
Insight into the Structure and Redox Chemistry of [Carbonatotetraamminecobalt(III)] Permanganate and Its Monohydrate as Co-Mn-Oxide Catalyst Precursors of the Fischer-Tropsch Synthesis

 , , , , ,
, , , , , 
Abstract
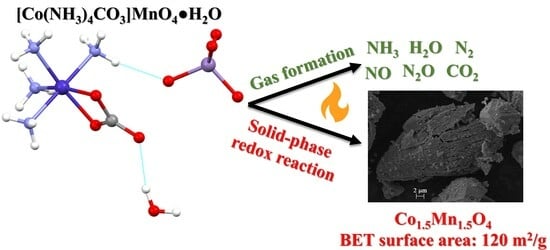
1. Introduction
2. Results and Discussion
2.1. Preparation and Properties of Compounds 1 and 2
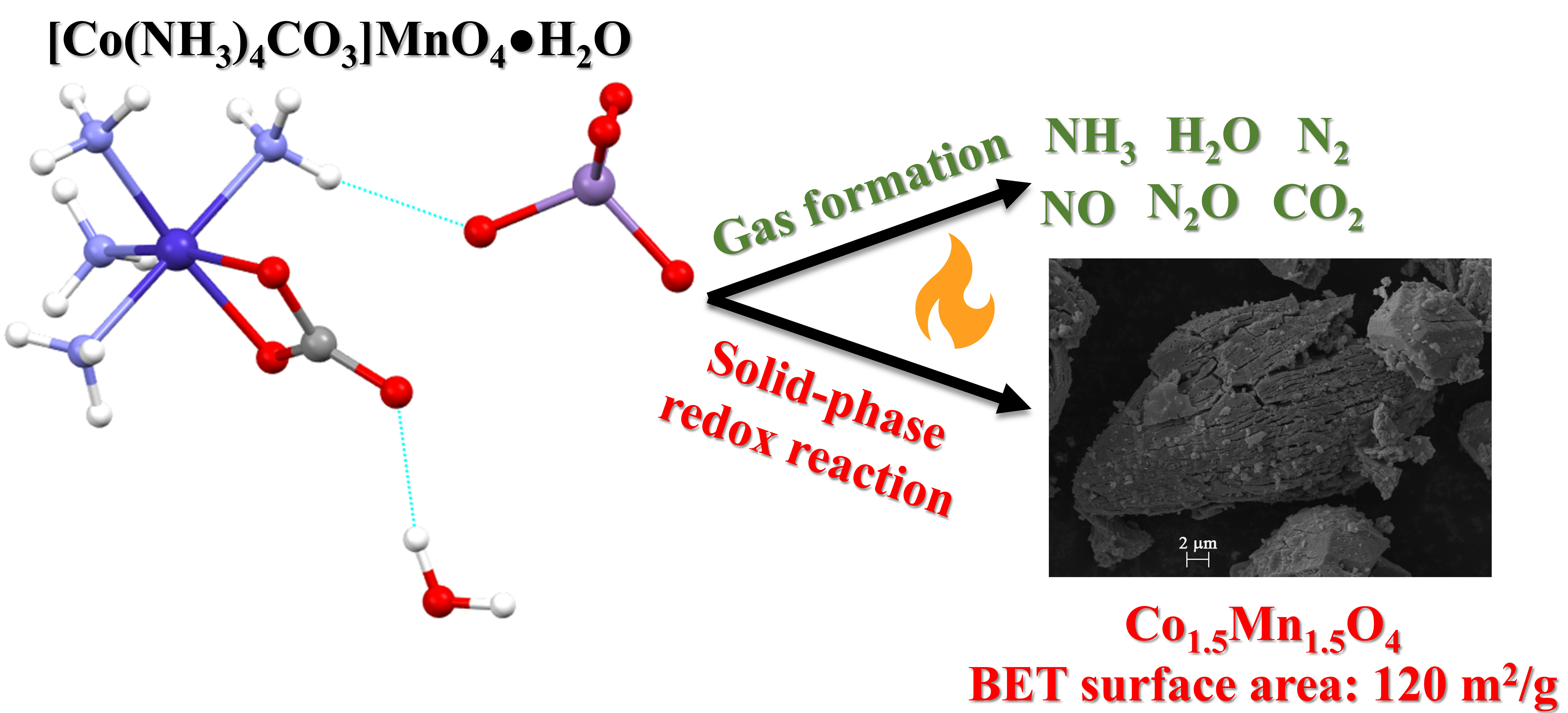
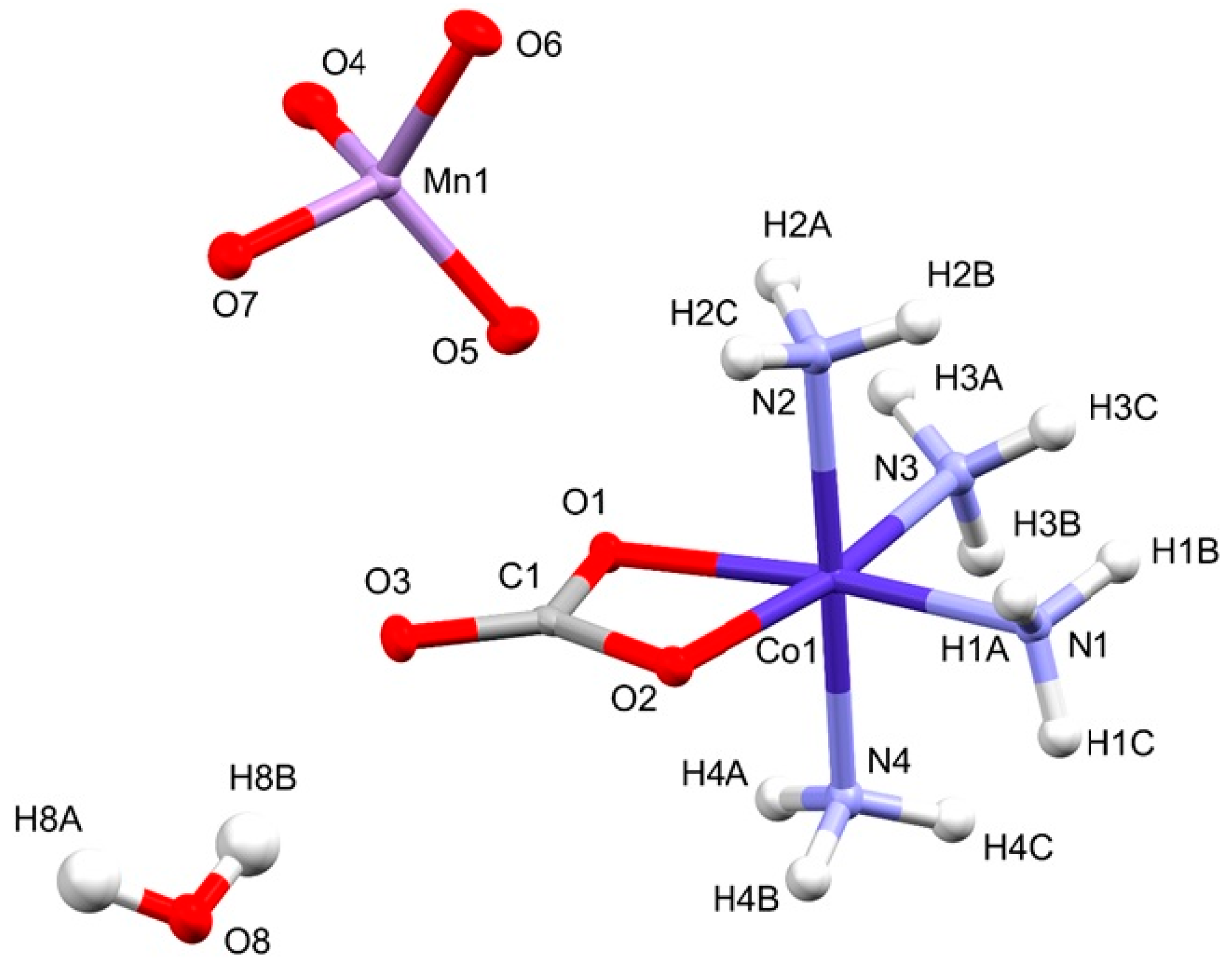
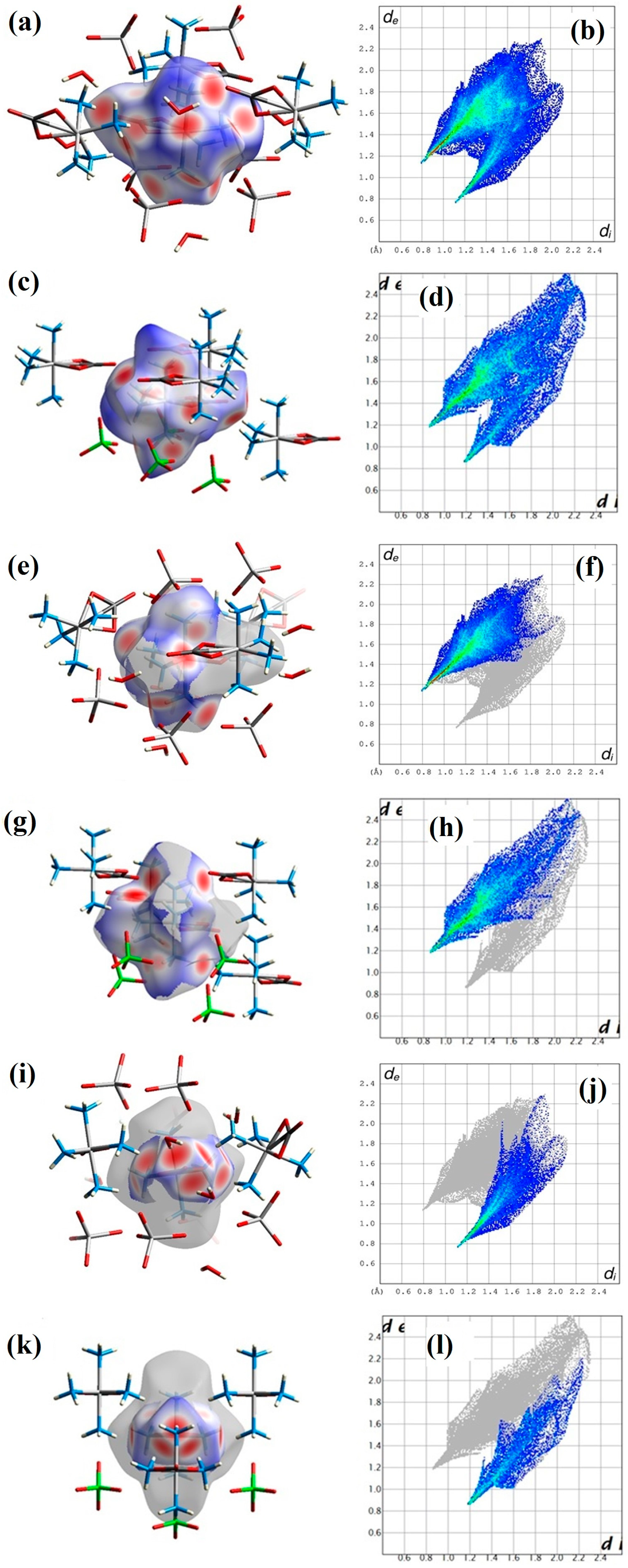
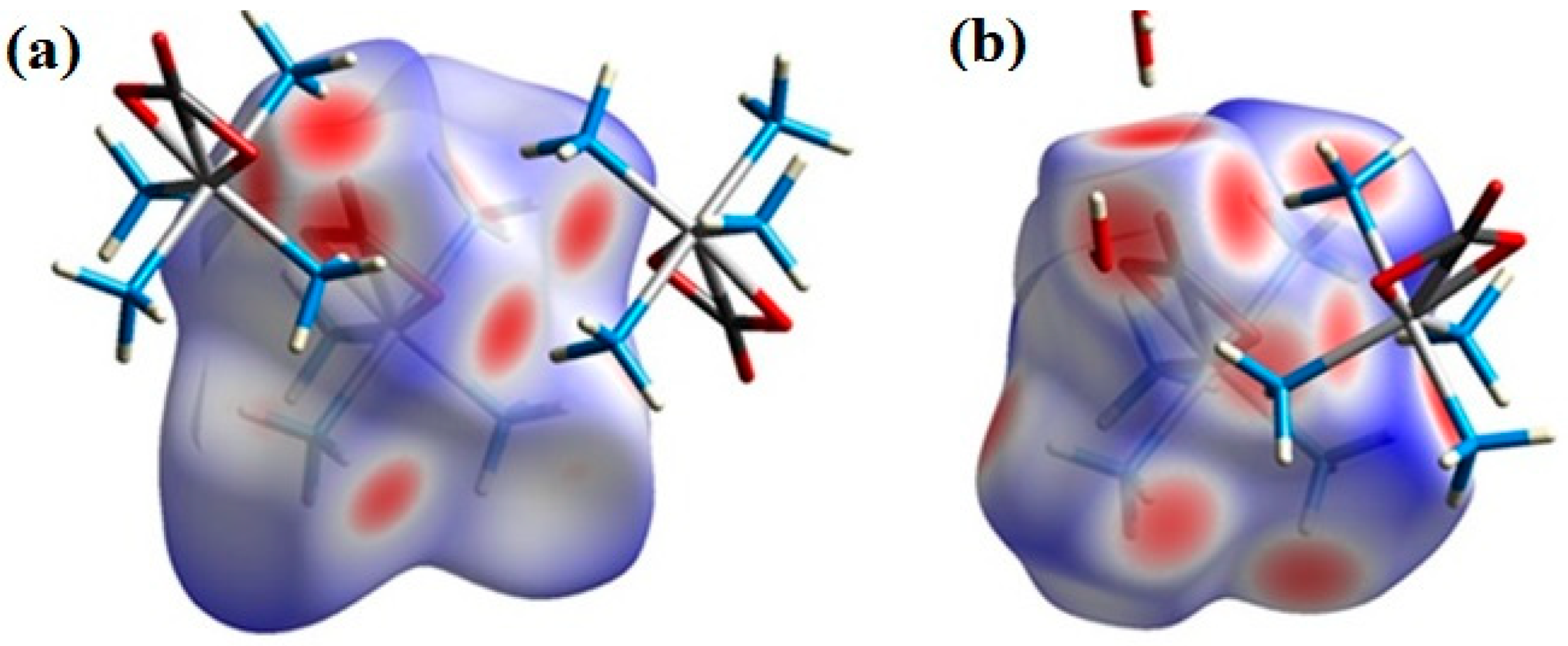
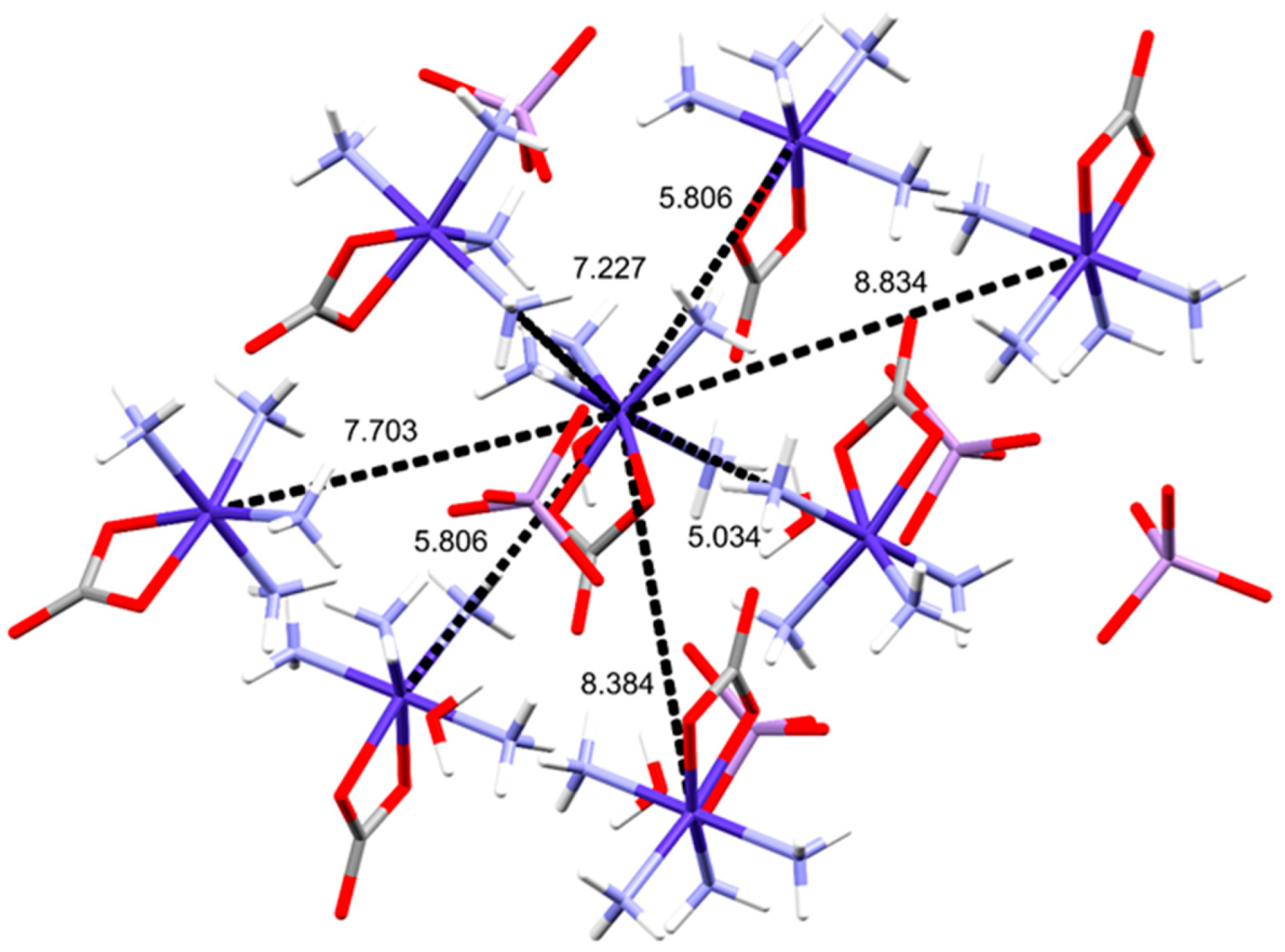
2.2. Structural Features of Compound 2

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nr | Donor—H···Acceptor | Symm. op. | D—H | H···A | D···A | D-H···A |
|---|---|---|---|---|---|---|
| 1 | N1--H1A···O5 | x, y, z | 0.89 | 2.52 | 3.371(6) | 159 |
| 2 | N1--H1B···O2 | x, y, −1 + z | 0.89 | 2.18 | 3.017(3) | 156 |
| 3 | N1--H1C···O3 | ½ − x, −y, 1/2 + z | 0.89 | 2.58 | 3.063(3) | 115 |
| 4 | N1--H1C···O2 | −x, −1/2 + y, 2 − z | 0.89 | 2.58 | 3.313(3) | 140 |
| 5 | N1--H1C···O1 | −x, −y, 2 − z | 0.89 | 2.59 | 3.311(3) | 139 |
| 6 | N4--H2···O5 | 1/2 − x, −y, 1/2 + z | 0.75(3) | 2.44(3) | 3.145(4) | 158(3) |
| 7 | N3--H4···O1 | −x, −y, 2 − z | 0.78(3) | 2.35(2) | 3.048(2) | 151(3) |
| 8 | N3--H5···O2 | x, y, −1 + z | 0.82(4) | 2.24(4) | 3.020(4) | 158(3) |
| Nr | Donor—H···Acceptor | Symm. op. | D—H | H···A | D···A | D-H···A |
|---|---|---|---|---|---|---|
| 1 | N1--H1A···O8 | 3/2 − x, −1/2 + y, z | 0.91 | 2.3 | 2.927(2) | 168 |
| 2 | N1--H1B···O4 | 3/2 − x, 1 − y, 1/2 + z | 0.91 | 2.53 | 2.967(2) | 110 |
| 3 | N1--H1B···O2 | 3/2 − x, −1/2 + y, z | 0.91 | 2.21 | 3.075(2) | 158 |
| 4 | N1--H1C···O7 | x, 3/2 − y, 1/2 + z | 0.91 | 2.13 | 2.993(2) | 158 |
| 5 | N2--H2A···O1 | 1 − x, 1 − y, 1 − z | 0.91 | 2.38 | 3.207(2) | 151 |
| 6 | N2--H2A···O7 | 3/2 − x, −1/2 + y, z | 0.91 | 2.59 | 3.229(2) | 127 |
| 7 | N2--H2B···O3 | 3/2 − x, −1/2 + y, z | 0.91 | 2.08 | 2.986(2) | 171 |
| 8 | N2--H2C···O5 | x, y, z | 0.91 | 2.10 | 2.960(2) | 157 |
| 9 | N3--H3A···O1 | 1 − x, 1 − y, 1 − z | 0.91 | 2.14 | 3.004(2) | 158 |
| 10 | N3--H3A···O3 | 1 − x, 1 − y, 1 − z | 0.91 | 2.59 | 3.377(2) | 145 |
| 11 | N3--H3B···O4 | 1 − x, 1 − y, 1 − z | 0.91 | 2.20 | 3.051(2) | 156 |
| 12 | N3--H3C···O2 | 3/2 − x, −1/2 + y, z | 0.91 | 2.24 | 3.131(2) | 165 |
| 13 | N4--H4A···O7 | −1/2 + x, 3/2 − y, 1 − z | 0.91 | 2.15 | 3.056(2) | 175 |
| 14 | N4--H4B···O4 | x, 3/2 − y, 1/2 + z | 0.91 | 2.17 | 3.079(2) | 172 |
| 15 | N4--H4C···O6 | 3/2 − x, 1 − y, 1/2 + z | 0.91 | 2.17 | 3.051(2) | 164 |
| 16 | O8--H8A···O3 | 1 − x, 2 − y, 1 − z | 0.87 | 1.99 | 2.836(2) | 165 |
| 17 | O8--H8B···O3 | x, y, z | 0.87 | 2.04 | 2.825(2) | 150 |
2.3. Spectroscopic Properties of Compounds 1 and 2
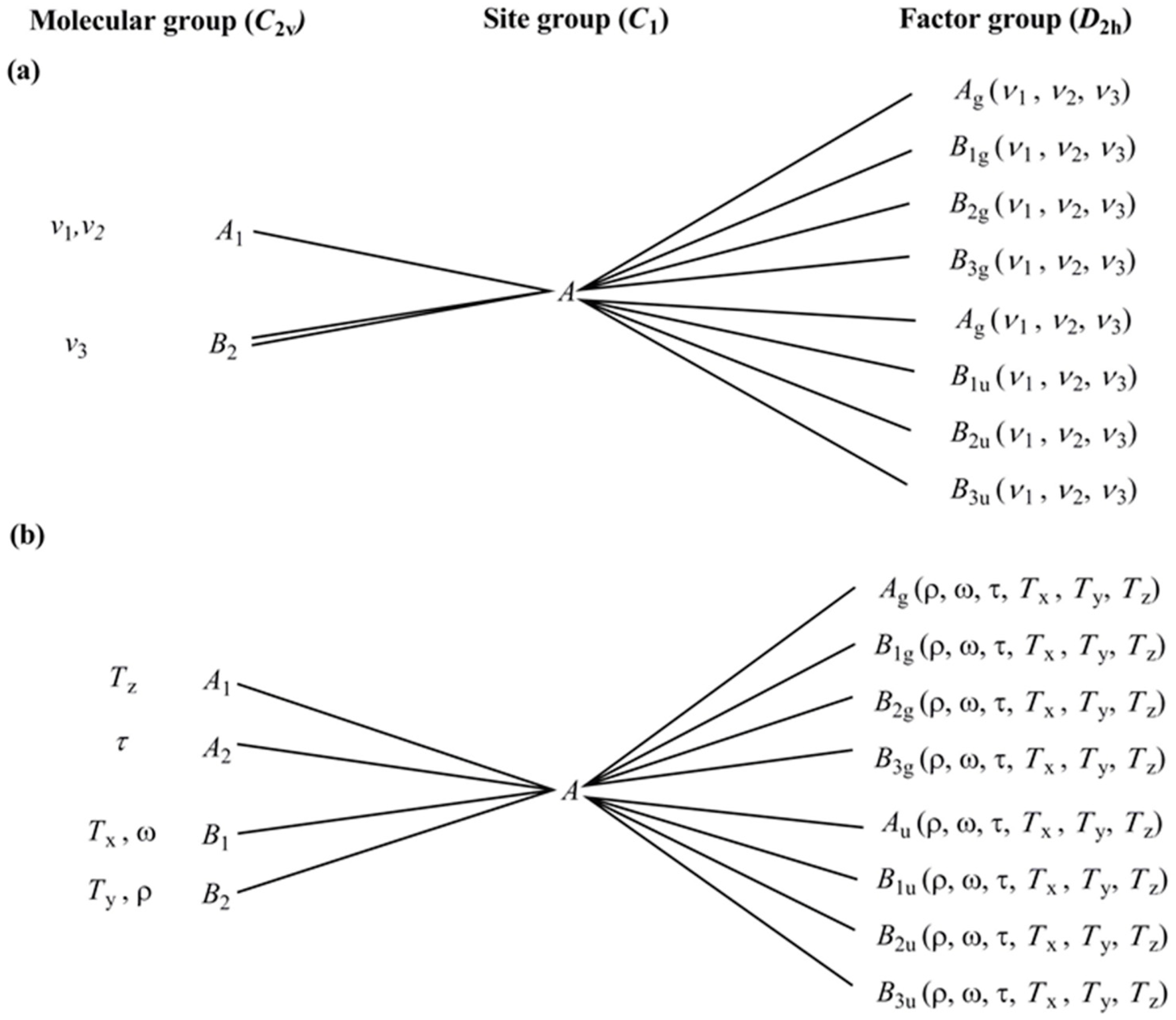
2.4. Vibrational Modes of the Ammonia and Carbonate Ligands in Compounds 1 and 2
| Band | [Co(NH3)4CO3]+, Calculated, cm−1 [54,55] | Measured, cm−1 | ||
|---|---|---|---|---|
| 1 | 2 | 1-ClO4 | ||
| ν(C=O)§ | 1577 | 1590 * | 1590 * | 1602 |
| νs(C-O) | 1052 | 1026 | 1035 | - |
| δ(O-C-O), in plane | 771 | 757 | 754 | 762 |
| νas(C-O) | 1274 | 1278 * | 1278 * | 1284 |
| δ(O-C=O§), in plane | 671 | 675 | 672 | 672 |
| π, out of plane | 859 | 824 | 828 | 836 |
2.5. Vibrational Modes of the cis-CoN4O2 Core in Compounds 1 and 2
2.6. Vibrational Modes of the Non-Coordinated Species (Permanganate Ion and Crystal Water) in Compounds 1 and 2
2.7. UV-Vis Spectroscopy
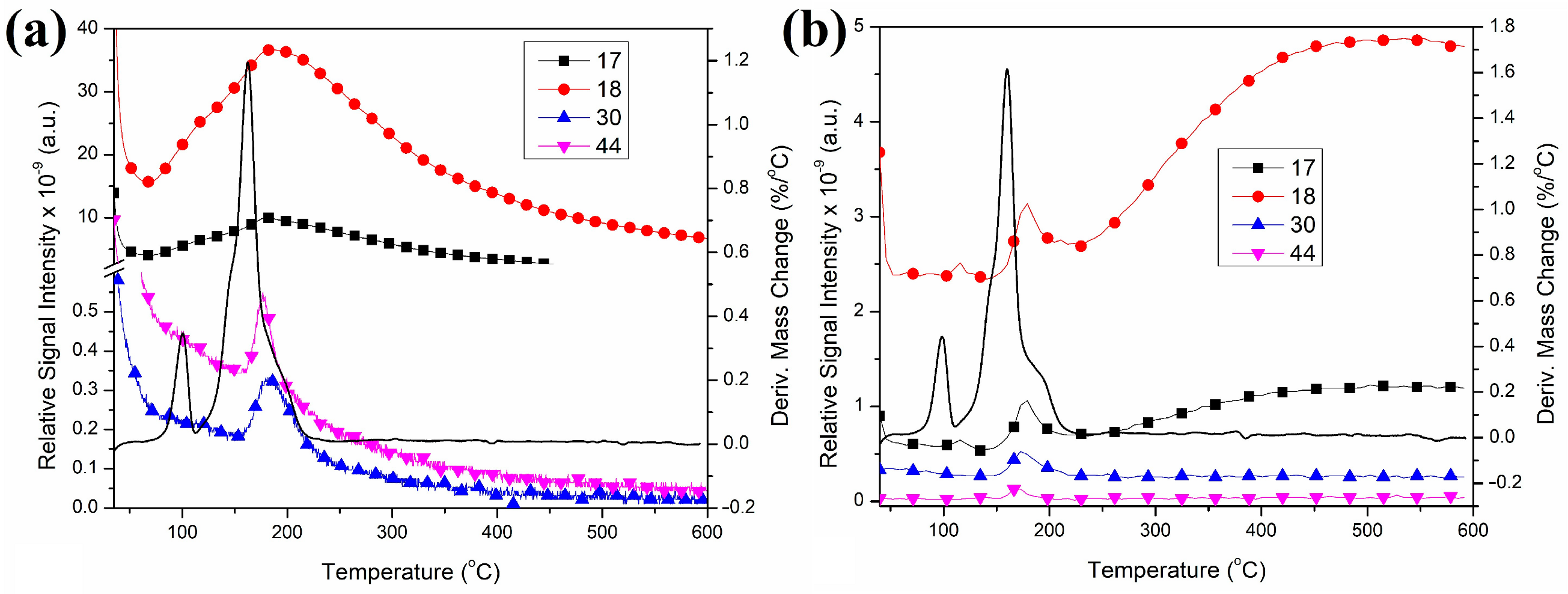
2.8. Thermal Decomposition of Compound 2
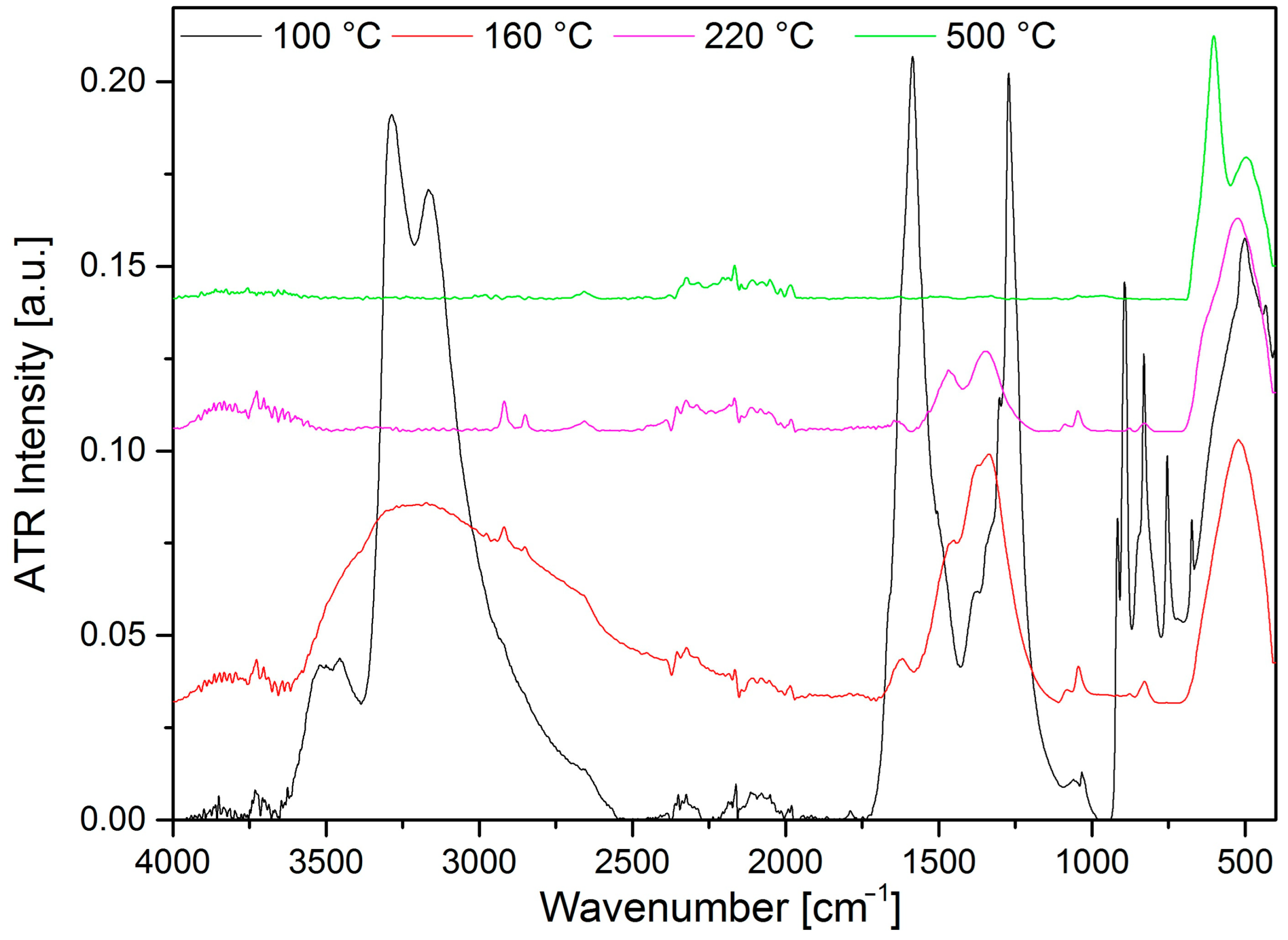
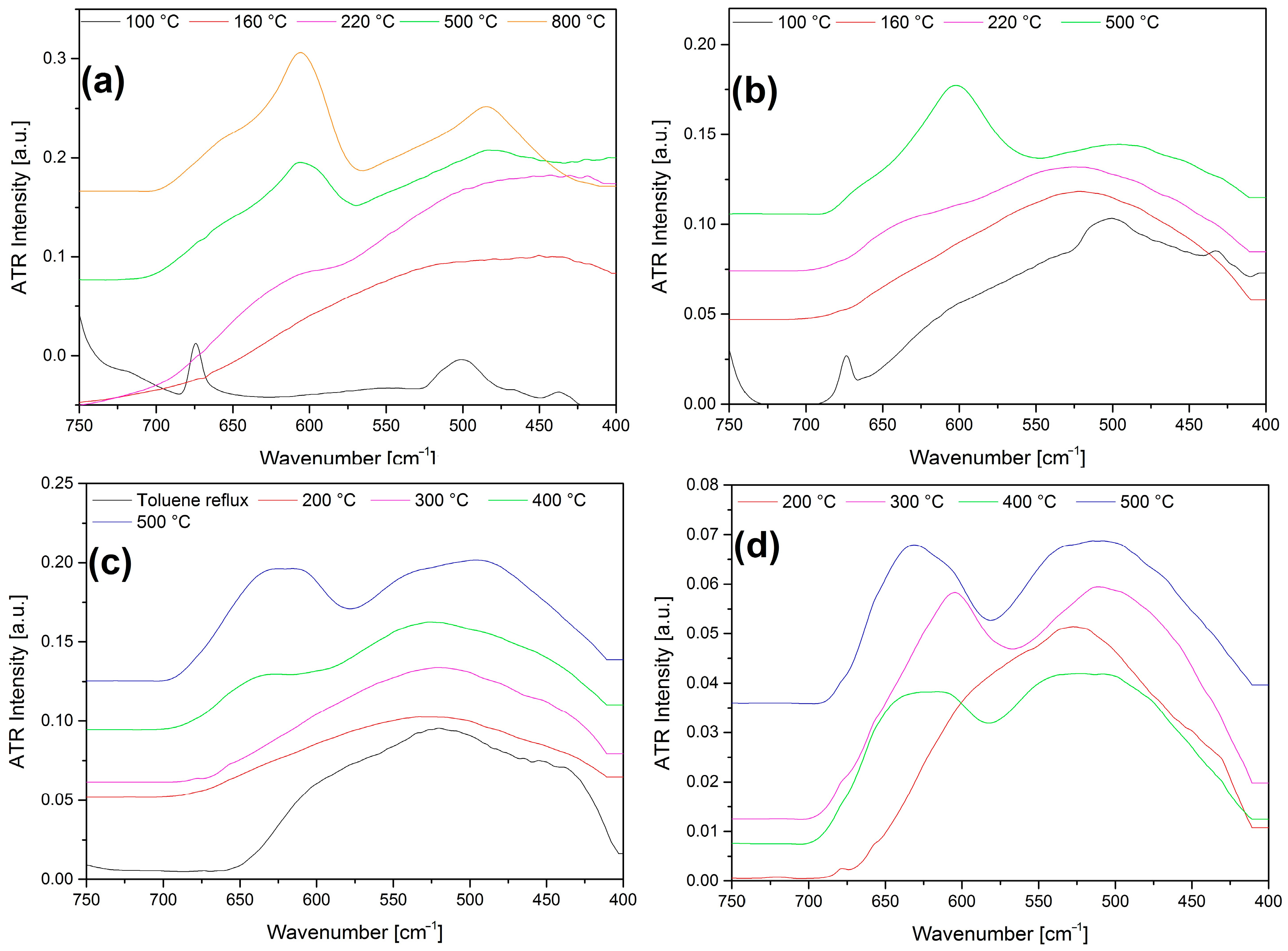
2.9. Characterization of the Solid Decomposition Products
3. Materials and Methods
3.1. Preparation of [Co(NH3)4CO3]MnO4·H2O (Compound 2)
3.2. Preparation of [Co(ND3)4CO3]MnO4·D2O (Compound 2-D)
3.3. Single Crystal X-ray Diffraction
3.4. Hirshfeld Surface Analysis
3.5. Powder X-ray Diffraction
3.6. Specific Surface Area Measurements
3.7. Vibrational Spectroscopy
3.8. UV-Vis Spectroscopy
3.9. Scanning Electron Microscopy
3.10. DSC Studies
3.11. Thermal Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Béres, K.A.; Homonnay, Z.; Kvitek, L.; Dürvanger, Z.; Kubikova, M.; Harmat, V.; Szilágyi, F.; Czégény, Z.; Németh, P.; Bereczki, L.; et al. Thermally Induced Solid-Phase Quasi-Intramolecular Redox Reactions of [Hexakis(urea-O)iron(III)] Permanganate: An Easy Reaction Route to Prepare Potential (Fe,Mn)Ox Catalysts for CO2 Hydrogenation. Inorg. Chem. 2022, 61, 14403–14418. [Google Scholar] [CrossRef]
- Fogaca, L.A.; Kováts, É.; Németh, G.; Kamarás, K.; Berés, K.A.; Németh, P.; Petruševski, V.; Bereczki, L.; Holló, B.B.; Sajó, I.E.; et al. Solid-Phase Quasi-Intramolecular Redox Reaction of [Ag(NH3)2]MnO4: An Easy Way to Prepare Pure AgMnO2. Inorg. Chem. 2021, 60, 3749–3760. [Google Scholar] [CrossRef]
- Flores-Lasluisa, J.X.; Huerta, F.; Cazorla-Amorós, D.; Morallón, E. Transition metal oxides with perovskite and spinel structures for electrochemical energy production applications. Environ. Res. 2022, 214, 113731. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, M.; Atashi, H.; Tabrizi, F.F.; Mirzaei, A.A.; Mansouri, G. Kinetics studies of nano-structured cobalt-manganese oxide catalysts in Fischer-Tropsch synthesis. J. Ind. Eng. Chem. 2013, 19, 1177–1183. [Google Scholar] [CrossRef]
- Homborg, H. Die Feinstruktur in den Schwingungsund elektronischen Absorptionsspektren von [CrO42− und [MnO4−]. Z. Anorg. Allgem. Chem. 1983, 498, 25–40. [Google Scholar] [CrossRef]
- Mueller, A.; Krebs, B. IR-Spektren kristalliner, wasserfreier Permanganate, Pertechnetate und Perrhenate. Z. Naturforsch. B 1966, 21b, 3–7. [Google Scholar] [CrossRef]
- Baran, E.J.; Aymonino, P.J. Über Hexamminkobalt(lll)-permanganat. Z. Anorg. Allgem. Chem. 1968, 362, 215–219. [Google Scholar] [CrossRef]
- Sacconi, B.L.; Sabatini, A.; Cans, P. Infrared Spectra from of Some Metal-Ammine Complexes. Inorg. Chem. 1964, 3, 1772–1774. [Google Scholar] [CrossRef]
- Mueller, A.; Boeschen, I.; Baran, E.J. Uber Hexamminmetallchalkogenometallate. Monatsh. Chem. 1973, 104, 821–835. [Google Scholar] [CrossRef]
- Lazarenko, G.A.; Neokladnova, L.N.; Shablovskaya, S.F. Effect of metals on the thermal decomposition of cobalt(III) hexaammine complexes with a metal-containing anion. Koord. Khimiya 1981, 7, 1485–1488. [Google Scholar]
- Lagunova, V.I.; Filatov, E.Y.; Plyusnin, P.E.; Korenev, S.V. In Situ and Ex Situ Studies of Tetrammineplatinum(II) Chromate Thermolysis. Russ. J. Inorg. Chem. 2020, 65, 1566–1570. [Google Scholar] [CrossRef]
- Serebrennikova, P.S.; Komarov, V.Y.; Sukhikh, A.S.; Khranenko, S.P.; Zadesenets, A.V.; Gromilov, S.A.; Yusenko, K.V. [Nien3](MoO4)0.5(WO4)0.5 co-crystals as single-source precursors for ternary refractory Ni–Mo–W alloys. Nanomaterials 2021, 11, 3272. [Google Scholar] [CrossRef]
- Müller, T.G.; Mogk, J.; Conrad, M.; Kraus, F. Octaammine EuII and YbII Azides and Their Thermal Decompositions to the Nitrides. Eur. J. Inorg. Chem. 2016, 26, 4162–4169. [Google Scholar] [CrossRef]
- Domonov, D.P.; Pechenyuk, S.I.; Semushina, Y.P.; Yusenko, K.V. Solid-state transformations in inner coordination sphere of [Co(NH3)6][Fe(C2O4)3]·3H2O as a route to access catalytically active Co-Fe materials. Materials 2019, 12, 221. [Google Scholar] [CrossRef]
- Yusenko, K.V.; Pechenyuk, S.I.; Vikulova, E.S.; Semushina, Y.P.; Baidina, I.A.; Filatov, E.Y. Isostructurality and Thermal Properties in the Series of Double Complex Salts [M1(NH3)6][M2(C2O4)3]·3H2O (M1 = Co, Ir, M2 = Fe, Cr). J. Struct. Chem. 2019, 60, 1062–1071. [Google Scholar] [CrossRef]
- Farhadi, S.; Pourzare, K.; Bazgir, S. Co3O4 nanoplates: Synthesis, characterization and study of optical and magnetic properties. J. Alloys Compd. 2014, 587, 632–637. [Google Scholar] [CrossRef]
- Garkul’, I.A.; Zadesenets, A.V.; Plyusnin, P.E.; Filatov, E.Y.; Asanova, T.I.; Kozlov, D.V.; Korenev, S.V. Zinc(II) and Manganese(II) Oxalatopalladates as Precursors of Bimetallic Nanomaterials. Russ. J. Inorg. Chem. 2020, 65, 1571–1576. [Google Scholar] [CrossRef]
- Ilyushin, M.A.; Shugalei, I.V.; Tverjanovich, A.S.; Smirnov, A.V. Influence of the Mechanism of the Initial Stages of the Ligand Decomposition on the Initiating Ability of Cobalt(III) Ammine Tetrazolate Complexes. Russ. J. Inorg. Chem. 2020, 90, 640–647. [Google Scholar] [CrossRef]
- Béres, K.A.; Homonnay, Z.; Barta Holló, B.; Gracheva, M.; Petruševski, V.M.; Farkas, A.; Dürvanger, Z.; Kótai, L. Synthesis, structure, and Mössbauer spectroscopic studies on the heat-induced solid-phase redox reactions of hexakis(urea-O)iron(III) peroxodisulfate. J. Mater. Res. 2023, 38, 1102–1118. [Google Scholar] [CrossRef]
- Mansouri, G.; Mansouri, M. Synthesis and characterization of Co-Mn nanocatalyst prepared by thermal decomposition for Fischer-Tropsch reaction. Iran. J. Chem. Eng. 2018, 37, 1–9. [Google Scholar]
- Kótai, L.; Petruševski, V.M.; Bereczki, L.; Béres, K.A. Catalytic Properties of the Spinel-Like CuxMn3−xO4 Copper Manganese Oxides—An Overview. Catalysts 2023, 13, 129. [Google Scholar] [CrossRef]
- Solt, H.E.; Németh, P.; Mohai, M.; Sajó, I.E.; Klébert, S.; Franguelli, F.P.; Fogaca, L.A.; Pawar, R.P.; Kótai, L. Temperature-Limited Synthesis of Copper Manganites along the Borderline of the Amorphous/Crystalline State and Their Catalytic Activity in CO Oxidation. ACS Omega 2021, 6, 1523–1533. [Google Scholar] [CrossRef]
- Li, J.; Zhao, D.; Zhang, L.; Ren, Y.; Yue, L.; Li, Z.; Sun, S.; Luo, Y.; Chen, Q.; Li, T.; et al. Boosting electrochemical nitrate-to-ammonia conversion by self-supported MnCo2O4 nanowire array. J. Colloid. Interface Sci. 2023, 629, 805–812. [Google Scholar] [CrossRef]
- Gavrikov, A.V.; Belova, E.V.; Ilyukhin, A.B.; Koroteev, P.S.; Sadovnikov, A.A. Preparation and properties of uncommon Cd-Mn carboxylate complexes—Per se and as precursors for CdMn2O4-based ceramics. App. Organomet. Chem. 2021, 35, e6190. [Google Scholar] [CrossRef]
- Packiaraj, S.; Jeyaraj, M.; Chandarasekaran, K.; Rawson, J.M.; Govindarajan, S. Simple and facile synthesis of pure nanocrystalline spinel cobaltite by thermal decomposition of solid solution precursors. J. Mater. Sci. Mater. Electron. 2019, 30, 18866–18877. [Google Scholar] [CrossRef]
- Dosaev, K.A.; Istomin, S.Y.; Strebkov, D.A.; Tsirlina, G.A.; Antipov, E.V.; Savinova, E.R. AMn2O4 spinels (A–Li, Mg, Mn, Cd) as ORR catalysts: The role of Mn coordination and oxidation state in the catalytic activity and their propensity to degradation. Electrochim. Acta 2022, 428, 140923. [Google Scholar] [CrossRef]
- Mansouri, M.; Atashi, H.; Khalilipour, M.M.; Setareshenas, N.; Shahraki, F. Rate expression of Fischer–Tropsch synthesis over Co–Mn nanocatalyst by response surface methodology (RSM). J. Korean Chem. Soc. 2013, 57, 769–777. [Google Scholar] [CrossRef]
- Chen, C.; Feng, C.; Wang, Y.; Li, J.; Liu, Z.; Wang, W.; Pan, Y.; Liu, Y. Design of robust Co-doped Mn3O4 spinel catalysts for selective catalytic reduction of NO with NH3 at low temperatures. Appl. Surf. Sci. 2022, 602, 154384. [Google Scholar] [CrossRef]
- Yang, X.; Wei, G.; Wu, P.; Liu, P.; Liang, X.; Chu, W. Novel halloysite nanotube-based ultrafine CoMn2O4 catalyst for efficient degradation of pharmaceuticals through peroxymonosulfate activation. Appl. Surf. Sci. 2022, 588, 152899. [Google Scholar] [CrossRef]
- Yang, X.; Wei, G.; Wu, P.; Liu, P.; Liang, X.; Chu, W. Controlling oxygen vacancies of CoMn2O4 by loading on planar and tubular clay minerals and its application for boosted PMS activation. J. Hazard. Mater. 2022, 436, 129060. [Google Scholar] [CrossRef]
- Zagorodnyaya, A.N.; Abisheva, Z.S.; Kvyatkovskaya, M.N.; Kokoveshnikova, T.A.; Kasymova, A.S.; Sadykanova, S.E. Thermal decomposition of tetraamminecopper diperrhenate. Dokl. Nats. Akad. Nauk Respub. Kazakh. 2004, 3, 89–96. [Google Scholar]
- Zagorodnyaya, A.N.; Abisheva, Z.S.; Aitekeyeva, S.N.; Bukurov, T.N.; Sapukov, I.A. Synthesis and properties of tetraamminecopper diperrhenate. Komplex Ispolz. Miner. Sirya 2003, 1, 16–23. [Google Scholar]
- Zadesenets, A.V.; Khranenko, S.P.; Shubin, Y.V.; Baidina, I.A.; Korenev, S.V. Complex Salts [Pd(NH3)4](ReO4)2 and [Pd(NH3)4](MnO4)2: Synthesis, Structure, and Thermal Properties. Russ. J. Coord. Chem. 2006, 32, 374–379. [Google Scholar] [CrossRef]
- Korolkov, I.V.; Zadesenets, A.V.; Gromilov, S.A.; Yusenko, K.V.; Baidina, I.A.; Korenev, S.V. Metal solid solutions obtained by thermolysis of Pt and Re salts. Crystal structure of [Pt(NH3)4](ReO4)2. J. Struct. Chem. 2006, 47, 489–498. [Google Scholar] [CrossRef]
- Pechenyuk, S.I.; Kuznetsov, V.Y.; Popova, R.A.; Zalkind, O.A. Synthesis and study of tetraammineplatinum(II) perrhenate. Zh. Neorg Khim. 1979, 24, 3306–3308. [Google Scholar]
- Franguelli, F.P.; Béres, K.A.; Kótai, L. Pyridinesilver Tetraoxometallate Complexes: Overview of the Synthesis, Structure, and Properties of Pyridine Complexed AgXO4 (X = Cl, Mn, Re) Compounds. Inorganics 2021, 9, 79. [Google Scholar] [CrossRef]
- Mehrotra, R.N. Review on the chemistry of [M(NH3)n](XO4)m (M = transition metal, X = Mn, Tc or Re, n = 1–6, m = 1–3) ammine complexes. Inorganics 2023, 11, 308. [Google Scholar] [CrossRef]
- Shubin, Y.V.; Filatov, E.Y.; Baidina, I.A.; Yusenko, K.V.; Zadesenetz, A.V.; Korenev, S.V. Synthesis of [M(NH3)5Cl](ReO4)2 (M = Cr, Co, Ru, Rh, Ir) and investigation of thermolysis products. crystal structure of [Rh(NH3)5Cl](ReO4)2. J. Struct. Chem. 2006, 47, 1103–1110. [Google Scholar] [CrossRef]
- Dahm, M.; Adam, A. [Co(NH3)6]ClCO3—Ein hexaammincobalt(III)-komplex mit einer ‘idealen’ carbonat-gruppe. Z. Naturforsch. B J. Chem. Sci. 2011, 56, 1117–1122. [Google Scholar] [CrossRef]
- Béres, K.A.; Szilágyi, F.; Homonnay, Z.; Dürvanger, Z.; Bereczki, L.; Trif, L.; Petruševski, V.M.; Farkas, A.; Bayat, N.; Kótai, L. Structural, Spectroscopic, and Thermal Decomposition Features of [Carbonatotetraamminecobalt(III)] Iodide—Insight into the Simultaneous Solid-Phase Quasi-Intramolecular Redox Reactions. Inorganics 2023, 11, 68. [Google Scholar] [CrossRef]
- Bereczki, L.; Petruševski, V.M.; Franguelli, F.P.; Béres, K.A.; Farkas, A.; Holló, B.B.; Czégény, Z.; Szilágyi, I.M.; Kótai, L. [Hexaamminecobalt(III)] Dichloride Permanganate—Structural Features and Heat-Induced Transformations into (CoII,MnII)(CoIII,MnIII)2O4 Spinels. Inorganics 2022, 10, 252. [Google Scholar] [CrossRef]
- Franguelli, F.P.; Kováts, É.; Czégény, Z.; Bereczki, L.; Petruševski, V.M.; Holló, B.B.; Béres, K.A.; Farkas, A.; Szilágyi, I.M.; Kótai, L. Multi-Centered Solid-Phase Quasi-Intramolecular Redox Reactions of [(Chlorido)Pentaamminecobalt(III)] Permanganate—An Easy Route to Prepare Phase Pure CoMn2O4 Spinel. Inorganics 2022, 10, 18. [Google Scholar] [CrossRef]
- Mohan, S.C.; Jenniefer, S.J.; Muthiah, P.T.; Jothivenkatachalam, K. Tetraammine(carbonato-k2O,O’)cobalt(III) perchlorate. Acta Cryst. Sect. E 2013, E69, i45–i46. [Google Scholar] [CrossRef]
- Wu, H.-X.; Zhu, J.-H.; Ma, L.-D.; Shen, X.-L.; Shi, G.-S. Powder diffraction of [Co(NH3)5CO3]NO3.1/2 H2O and [Co(NH3)4CO3]NO3.1/2H2O. Powder Diffr. 1998, 13, 32–34. [Google Scholar] [CrossRef]
- Christensen, A.N.; Hazell, R.G. X-ray Crystallographic Study and Thermogravimetric Analysis of Tetrammine-Carbonatocobalt(III) Nitrate Hemihydrate, [Co(NH3)4CO3]NO3∙0.5H2O. Acta Chem. Scand. 1999, 53, 399–402. [Google Scholar] [CrossRef]
- Bernal, I.; Cetrullo, J. The Phenomenon of Conglomerate Crystallization. XVIII. Clavic Dissymmetry in Coordination Compounds. XVI. The Crystal Structure of Racemic {[Co(NH3)4(CO3)]NO3}2·H2O. Struct. Chem. 1990, 1, 227–234. [Google Scholar] [CrossRef]
- Macikenas, D.; Hazell, R.G.; Christensen, A.N. X-ray Crystallographic Study of Tetrammine-CarbonatocobaIt(III) Sulfate Trihydratef [Co(NH3)4CO3]2SO4∙3H2O. Acta Chem. Scand. 1995, 49, 636–639. [Google Scholar] [CrossRef][Green Version]
- Franguelli, F.P.; Barta Holló, B.; Petruševski, V.M.; Sajó, I.E.; Klébert, S.; Farkas, A.; Bódis, E.; Szilágyi, I.M.; Pawar, R.P.; Kótai, L. Thermal decomposition and spectral characterization of di[carbonatotetraamminecobalt(III)] sulfate trihydrate and the nature of its thermal decomposition products. J. Therm. Anal. Calorim. 2021, 145, 2907–2923. [Google Scholar] [CrossRef]
- Kótai, L.; Gács, I.; Sajó, I.E.; Sharma, P.K.; Banerji, K.K. Beliefs and facts in permanganate chemistry—An overview on the synthesis and the reactivity of simple and complex permanganates. Trends Inorg. Chem. 2009, 11, 25–104. [Google Scholar] [CrossRef]
- Kótai, L.; Keszler, Á.; Pató, J.; Holly, S.; Banerji, K.K. The reaction of barium manganate with acids and their precursors. Indian J. Chem. Sect. A 1999, 38, 966–968. [Google Scholar]
- Strock, L.W.; McCutcheon, T.P. The crystalline form of some new cobaltammines. J. Am. Chem. Soc. 1931, 53, 2852–2866. [Google Scholar] [CrossRef]
- Siebert, H. Ultrarotspektren von Kobalt (III)-Komplexen mit Ammoniak und Resten von Sauerstofsäuren als Liganden. Z. Anorg. Allg. Chem. 1959, 298, 51–63. [Google Scholar] [CrossRef]
- Goldsmith, J.A.; Hezel, A.; Ross, S.D. The skeletal vibrations of some cobalt (III) carbonato-, phosphato- and sulphato-complexes. Spectrochim. Acta A 1968, 24, 1139–1147. [Google Scholar] [CrossRef]
- Goldsmith, J.A.; Ross, S.D. Factors afecting the infra-red spectra of planar anions with D3h symmetry—IV the vibrational spectra of some complex carbonates in the region 4000–400 cm−1. Spectrochim. Acta A 1968, 24, 993–998. [Google Scholar] [CrossRef]
- Fujita, J.; Martell, A.E.; Nakamoto, K. Infrared spectra of metal chelate compounds. VIII. Infrared spectra of Co(III) carbonato complexes. J. Chem. Phys. 1962, 36, 339–345. [Google Scholar] [CrossRef]
- Chen, Y.; Christensen, D.H.; Sorensen, G.O.; Nielsen, O.F.; Pedersen, E.B. The skeletal vibrational spectra and metal-ligand force constants of cobalt(III) ammine complexes. J. Mol. Struct. 1993, 299, 61–72. [Google Scholar] [CrossRef]
- Sajo, I.E.; Bakos, L.P.; Szilagyi, I.M.; Lendvay, G.; Magyari, J.; Mohai, M.; Szegedi, A.; Farkas, A.; Janosity, A.; Klebert, S.; et al. Unexpected Sequential NH3/H2O Solid/Gas Phase Ligand Exchange and Quasi-Intramolecular Self-Protonation Yield [NH4Cu(OH)MoO4], a Photocatalyst Misidentified before as (NH4)2Cu(MoO4)2. Inorg. Chem. 2018, 57, 13679–13692. [Google Scholar] [CrossRef] [PubMed]
- Grinberg, A.A.; Varshavskii, Y.S. The frequency of coordinated ammonia deformation mode and its relationship with the chemical properties of transition metal ammonia complexes. Primen. Molekul. Spektr. Khim. 1966, 1, 104–107. [Google Scholar]
- Barsan, M.M.; Butler, I.S.; Gilson, D.F.R. Vibrational spectroscopy at high pressure of the permanganate anion trapped in potassium bromide and potassium perchlorate matrices. J. Phys. Chem. B 2006, 110, 9291–9297. [Google Scholar] [CrossRef] [PubMed]
- Carassiti, V.; Martelli, R. Absorption spectra of solutions of complex salts. II. Cobalt tetra- and pentaammines. Ann. Chim. 1957, 47, 402–409. [Google Scholar]
- Burkov, V.I.; Egorsyheva, A.V.; Kargin, Y.F. Electronic structure and optical spectra of tetrahedral [MO4]n− complexes of 3d metals. Russ. J. Inorg. Chem. 2003, 48, 537–555, Translated from Zh. Neorg. Khim. 2003, 4, 620–638. [Google Scholar]
- Sastri, V.S. Studies on the disposition of carbonato group in cobalt(III) complexes. Inorg. Chim. Acta 1972, 6, 264–266. [Google Scholar] [CrossRef]
- Sastri, V.S.; Langford, C.H. Electronic spectra of carbonato and oxalato ammine complexes of cobalt(III); average feld model for singlet, triplet, and charge transfer bands. Can. J. Chem. 1969, 47, 4237–4240. [Google Scholar] [CrossRef]
- Mustafaev, R.A.; Tagiev, S.I.; Gabulov, D.M.; Musaev, T.P. Method of calculating the thermophysical parameters of liquid aromatic hydrocarbons. High Temp. 1985, 23, 837–841. [Google Scholar]
- Zhu, M.-L.; Liu, X.; Niu, Y.; Yang, G.-Q.; Bao, J.; Song, Y.-H.; Yan, Y.-F.; Du, N.; Shui, M.-L.; Zhu, K.; et al. Long-chain α-olefins production over Co-MnOx catalyst with optimized interface. Appl. Catal. B Environ. 2024, 346, 123783. [Google Scholar]
- Van Koppen, L.M.; Iulian, D.A.; Leendert, B.G.; Hensen, E.J.M. Manganese as a structural promoter in silica-supported cobalt Fischer-Tropsch catalysts under simulated high conversion conditions. J. Catal. 2023, 428, 115173. [Google Scholar] [CrossRef]
- Li, B.-X.; Wang, J.-G.; Wang, Q.; Zhang, W.; Liu, Y.; Ma, Z.-C.; Ma, Z.-Y.; Hou, B. Study on the effect of different metal oxides on the performance of cobalt-based Fischer-Tropsch catalysts. J. Fuel. Chem. Technol. 2023, 51, 1411–1420. [Google Scholar]
- Van Koppen, L.M.; Iulian, D.A.; Leendert, B.G.; Hensen, E.J.M. Sintering and carbidization under simulated high conversion on a cobalt-based Fischer-Tropsch catalyst; manganese oxide as a structural promotor. J. Catal. 2022, 413, 106–118. [Google Scholar] [CrossRef]
- Tucker, C.L.; Ragoo, Y.; Mathe, S.; Macheli, L.; Bordoloi, A.; Rocha, T.C.R.; Govender, S.; Kooyman, P.J.; van Steen, E. Manganese promotion of a cobalt Fischer-Tropsch catalyst to improve operation at high conversion. J. Catal. 2022, 411, 97–108. [Google Scholar] [CrossRef]
- Zafari, R.; Abdouss, M.; Zamani, Y. Effect of Mn and reduced graphene oxide for the Fischer–Tropsch reaction: An efficient catalyst for the production of light olefins from syngas. React. Kinet. Mech. Catal. 2020, 129, 707–724. [Google Scholar] [CrossRef]
- Gong, K.; Lin, T.; An, Y.; Wang, X.; Yu, F.; Wu, B.; Li, X.; Li, S.; Lu, Y.; Zhong, L.; et al. Fischer-Tropsch to olefins over CoMn-based catalysts: Effect of preparation methods. Appl. Catal. A Gen. 2020, 592, 117414. [Google Scholar] [CrossRef]
- Schneider, J.; Struve, M.; Trommler, U.; Schlüter, M.; Seidel, L.; Dietrich, S.; Rönsch, S. Performance of supported and unsupported Fe and Co catalysts for the direct synthesis of light alkenes from synthesis gas. Fuel Process. Technol. 2018, 170, 64–78. [Google Scholar] [CrossRef]
- Sun, F.; Yang, R.; Xia, Z.; Yang, Y.; Zhao, Z.; Gu, S.; Wu, D.; Ding, Y.; Jiang, Z. Effects of cobalt carbide on Fischer–Tropsch synthesis with MnO supported Co-based catalysts. J. Energy Chem. 2020, 42, 227–232. [Google Scholar] [CrossRef]
- Nawaz, M.A.; Li, M.; Saif, M.; Song, G.; Wang, Z.; Liu, D. Harnessing the Synergistic Interplay of Fischer-Tropsch Synthesis (Fe-Co) Bimetallic Oxides in Na-FeMnCo/HZSM-5 Composite Catalyst for Syngas Conversion to Aromatic Hydrocarbons. ChemCatChem 2021, 13, 1966–1980. [Google Scholar] [CrossRef]
- Barbieri, G.A. Sui composti di ferriurea. Rend. Atti Real. Accad Rend. Lincei. 1913, 22, 867–870. [Google Scholar]
- Barbieri, G.A. Nuove ricerche sulle metalli-uree. Rend. Accad. Linzei Ser.5 1915, 24, 916–921. [Google Scholar]
- Penland, R.B.; Mizushima, S.; Columba, C.; Quagliano, J.V. Infrared Absorption Spectra of Inorganic Coordination Complexes. X. Studies of Some Metal-Urea Complexes. J. Am. Chem. Soc. 1957, 79, 1575–1578. [Google Scholar] [CrossRef]
- Lupin, M.S.; Peters, G.E. Thermal decomposition of aluminum, iron and manganese complexes of urea. Thermochim. Acta 1984, 73, 79–87. [Google Scholar] [CrossRef]
- Li, X.; Wang, S.; Shi, X. Room temperature solid-phase synthesis and characterization of urea Fe(III) complexes. Zhànjiāng Shīfàn Xuéyuàn Xuébào 2005, 6, 60–64. [Google Scholar]
- Bala, R.; Sachdeva, D.; Kumar, M.; Prakash, V. Advances in coordination chemistry of hexaurea complexes of chromium(III). J. Coord. Chem. 2020, 73, 2801–2837. [Google Scholar] [CrossRef]
- Sell, W.J. On a Series of Salts of a Base Containing Chromium and Urea. No. 1. Proc. R. Soc. Lond. 1882, 33, 267–274. [Google Scholar]
- Sell, W.J.; Lewis, W.J. On a Series of Salts of a Base Containing Chromium and Urea. No. 2. Proc. R. Soc. Lond. 1889, 45, 321–345. [Google Scholar]
- Klobb, T. Contribuiton a l’etude des sels luteocobaltique. Bull. Soc. Chim. 1901, 25, 1022–1031. [Google Scholar]
- Wilke-Doerfurt, E.; Guenzert, T. Über neue Salze der Perrheniumsäure. Z. Anorg. Allgem. Chem. 1933, 215, 369–387. [Google Scholar] [CrossRef]
- Wilke-Doerfurt, E.; Pfau, R. Über den Isomorphismus zwischen Chrom(III)Hexaharnstoff-Permanganat, -Perchlorat, -Borfluorid, -Fluorsulfonat und -Jodid. Electrochem. Angew. Phys. Chem. 1930, 36, 118–122. [Google Scholar]
- Béres, K.A.; Homonnay, Z.; Kótai, L. Hexakis(urea-O)iron Complex Salts as a Versatile Material Family: Overview of Their Properties and Applications. ACS Omega 2024, 9, 11148–11167. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT– Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. Structure validation in chemical crystallography. Acta Cryst. 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]










| Compound | Label | Ref. |
|---|---|---|
| [Co(NH3)4CO3]MnO4 | 1 | [4,27] |
| [Co(NH3)4CO3]ClO4 | 1-ClO4 | [43] |
| [Co(NH3)4CO3]MnO4∙H2O | 2 | New compound |
| [Co(NH3)4CO3]NO3 | 3 | [44] |
| [Co(NH3)4CO3]NO3∙0.5H2O | 4 | [45,46] |
| [Co(NH3)4CO3]2SO4∙3H2O | 5 | [47,48] |
| Compound | Compound 1 | Compound 2 | Compound 1-ClO4 [43] |
|---|---|---|---|
| Temperature, K | 296 | 101.1(6) | 296 |
| Crystal system | Triclinic | Orthorhombic | Orthorhombic |
| Space group | P-1 | Pbca | Pnma |
| Lattice constants, Å | a = 7.09040(5) | a = 11.10180(10) | a = 17.8961(5) |
| b = 8.77177(5) | b = 10.57700(10) | b = 8.0768(2) | |
| c = 9.76420(5) | c = 17.06750(10) | c = 6.8871(2) | |
| Volume, Å3 | 588.66 | 2004.13(3) | 995.48(5) |
| Dc, mg/m3 | 3.106 | 1.139 | 1.147 |
| Z | Z = 1 | 8 | 4 |
| Mode | [Co(NH3)4CO3]+, cm−1 | Measured Values in cm−1 at Room Temperature | Assignment | |||
|---|---|---|---|---|---|---|
| Ref. [55] | Our Results | Ref. [48] | ||||
| 1 | 2 | 2-D | 5 | |||
| ν1 | - | 3242, 3168 | 3151 | 2347sh, 2333sh, 2303 | 3289, 3192 | νs(N-H) |
| ν2 | 1300 | 1298 | 1346 | 997 | 1300 | δs(H-N-H |
| ν3 | - | 3288 | 3281 | 2462, 2450 | 3425 | νas(N-H) |
| ν4 | 1638 * | 1579 * | 1621 * | 1187, 11154 | 1645 * | δas(H-N-H) |
| ν5 | 810 | ~800 | ~800 | 664, 651 | 828 | ρ(NH3) |
| Calcination Circumstances | BET Surface Area [m2/g] |
|---|---|
| In air at 400 °C (2 h) | 120 |
| In air at 500 °C (2 h) | 52 |
| In air at 800 °C (2 h) | 1.8 |
| In inert atm. at 500 °C (2 h) | 10 |
| At toluene reflux temperature (2 h), washing with water and then in air at 400 °C (2 h) | 36 |
| At toluene reflux temperature (2 h), then in air at 400 °C (2 h) without washing | 37 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Béres, K.A.; Dürvanger, Z.; Homonnay, Z.; Bereczki, L.; Barta Holló, B.; Farkas, A.; Petruševski, V.M.; Kótai, L. Insight into the Structure and Redox Chemistry of [Carbonatotetraamminecobalt(III)] Permanganate and Its Monohydrate as Co-Mn-Oxide Catalyst Precursors of the Fischer-Tropsch Synthesis. Inorganics 2024, 12, 94. https://doi.org/10.3390/inorganics12040094
Béres KA, Dürvanger Z, Homonnay Z, Bereczki L, Barta Holló B, Farkas A, Petruševski VM, Kótai L. Insight into the Structure and Redox Chemistry of [Carbonatotetraamminecobalt(III)] Permanganate and Its Monohydrate as Co-Mn-Oxide Catalyst Precursors of the Fischer-Tropsch Synthesis. Inorganics. 2024; 12(4):94. https://doi.org/10.3390/inorganics12040094
Chicago/Turabian StyleBéres, Kende Attila, Zsolt Dürvanger, Zoltán Homonnay, Laura Bereczki, Berta Barta Holló, Attila Farkas, Vladimir M. Petruševski, and László Kótai. 2024. "Insight into the Structure and Redox Chemistry of [Carbonatotetraamminecobalt(III)] Permanganate and Its Monohydrate as Co-Mn-Oxide Catalyst Precursors of the Fischer-Tropsch Synthesis" Inorganics 12, no. 4: 94. https://doi.org/10.3390/inorganics12040094
APA StyleBéres, K. A., Dürvanger, Z., Homonnay, Z., Bereczki, L., Barta Holló, B., Farkas, A., Petruševski, V. M., & Kótai, L. (2024). Insight into the Structure and Redox Chemistry of [Carbonatotetraamminecobalt(III)] Permanganate and Its Monohydrate as Co-Mn-Oxide Catalyst Precursors of the Fischer-Tropsch Synthesis. Inorganics, 12(4), 94. https://doi.org/10.3390/inorganics12040094