the related chemistry where the THF analogous adduct of 2 could not be isolated and evolved into a mixture of species. Gratifyingly, we identified the major compound resulting from the reaction of 1 with THF as being a tetranuclear Al2Mo2 complex, [Cp*Mo(CO)3(μ-CH2CH2CH2CH2O)Al(iBu)2]2, (3). This complex features a bridging alkoxybutyl fragment bridging the Al and the Mo centers, originating from the ring opening of THF. Such reactivity is quite unexpected, as no external reducing agents or aggressive experimental conditions were used, and therefore adds to the very rare occurrences of THF scission by heterobimetallic complexes. While the exact mechanism remains unidentified, it is reminiscent to that of a masked metal-only frustrated Lewis pair. This highlights the high reactivity of these Al/Mo assemblies, which hold promise for future investigations. Note, however, that these species do not feature unsupported direct Mo–Al interactions. To get around this limitation, we are currently exploring alternative strategies to prepare molybdenum-based heterobimetallic binuclear species in the absence of carbonyl ligands, wishing to augment the metal–metal interactions in the resulting compounds.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/inorganics12030072/s1, Figures S1–S11: NMR spectra for compounds 1–3; Figures S12 and S13: DRIFT spectra; Table S1: Crystallographic parameters for compounds 1, 2 and 3.
Author Contributions
C.C. conceptualized the work; L.E. performed the synthesis and the spectroscopic characterization of the complexes; E.J. performed the XRD studies; C.C. and C.T. supervised the work; L.E. wrote the experimental section; C.C. wrote the manuscript; E.J., C.T. and L.E. corrected the manuscript; C.C. managed the project and acquired funding. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the CNRS MOMENTUM program.
Data Availability Statement
CCDC 2330941 to 2330943 contain the supplementary crystallographic data for this paper. These data are provided free of charge by the Cambridge Crystallographic Data Centre. Other data can be obtained upon reasonable request directly from the authors.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Mears, K.L.; Stennett, C.R.; Taskinen, E.K.; Knapp, C.E.; Carmalt, C.J.; Tuononen, H.M.; Power, P.P. Molecular Complexes Featuring Unsupported Dispersion-Enhanced Aluminum-Copper and Gallium-Copper Bonds. J. Am. Chem. Soc. 2020, 142, 19874–19878. [Google Scholar] [CrossRef]
- McManus, C.; Hicks, J.; Cui, X.; Zhao, L.; Frenking, G.; Goicoechea, J.M.; Aldridge, S. Coinage Metal Aluminyl Complexes: Probing Regiochemistry and Mechanism in the Insertion and Reduction of Carbon Dioxide. Chem. Sci. 2021, 12, 13458–13468. [Google Scholar] [CrossRef]
- Fernández, S.; Fernando, S.; Planas, O. Cooperation towards Nobility: Equipping First-Row Transition Metals with an Aluminium Sword. Dalton Trans. 2023, 52, 14259–14286. [Google Scholar] [CrossRef]
- Lachguar, A.; Pichugov, A.V.; Neumann, T.; Dubrawski, Z.; Camp, C. Cooperative Activation of Carbon-Hydrogen Bonds by Heterobimetallic Systems. Dalton Trans. 2023, 53, 1393–1409. [Google Scholar] [CrossRef]
- Weiss, J.; Stetzkamp, D.; Nuber, B.; Fischer, R.A.; Boehme, C.; Frenking, G. [(η5-C5Me5)Al-Fe(CO)4] Synthesis, Structure, and Bonding. Angew. Chem. Int. Ed. 1997, 36, 70–72. [Google Scholar] [CrossRef]
- Gorgas, N.; Stadler, B.; White, A.J.P.; Crimmin, M.R. Vinylic C–H Activation of Styrenes by an Iron–Aluminum Complex. J. Am. Chem. Soc. 2024, 146, 4252–4259. [Google Scholar] [CrossRef]
- Gorgas, N.; White, A.J.P.; Crimmin, M.R. Cooperative C-H Bond Activation by a Low-Spin D6Iron-Aluminum Complex. J. Am. Chem. Soc. 2022, 144, 8770–8777. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.Y.; Schwamm, R.J.; Hill, M.S.; Mahon, M.F.; McMullin, C.L.; Rajabi, N.A. Ambiphilic Al−Cu Bonding. Angew. Chem. Int. Ed. 2021, 60, 14390–14393. [Google Scholar] [CrossRef] [PubMed]
- Escomel, L.; Del Rosal, I.; Maron, L.; Jeanneau, E.; Veyre, L.; Thieuleux, C.; Camp, C. Strongly Polarized Iridiumδ−-Aluminumδ+Pairs: Unconventional Reactivity Patterns Including CO2 Cooperative Reductive Cleavage. J. Am. Chem. Soc. 2021, 143, 4844–4856. [Google Scholar] [CrossRef] [PubMed]
- Escomel, L.; Soulé, N.; Robin, E.; Del Rosal, I.; Maron, L.; Jeanneau, E.; Thieuleux, C.; Camp, C. Rational Preparation of Well-Defined Multinuclear Iridium-Aluminum Polyhydride Clusters and Comparative Reactivity. Inorg. Chem. 2022, 61, 5715–5730. [Google Scholar] [CrossRef] [PubMed]
- Sinhababu, S.; Radzhabov, M.R.; Telser, J.; Mankad, N.P. Cooperative Activation of CO2 and Epoxide by a Heterobinuclear Al-Fe Complex via Radical Pair Mechanisms. J. Am. Chem. Soc. 2022, 144, 3210–3221. [Google Scholar] [CrossRef]
- Hicks, J.; Mansikkamäki, A.; Vasko, P.; Goicoechea, J.M.; Aldridge, S. A Nucleophilic Gold Complex. Nat. Chem. 2019, 11, 237–241. [Google Scholar] [CrossRef]
- Lai, Q.; Bhuvanesh, N.; Ozerov, O.V. Unexpected B/Al Transelementation within a Rh Pincer Complex. J. Am. Chem. Soc. 2020, 142, 20920–20923. [Google Scholar] [CrossRef]
- Cammarota, R.C.; Lu, C.C. Tuning Nickel with Lewis Acidic Group 13 Metalloligands for Catalytic Olefin Hydrogenation. J. Am. Chem. Soc. 2015, 137, 12486–12489. [Google Scholar] [CrossRef]
- Butler, M.J.; Crimmin, M.R. Magnesium, Zinc, Aluminium and Gallium Hydride Complexes of the Transition Metals. Chem. Commun. 2017, 53, 1348–1365. [Google Scholar] [CrossRef] [PubMed]
- Takaya, J.; Iwasawa, N. Synthesis, Structure, and Catalysis of Palladium Complexes Bearing a Group 13 Metalloligand: Remarkable Effect of an Aluminum-Metalloligand in Hydrosilylation of CO2. J. Am. Chem. Soc. 2017, 139, 6074–6077. [Google Scholar] [CrossRef] [PubMed]
- Morisako, S.; Watanabe, S.; Ikemoto, S.; Muratsugu, S.; Tada, M.; Yamashita, M. Synthesis of A Pincer-IrV Complex with A Base-Free Alumanyl Ligand and Its Application toward the Dehydrogenation of Alkanes. Angew. Chem.-Int. Ed. 2019, 58, 15031–15035. [Google Scholar] [CrossRef]
- Hara, N.; Aso, K.; Li, Q.Z.; Sakaki, S.; Nakao, Y. C2-Selective Alkylation of Pyridines by Rhodium–Aluminum Complexes. Tetrahedron 2021, 95, 132339. [Google Scholar] [CrossRef]
- Hara, N.; Saito, T.; Semba, K.; Kuriakose, N.; Zheng, H.; Sakaki, S.; Nakao, Y. Rhodium Complexes Bearing PAlP Pincer Ligands. J. Am. Chem. Soc. 2018, 140, 7070–7073. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.Z.; Hara, N.; Semba, K.; Nakao, Y.; Sakaki, S. Rh Complex with Unique Rh–Al Direct Bond: Theoretical Insight into Its Characteristic Features and Application to Catalytic Reaction via σ-Bond Activation. Top. Catal. 2022, 65, 392–417. [Google Scholar] [CrossRef]
- Hicks, J.; Vasko, P.; Goicoechea, J.M.; Aldridge, S. The Aluminyl Anion: A New Generation of Aluminium Nucleophile. Angew. Chem. Int. Ed. 2021, 60, 1702–1713. [Google Scholar] [CrossRef]
- Coles, M.P.; Evans, M.J. The Emerging Chemistry of the Aluminyl Anion. Chem. Commun. 2023, 59, 503–519. [Google Scholar] [CrossRef]
- Kong, R.Y.; Crimmin, M.R. 1strow Transition Metal Aluminylene Complexes: Preparation, Properties and Bonding Analysis. Dalton Trans. 2021, 50, 7810–7817. [Google Scholar] [CrossRef]
- Butovskii, M.V.; Döring, C.; Bezugly, V.; Wagner, F.R.; Grin, Y.; Kempe, R. Molecules Containing Rare-Earth Atoms Solely Bonded by Transition Metals. Nat. Chem. 2010, 2, 741–744. [Google Scholar] [CrossRef]
- Sobaczynski, A.P.; Obenauf, J.; Kempe, R. Alkane Elimination Reactions between Yttrium Alkyls and Tungsten Hydrides. Eur. J. Inorg. Chem. 2014, 2014, 1211–1217. [Google Scholar] [CrossRef]
- Lassalle, S.; Jabbour, R.; Schiltz, P.; Berruyer, P.; Todorova, T.K.; Veyre, L.; Gajan, D.; Lesage, A.; Thieuleux, C.; Camp, C. Metal–Metal Synergy in Well-Defined Surface Tantalum–Iridium Heterobimetallic Catalysts for H/D Exchange Reactions. J. Am. Chem. Soc. 2019, 141, 19321–19335. [Google Scholar] [CrossRef]
- Lassalle, S.; Petit, J.; Falconer, R.L.; Hérault, V.; Jeanneau, E.; Thieuleux, C.; Camp, C. Reactivity of Tantalum/Iridium and Hafnium/Iridium Alkyl Hydrides with Alkyl Lithium Reagents: Nucleophilic Addition, Alpha-H Abstraction, or Hydride Deprotonation? Organometallics 2022, 41, 1675–1687. [Google Scholar] [CrossRef]
- Ye, C.Z.; Del Rosal, I.; Boreen, M.A.; Ouellette, E.T.; Russo, D.R.; Maron, L.; Arnold, J.; Camp, C. A Versatile Strategy for the Formation of Hydride-Bridged Actinide-Iridium Multimetallics. Chem. Sci. 2022, 14, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Stender, M.; Oesen, H.; Blaurock, S.; Hey-Hawkins, E. Synthese und Molekülstruktur von [{Cp’(μ-η1:η5-C5H3Me)Mo(μ-AlRH)}2] (Cp’ = C5H4Me, R = iBu, Et). Z. Anorg. Allg. Chem. 2001, 627, 980–984. [Google Scholar] [CrossRef]
- Skupiński, W.A.; Huffman, J.C.; Bruno, J.W.; Caulton, K.G. Dinuclear Elimination from Rhenium Hydrides and AlMe3: Rhenium/Aluminum Polyhydrides. J. Am. Chem. Soc. 1984, 106, 8128–8136. [Google Scholar] [CrossRef]
- Ohashi, M.; Matsubara, K.; Iizuka, T.; Suzuki, H. Trinuclear Ruthenium Polyhydride Complexes with a Triply Bridging Ligand: [{(η5-C5Me5)Ru}3(μ3-M)(μ-H)3(μ3-H)] (M = Li, MgiPr, and ZnEt) and [{(H5-C5Me5)Ru}3 (μ3-M)(μ-H)3] (M = AlEt and GaMe). Angew. Chem.-Int. Ed. 2003, 42, 937–940. [Google Scholar] [CrossRef]
- Golden, J.T.; Peterson, T.H.; Holland, P.L.; Bergman, R.G.; Andersen, R.A. Adduct Formation and Single and Double Deprotonation of Cp*(PMe3)Ir(H)2 with Main Group Metal Alkyls and Aryls: Synthesis and Structure of Three Novel Ir-Al and Ir-Mg Heterobimetallics. J. Am. Chem. Soc. 1998, 120, 223–224. [Google Scholar] [CrossRef]
- Isaac, C.J.; Miloserdov, F.M.; Pécharman, A.F.; Lowe, J.P.; McMullin, C.L.; Whittlesey, M.K. Structure and Reactivity of [Ru-Al] and [Ru-Sn] Heterobimetallic PPh3-Based Complexes. Organometallics 2022, 41, 2716–2730. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.; Quadrelli, E.A.; Camp, C. Lability of Ta-NHC Adducts as a Synthetic Route towards Heterobimetallic Ta/Rh Complexes. Dalton Trans. 2020, 49, 3120–3128. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.; Moneuse, R.; Petit, J.; Pavard, P.-A.A.; Dardun, V.; Rivat, M.; Schiltz, P.; Solari, M.; Jeanneau, E.; Veyre, L.; et al. Early/Late Heterobimetallic Tantalum/Rhodium Species Assembled Through a Novel Bifunctional NHC-OH Ligand. Chem.-Eur. J. 2018, 24, 4361–4370. [Google Scholar] [CrossRef] [PubMed]
- Lassalle, S.; Jabbour, R.; Del Rosal, I.; Maron, L.; Fonda, E.; Veyre, L.; Gajan, D.; Lesage, A.; Thieuleux, C.; Camp, C. Stepwise Construction of Silica-Supported Tantalum/Iridium Heteropolymetallic Catalysts Using Surface Organometallic Chemistry. J. Catal. 2020, 392, 287–301. [Google Scholar] [CrossRef]
- Del Rosal, I.; Lassalle, S.; Dinoi, C.; Thieuleux, C.; Maron, L.; Camp, C. Mechanistic Investigations via DFT Support the Cooperative Heterobimetallic C-H and O-H Bond Activation across TaIr Multiple Bonds. Dalton Trans. 2021, 50, 504–510. [Google Scholar] [CrossRef]
- Weberg, R.T.; Norton, J.R. Kinetic and Thermodynamic Acidity of Hydrido Transition-Metal Complexes. 6. Interstitial Hydrides. J. Am. Chem. Soc. 1990, 112, 1105–1108. [Google Scholar] [CrossRef]
- Morris, R.H. Estimating the Acidity of Transition Metal Hydride and Dihydrogen Complexes by Adding Ligand Acidity Constants. J. Am. Chem. Soc. 2014, 136, 1948–1959. [Google Scholar] [CrossRef]
- Liddle, S.T.; Gardner, B.M. Reactions of [Y(BDI)(I)2(THF)] [BDI = {HC(CMeNAr)2}-, Ar = 2,6-Diisopropylphenyl] with Na[M(Cp)(CO)3] (M = Cr, W): X-Ray Crystal Structures of [{Y(BDI)[Cr(Cp)(CO)3]2(THF)}2] and [W(Cp)(CO)3][Na(THF)2]. J. Organomet. Chem. 2009, 694, 1581–1585. [Google Scholar] [CrossRef]
- Arleth, N.; Gamer, M.T.; Köppe, R.; Pushkarevsky, N.A.; Konchenko, S.N.; Fleischmann, M.; Bodensteiner, M.; Scheer, M.; Roesky, P.W. The Approach to 4d/4f-Polyphosphides. Chem. Sci. 2015, 6, 7179–7184. [Google Scholar] [CrossRef]
- Reinfandt, N.; Schoo, C.; Dütsch, L.; Köppe, R.; Konchenko, S.N.; Scheer, M.; Roesky, P.W. Synthesis of Unprecedented 4d/4f-Polypnictogens. Chem.-Eur. J. 2021, 27, 3974–3978. [Google Scholar] [CrossRef]
- Abdalla, J.A.B.; Riddlestone, I.M.; Tirfoin, R.; Phillips, N.; Bates, J.I.; Aldridge, S. Al-H σ-Bond Coordination: Expanded Ring Carbene Adducts of AlH3 as Neutral Bi- and Tri-Functional Donor Ligands. Chem. Commun. 2013, 49, 5547–5549. [Google Scholar] [CrossRef]
- Abdalla, J.A.B.; Riddlestone, I.M.; Turner, J.; Kaufman, P.A.; Tirfoin, R.; Phillips, N.; Aldridge, S. Coordination and Activation of Al-H and Ga-H Bonds. Chem.-Eur. J. 2014, 20, 17624–17634. [Google Scholar] [CrossRef]
- Dardun, V.; Escomel, L.; Jeanneau, E.; Camp, C. On the Alcoholysis of Alkyl-Aluminum(Iii) Alkoxy-NHC Derivatives: Reactivity of the Al-Carbene Lewis Pair versus Al-Alkyl. Dalton Trans. 2018, 47, 10429–10433. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.L.; Lukens, W.W.; Lu, C.C.; Arnold, J. Photochemical Route to Actinide-Transition Metal Bonds: Synthesis, Characterization and Reactivity of a Series of Thorium and Uranium Heterobimetallic Complexes. J. Am. Chem. Soc. 2014, 136, 3647–3654. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.; Schulten, C.; Nembenna, S.; Stasch, A. Synthesis and Crystal Structures of Bulky Guanidinato Zirconium(IV) and Hafnium(IV) Chloride Complexes. J. Chem. Crystallogr. 2012, 42, 866–870. [Google Scholar] [CrossRef]
- Woen, D.H.; Huh, D.N.; Ziller, J.W.; Evans, W.J. Reactivity of Ln(II) Complexes Supported by (C5H4Me)1- Ligands with THF and PhSiH3: Isolation of Ring-Opened, Bridging Alkoxyalkyl, Hydride, and Silyl Products. Organometallics 2018, 37, 3055–3063. [Google Scholar] [CrossRef]
- Mulvey, R.E.; Blair, V.L.; Clegg, W.; Kennedy, A.R.; Klett, J.; Russo, L. Cleave and Capture Chemistry Illustrated through Bimetallic-Induced Fragmentation of Tetrahydrofuran. Nat. Chem. 2010, 2, 588–591. [Google Scholar] [CrossRef] [PubMed]
- Kronig, S.; Theuergarten, E.; Holschumacher, D.; Bannenberg, T.; Daniliuc, C.G.; Jones, P.G.; Tamm, M. Dihydrogen Activation by Frustrated Carbene-Borane Lewis Pairs: An Experimental and Theoretical Study of Carbene Variation. Inorg. Chem. 2011, 50, 7344–7359. [Google Scholar] [CrossRef] [PubMed]
- Holschumacher, D.; Bannenberg, T.; Hrib, C.G.; Jones, P.G.; Tamm, M. Heterolytic Dihydrogen Activation by a Frustrated Carbene–Borane Lewis Pair. Angew. Chem. Int. Ed. 2008, 47, 7428–7432. [Google Scholar] [CrossRef]
- Geier, S.J.; Stephan, D.W. Lutidine/B(C6F5)3: At the Boundary of Classical and Frustrated Lewis Pair Reactivity. J. Am. Chem. Soc. 2009, 131, 3476–3477. [Google Scholar] [CrossRef] [PubMed]
- Krachko, T.; Nicolas, E.; Ehlers, A.W.; Nieger, M.; Slootweg, J.C. Ring-Opening of Epoxides Mediated by Frustrated Lewis Pairs. Chem. Eur. J. 2018, 24, 12669–12677. [Google Scholar] [CrossRef] [PubMed]
- Birkmann, B.; Voss, T.; Geier, S.J.; Ullrich, M.; Kehr, G.; Erker, G.; Stephan, D.W. Frustrated Lewis Pairs and Ring-Opening of THF, Dioxane, and Thioxane. Organometallics 2010, 29, 5310–5319. [Google Scholar] [CrossRef]
- Yolsal, U.; Horton, T.A.R.; Wang, M.; Shaver, M.P. Cyclic Ether Triggers for Polymeric Frustrated Lewis Pair Gels. J. Am. Chem. Soc. 2021, 143, 12980–12984. [Google Scholar] [CrossRef] [PubMed]
- Welch, G.C.; Prieto, R.; Dureen, M.A.; Lough, A.J.; Labeodan, O.A.; Höltrichter-Rössmann, T.; Stephan, D.W. Reactions of Phosphines with Electron Deficient Boranes. Dalton Trans. 2009, 1559–1570. [Google Scholar] [CrossRef] [PubMed]
- Wallach, C.; Geitner, F.S.; Fässler, T.F. FLP-Type Nitrile Activation and Cyclic Ether Ring-Opening by Halo-Borane Nonagermanide-Cluster Lewis Acid–Base Pairs. Chem. Sci. 2021, 12, 6969–6976. [Google Scholar] [CrossRef]
- Bortoluzzi, M.; Bresciani, G.; Marchetti, F.; Pampaloni, G.; Zacchini, S. Synthesis and Structural Characterization of Mixed Halide–N,N-Diethylcarbamates of Group 4 Metals, Including a Case of Unusual Tetrahydrofuran Activation. New J. Chem. 2017, 41, 1781–1789. [Google Scholar] [CrossRef]
- Liddle, S.T.; Arnold, P.L. Synthesis and Characterisation of Yttrium Complexes Supported by the Beta-Diketiminate Ligand {ArNC(CH3)CHC(CH3)NAr}- (Ar = 2,6-iPr2C6H3). Dalton Trans. 2007, 3305–3313. [Google Scholar] [CrossRef]
- Breen, T.L.; Stephan, D.W. Substitution or Nucleophilic Attack by Phosphines on ZrCl4(THF)2. Inorg. Chem. 1992, 31, 4019–4022. [Google Scholar] [CrossRef]
- Marchetti, F.; Pampaloni, G.; Zacchini, S. Reactivity of Niobium and Tantalum Pentahalides with Cyclic Ethers and the Isolation and Characterization of Intermediates in the Polymerization of Tetrahydrofuran. Inorg. Chem. 2008, 47, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Travia, N.E.; Monreal, M.J.; Scott, B.L.; Kiplinger, J.L. Thorium-Mediated Ring-Opening of Tetrahydrofuran and the Development of a New Thorium Starting Material: Preparation and Chemistry of ThI4(DME)2. Dalton Trans. 2012, 41, 14514–14523. [Google Scholar] [CrossRef] [PubMed]
- Avens, L.R.; Barnhart, D.M.; Burns, C.J.; Mckee, S.D. Uranium-Mediated Ring Opening of Tetrahydrofuran. Crystal Structure of UI2(OCH2CH2CH2CH2I)2(Ph3PO)2. Inorg. Chem. 1996, 35, 537–539. [Google Scholar] [CrossRef] [PubMed]
- Sinhababu, S.; Singh, R.P.; Radzhabov, M.R.; Kumawat, J.; Ess, D.H.; Mankad, N.P. Coordination-Induced O-H/N-H Bond Weakening by a Redox Non-Innocent, Aluminum-Containing Radical. Nat. Commun. 2024, 15, 1315. [Google Scholar] [CrossRef] [PubMed]
- Campos, J. Dihydrogen and Acetylene Activation by a Gold(I)/Platinum(0) Transition Metal Only Frustrated Lewis Pair. J. Am. Chem. Soc. 2017, 139, 2944–2947. [Google Scholar] [CrossRef]
- Bauer, J.; Braunschweig, H.; Dewhurst, R.D. Metal-Only Lewis Pairs with Transition Metal Lewis Bases. Chem. Rev. 2012, 112, 4329–4346. [Google Scholar] [CrossRef]
- Navarro, M.; Campos, J. Bimetallic Frustrated Lewis Pairs. Adv. Organomet. Chem. 2021, 75, 95–148. [Google Scholar] [CrossRef]
- Sinhababu, S.; Mankad, N.P. Diverse Thermal and Photochemical Reactivity of an Al-Fe Bonded Heterobimetallic Complex. Organometallics 2022, 41, 1917–1921. [Google Scholar] [CrossRef]
- Nolan, S.P.; Hoff, C.D.; Landrum, J.T. Synthesis and Thermochemistry of HMo(CO)3C5Me5; Comparison of Cyclopentadienyl and Pentamethylcyclopentadienyl Ligands. J. Organomet. Chem. 1985, 282, 357–362. [Google Scholar] [CrossRef]
- Rigaku Oxford Diffraction. CrysAlisPro Software System; Rigaku Corporation: Oxford, UK, 2019. [Google Scholar]
- Clark, R.C.; Reid, J.S. The Analytical Calculation of Absorption in Multifaceted Crystals. Acta Crystallogr. Sect. A Found. Crystallogr. 1995, 51, 887–897. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT-Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. A Found. Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
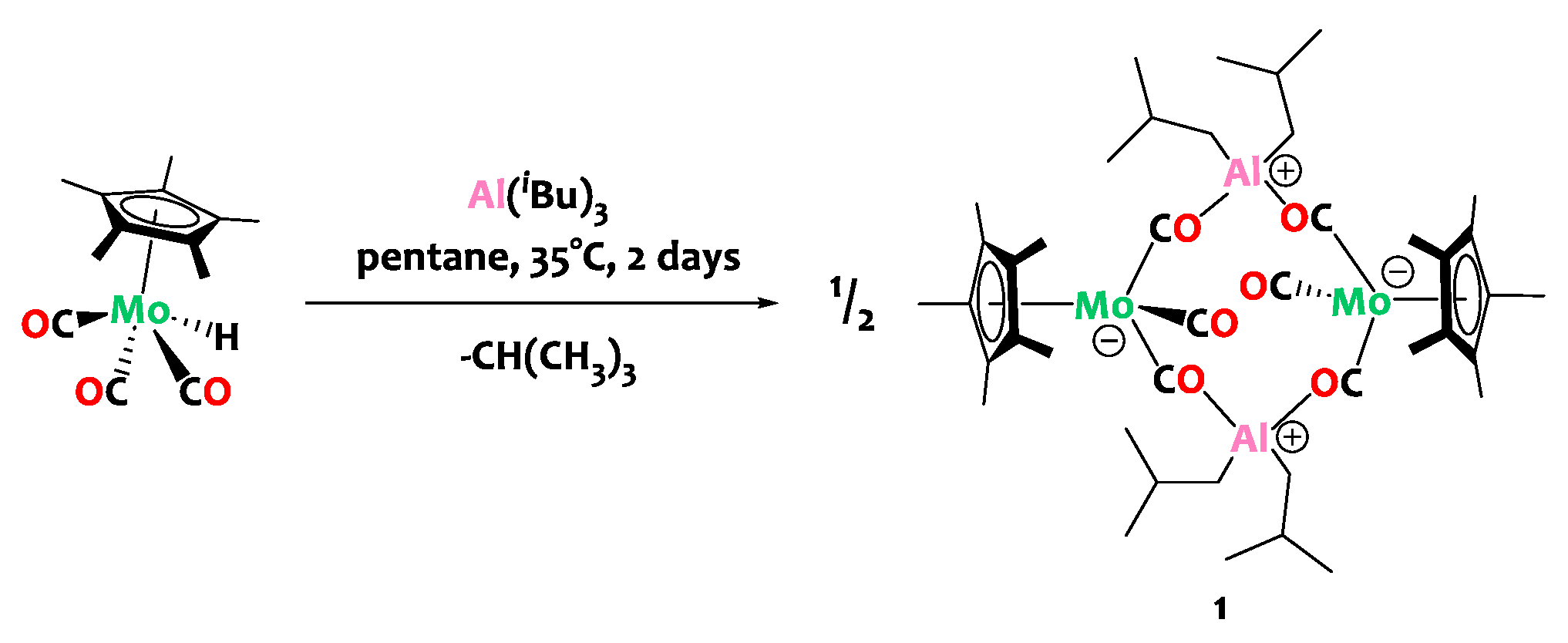
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).