
Direct Synthesis of C-Substituted [RC(O)CH2-CB11H11]− Carborate Anions


Abstract

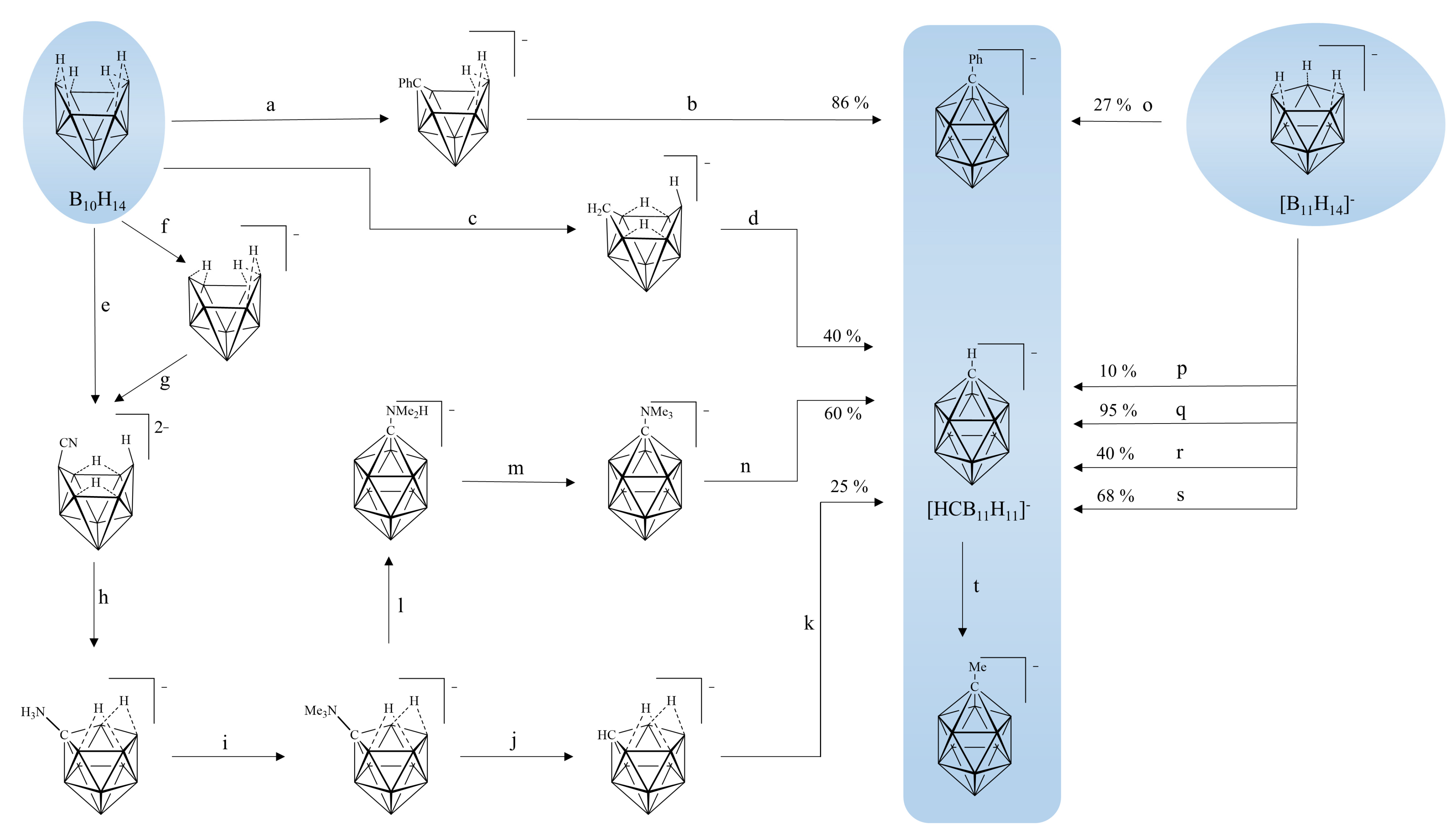
1. Introduction
2. Results and Discussion
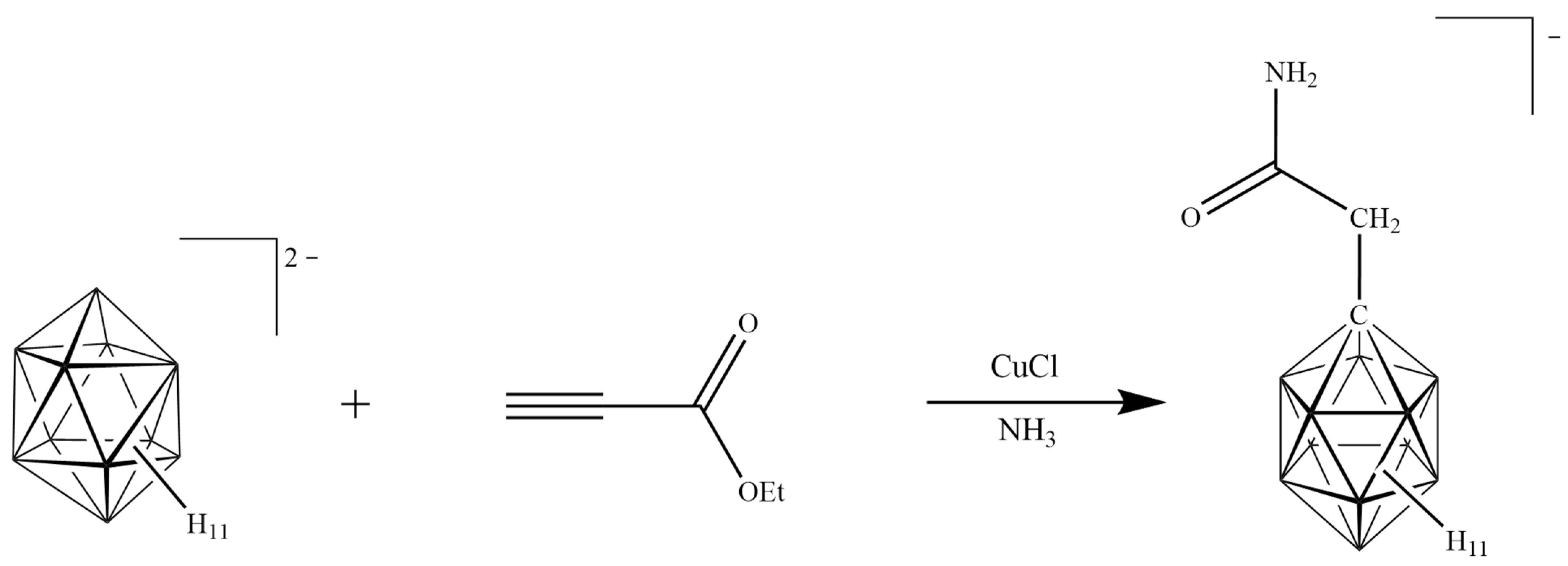
2.1. Reaction Design
2.2. Synthesis of [NH2C(O)CH2-CB11H11]−
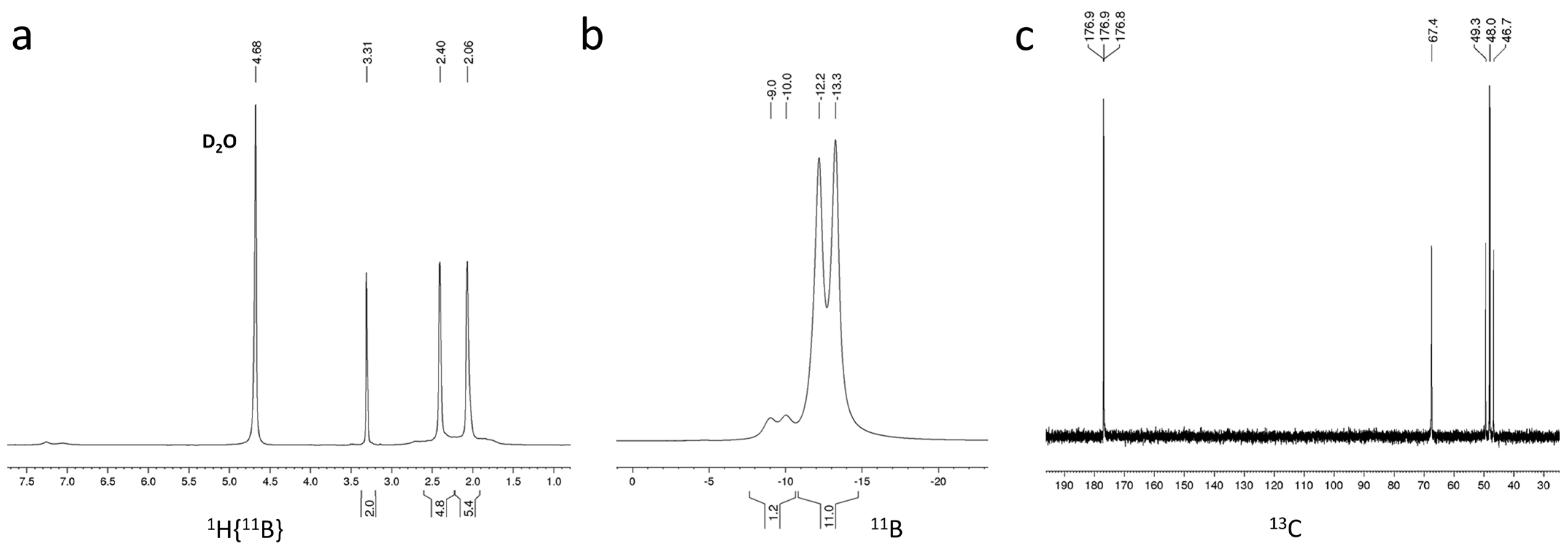
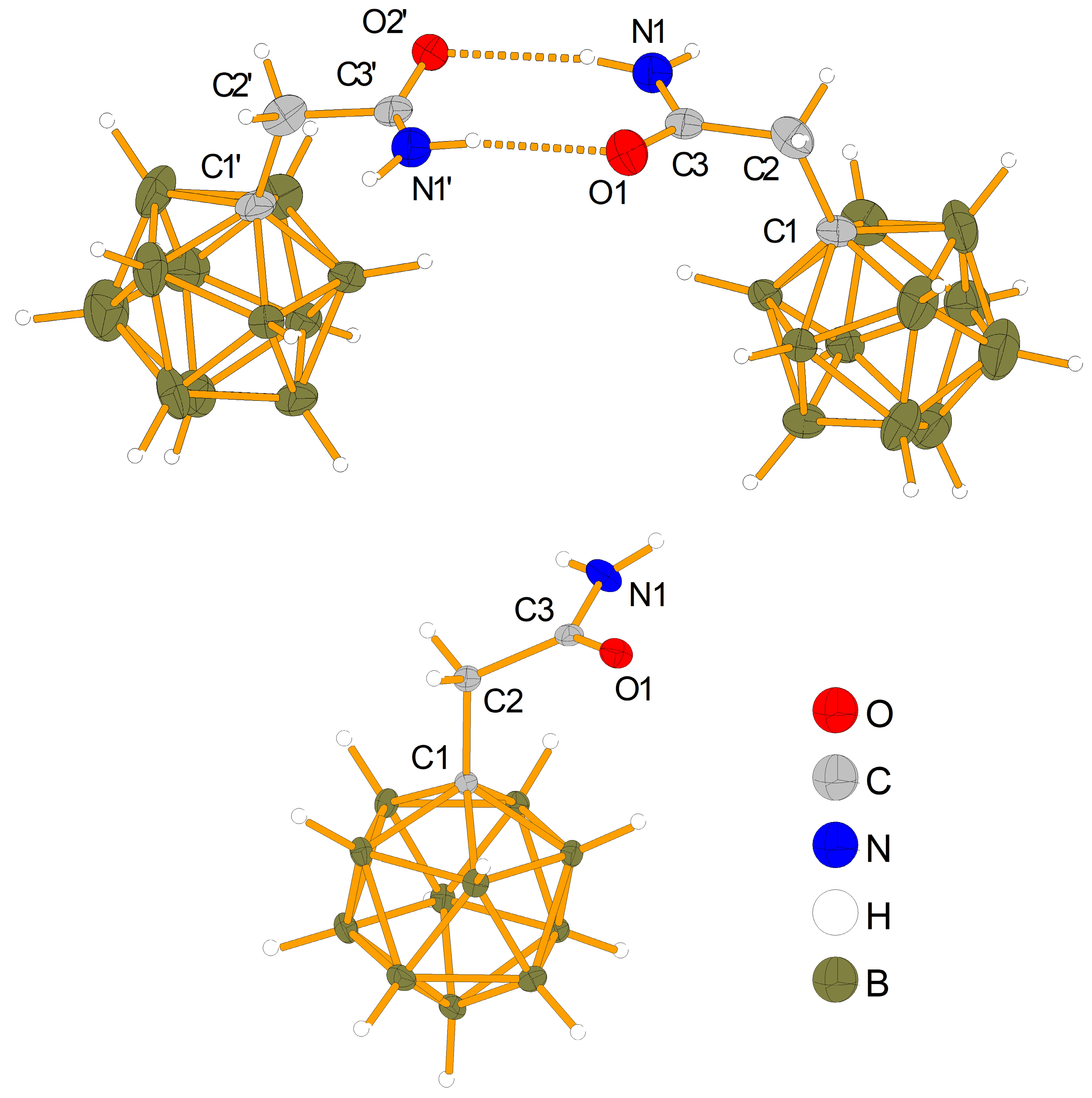
2.3. Characterization and Properties of K[NH2C(O)CH2-CB11H11]
2.4. Reactions with Other Terminal Alkynes
3. Experimental Section
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Körbe, S.; Schreiber, P.J.; Michl, J. Chemistry of the Carba-closo-dodecaborate(-) Anion, CB11H12−. Chem. Rev. 2006, 106, 5208–5249. [Google Scholar] [CrossRef]
- Kanazawa, J.; Kitazawa, Y.; Uchiyama, M. Recent Progress in the Synthesis of the Monocarba-closo-dodecaborate(−) Anions. Chem. Eur. J. 2019, 25, 9123–9132. [Google Scholar] [CrossRef]
- Douvris, C.; Michl, J. Update 1 of: Chemistry of the Carba-closo-dodecaborate(−) Anion, CB11H12−. Chem. Rev. 2013, 113, PR179–PR233. [Google Scholar] [CrossRef]
- Fisher, S.P.; Tomich, A.W.; Lovera, S.O.; Kleinsasser, J.F.; Guo, J.; Asay, M.J.; Nelson, H.M.; Lavallo, V. Nonclassical Applications of closo-Carborane Anions: From Main Group Chemistry and Catalysis to Energy Storage. Chem. Rev. 2019, 119, 8262–8290. [Google Scholar] [CrossRef]
- Strauss, S.H. The search for larger and more weakly coordinating anions. Chem. Rev. 1993, 93, 927–942. [Google Scholar] [CrossRef]
- Seppelt, K. “Noncoordinating” Anions, II. Angew. Chem. Int. Ed. Engl. 1993, 32, 1025–1027. [Google Scholar] [CrossRef]
- Reed, C.A. Carboranes: A New Class of Weakly Coordinating Anions for Strong Electrophiles, Oxidants, and Superacids. Acc. Chem. Res. 1998, 31, 133–139. [Google Scholar] [CrossRef]
- Riddlestone, I.M.; Kraft, A.; Schaefer, J.; Krossing, I. Taming the Cationic Beast: Novel Developments in the Synthesis and Application of Weakly Coordinating Anions. Angew. Chem. Int. Ed. 2018, 57, 13982–14024. [Google Scholar] [CrossRef]
- Krossing, I.; Raabe, I. Noncoordinating Anions—Fact or Fiction? A Survey of Likely Candidates. Angew. Chem. Int. Ed. 2004, 43, 2066–2090. [Google Scholar] [CrossRef]
- Bolli, C.; Köchner, T.; Knapp, C. [NO][HCB11Cl11]—Synthesis, Characterization, Crystal Structure, and Reaction with P4. Z. Anorg. Allg. Chem. 2012, 638, 559–564. [Google Scholar] [CrossRef]
- Knapp, C. Weakly Coordinating Anions: Halogenated Borates and Dodecaborates. In Comprehensive Inorganic Chemistry II; Reedijk, J., Poeppelmeier, K., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; Volume 1, pp. 651–679. [Google Scholar]
- Boeré, R.T.; Bolli, C.; Finze, M.; Himmelspach, A.; Knapp, C.; Roemmele, T.L. Quantum-Chemical and Electrochemical Investigation of the Electrochemical Windows of Halogenated Carborate Anions. Chem. Eur. J. 2013, 19, 1784–1795. [Google Scholar] [CrossRef]
- Dunks, G.B.; Palmer Ordonez, K. A One-Step Synthesis of B11H14− Ion from NaBH4. Inorg. Chem. 1978, 17, 1514–1516. [Google Scholar] [CrossRef]
- Dunks, G.B.; Barker, K.; Hedaya, E.; Hefner, C.; Palmer-Ordonez, K.; Remec, P. Simplified Synthesis of B10H14 from NaBH4 via B11H14− Ion. Inorg. Chem. 1981, 20, 1692–1697. [Google Scholar] [CrossRef]
- Knoth, W.H. 1-B9H9CH− and B11H11CH. J. Am. Chem. Soc. 1967, 89, 1274–1275. [Google Scholar] [CrossRef]
- Knoth, W.H. B10H12CNH3, B9H9CH−, B11H11CH−, and Metallomonocarboranes. Inorg. Chem. 1971, 10, 598–605. [Google Scholar] [CrossRef]
- Plešek, J.; Jelínek, T.; Drdáková, E.; Heřmánek, S.; Štíbr, B. A Convenient Preparation of 1-CB11H12− and Its C-Amino Derivatives. Collect. Czech. Chem. Commun. 1984, 49, 1559–1562. [Google Scholar] [CrossRef]
- Tanaka, N.; Shoji, Y.; Fukushima, T. Convenient Route to Monocarba-closo-dodecaborate Anions. Organometallics 2016, 35, 2022–2025. [Google Scholar] [CrossRef]
- Franken, A.; King, B.T.; Michl, J. Method for Preparation of Carborane Anions (WO002002079210). U.S. Patent 20040242934, 1 April 2002. [Google Scholar]
- Körbe, S.; Sowers, D.B.; Franken, A.; Michl, J. Preparation of 1-p-Halophenyl and 1-p-Biphenylyl Substituted Monocarbadodecaborate Anions [closo-1-Ar-CB11H11]− by Insertion of Arylhalocarbenes into [nido-B11H14]−. Inorg. Chem. 2004, 43, 8158–8161. [Google Scholar] [CrossRef]
- Franken, A.; King, B.T.; Rudolph, J.; Rao, P.; Noll, B.C.; Michl, J. Preparation of [closo-CB11H12]− by Dichlorocarbene Insertion into [nido-B11H14]−. Collect. Czech. Chem. Commun. 2001, 66, 1238–1249. [Google Scholar] [CrossRef]
- Berger, A.; Buckley, C.E.; Paskevicius, M. Synthesis of closo-CB11H12− Salts Using Common Laboratory Reagents. Inorg. Chem. 2021, 60, 14744–14751. [Google Scholar] [CrossRef]
- Toom, L.; Kütt, A.; Leito, I. Simple and scalable synthesis of the carborane anion CB11H12−. Dalton Trans. 2019, 48, 7499–7502. [Google Scholar] [CrossRef]
- Pecyna, J.; Rončević, I.; Michl, J. Insertion of Carbenes into Deprotonated nido-Undecaborane, B11H13(2-). Molecules 2019, 24, 3779. [Google Scholar] [CrossRef]
- Han, H.; Wang, Y.-Y.; Yu, X.-C.; Ma, Y.-N.; Chen, X. A Simple and Efficient Way to Directly Synthesize Unsolvated Alkali Metal (M = Na, K) Salts of [CB11H12]−. Crystals 2022, 12, 1339. [Google Scholar] [CrossRef]
- Grimes, R.N. Carboranes; Elsevier Inc.: London, UK, 2011. [Google Scholar]
- Michl, J. Chemistry of the three-dimensionally aromatic CB11 cage. Pure Appl. Chem. 2008, 80, 429–446. [Google Scholar] [CrossRef]
- Clayton, J.R.; King, B.T.; Zharov, I.; Fete, M.G.; Volkis, V.; Douvris, C.; Valášek, M.; Michl, J. Salts of dodecamethylcarba-closo-dodecaborate(-) anion, CB11Me12−, and the radical dodecamethylcarba-closo-dodecaboranyl, CB11Me12. Inorg. Synth. 2010, 35, 56–63. [Google Scholar]
- Nava, M.J.; Reed, C.A. High Yield C-Derivatization of Weakly Coordinating Carborane Anions. Inorg. Chem. 2010, 49, 4726–4728. [Google Scholar] [CrossRef]
- Tomich, A.W.; Chen, J.; Carta, V.; Guo, J.; Lavallo, V. Electrolyte Engineering with Carboranes for Next-Generation Mg Batteries. ACS Cent. Sci. 2024, 10, 264–271. [Google Scholar] [CrossRef]
- Lovera, S.O.; Bagsdasarian, A.L.; Guo, J.; Nelson, H.M.; Lavallo, V. Cesium carbonate mediated C–H functionalization of perhalogenated 12-vertex carborane anions. Chem. Commun. 2022, 58, 4060–4062. [Google Scholar] [CrossRef]
- Jelínek, T.; Baldwin, P.; Scheidt, W.R.; Reed, C.A. New weakly coordinating anions. 2. Derivatization of the carborane anion CB11H12−. Inorg. Chem. 1993, 32, 1982–1990. [Google Scholar] [CrossRef]
- Jelínek, T.; Kilner, C.A.; Thornton-Pett, M.; Kennedy, J.D. Monocarbaborane anion chemistry. The elusive C-arylated [PhCB11H11]−, [PhCB9H9]− and [PhCB8H8]− anions. Chem. Commun. 2001, 1790–1791. [Google Scholar]
- Volkov, O.; Paetzold, P. The chemistry of the undecaborates. J. Organomet. Chem. 2003, 680, 301–311. [Google Scholar] [CrossRef]
- Sivaev, I.B. Chemistry of 11-Vertex Polyhedral Boron Hydrides (Review). Russ. J. Inorg. Chem. 2019, 64, 955–976. [Google Scholar] [CrossRef]
- Aftandilian, V.D.; Miller, H.C.; Parshall, G.W.; Muetterties, E.L. Chemistry of Boranes. V. First Example of a B11 Hydride, the B11H14− Anion. Inorg. Chem. 1962, 1, 734–737. [Google Scholar] [CrossRef]
- Fritchie, C.J. The Crystal Structure of Cesium Tetramethylammonium Tridecahydroundecaborate. Inorg. Chem. 1967, 6, 1199–1203. [Google Scholar] [CrossRef]
- Brellochs, B. New Routes to Carboranes. In Contemporary Boron Chemistry; The Royal Society of Chemistry: London, UK, 2000; pp. 212–214. [Google Scholar]
- Bernhardt, E.; Willner, H. Verfahren zur Herstellung von closo-Boraten. Disclosure document DE 102008004530 A1, 15 January 2008. Available online: https://worldwide.espacenet.com/patent/search/family/040758545/publication/DE102008004530A1?q=DE102008004530A1 (accessed on 12 June 2024).
- Preetz, W.; Peters, G. The Hexahydro-closo-hexaborate Dianion [B6H6]2− and Its Derivatives. Eur. J. Inorg. Chem. 1999, 1999, 1831–1846. [Google Scholar] [CrossRef]
- Sivaev, I.B.; Prikaznov, A.V.; Naoufal, D. Fifty years of the closo-decaborate anion chemistry. Collect. Czech. Chem. Commun. 2010, 75, 1149–1199. [Google Scholar] [CrossRef]
- Sivaev, I.B.; Bregadze, V.V.; Sjöberg, S. Chemistry of closo-Dodecaborate Anion [B12H12]2−: A Review. Collect. Czech. Chem. Commun. 2002, 67, 679–727. [Google Scholar] [CrossRef]
- Knoth, W.H.; Muetterties, E.L. Chemistry of Boranes—II. Decaborane derivatives based on the B10H12 structural unit. J. Inorg. Nucl. Chem. 1961, 20, 66–72. [Google Scholar] [CrossRef]
- Baše, K.; Alcock, N.W.; Howarth, O.W.; Powell, H.R.; Harrison, A.T.; Wallbridge, M.G.H. The structure of the arachno-[B10H13CN]2− anion; an example of endo substitution in the decaborane(14) framework. J. Chem. Soc. Chem. Commun. 1988, 341–342. [Google Scholar] [CrossRef]
- Schlüter, F. Die Chemie der closo-Borate [BnHn]2− (n = 6–9, 11) und [B21H18]−. Doctoral Thesis, Bergische Universität Wuppertal, Wuppertal, Germany, 2012. [Google Scholar]
- Konieczka, S.Z.; Schlüter, F.; Sindorf, C.; Kerpen, C.; Bernhardt, E.; Finze, M. Stepwise Introduction of Cyano Groups into nido- and closo-Undecaborate Clusters. Chem. Eur. J. 2018, 24, 3528–3538. [Google Scholar] [CrossRef]
- Onak, T.P.; Williams, R.E.; Weiss, H.G. The Synthesis of B4CnH2n+4 Compounds from Pentaborane-9 and Alkynes Catalyzed by 2,6-Dimethylpyridine. J. Am. Chem. Soc. 1962, 84, 2830–2831. [Google Scholar] [CrossRef]
- Zakharkin, L.I.; Stanko, V.I.; Brattsev, V.A.; Chapovskii, Y.A.; Okhlobystin, O.Y. Synthesis of a new class of organoboron compounds, B10C2H12 (barene) and its derivatives. Bull. Acad. Sci. USSR Div. Chem. Sci. 1963, 12, 2074. [Google Scholar] [CrossRef]
- Beall, H. Icosahedral Carboranes. XVII. A Simplified Preparation of o-Carborane. Inorg. Chem. 1972, 11, 637–638. [Google Scholar] [CrossRef]
- Bicerano, J.; Marynick, D.S.; Lipscomb, W.N. Molecular Orbital Studies on Large Closo Boron Hydrides. Inorg. Chem. 1978, 17, 3443–3453. [Google Scholar] [CrossRef]
- Pearson, R.G. Ionization potentials and electron affinities in aqueous solution. J. Am. Chem. Soc. 1986, 108, 6109–6114. [Google Scholar] [CrossRef]
- Mordini, A.; Valacchi, M. Product subclass 8: Sodium acetylides. In Science of Synthesis, 8b: Category 1, Organometallics; Science of Synthesis; Georg Thieme Verlag KG: Stuttgart, Germany, 2006; Volume 8b, pp. 1209–1214. [Google Scholar]
- Castro, C.E.; Gaughan, E.J.; Owsley, D.C. Indoles, Benzofurans, Phthalides, and Tolanes via Copper(I) Acetylides. J. Org. Chem. 1966, 31, 4071–4078. [Google Scholar] [CrossRef]
- Kraus, G.A.; Taschner, M.J. Timed Diels-Alder reactions. J. Am. Chem. Soc. 1980, 102, 1974–1977. [Google Scholar] [CrossRef]
- Balsamo, A.; Macchia, B.; Macchia, F.; Rossello, A.; Domiano, P. 3-(4-Iodomethyl-2-oxo-1-azetidinyl)propynoic acid t-butyl ester: A new β-lactam derivative. Synthesis of an ynamide by reaction of 4-iodomethylazetidin-2-one with the t-butyl ester of propiolic acid in the presence of copper(I). Tetrahedron Lett. 1985, 26, 4141–4144. [Google Scholar] [CrossRef]
- Fujimori, S.; Carreira, E.M. CuI-Catalyzed Conjugate Addition of Ethyl Propiolate. Angew. Chem. Int. Ed. 2007, 46, 4964–4967. [Google Scholar] [CrossRef]
- Jouvin, K.; Heimburger, J.; Evano, G. Click-alkynylation of N- and P-nucleophiles by oxidative cross-coupling with alkynylcopper reagents: A general synthesis of ynamides and alkynylphosphonates. Chem. Sci. 2012, 3, 756–760. [Google Scholar] [CrossRef]
- Jouvin, K.; Veillard, R.; Theunissen, C.; Alayrac, C.; Gaumont, A.-C.; Evano, G. Unprecedented Synthesis of Alkynylphosphine-boranes through Room-Temperature Oxidative Alkynylation. Org. Lett. 2013, 15, 4592–4595. [Google Scholar] [CrossRef]
- Theunissen, C.; Lecomte, M.; Jouvin, K.; Laouiti, A.; Guissart, C.; Heimburger, J.; Loire, E.; Evano, G. Convenient and practical alkynylation of heteronucleophiles with copper acetylides. Synthesis 2014, 46, 1157–1166. [Google Scholar]
- Chen, W.; Wang, B.; Liu, N.; Huang, D.; Wang, X.; Hu, Y. Tandem Synthesis of 3-Halo-5-Substituted Isoxazoles from 1-Copper(I) Alkynes and Dihaloformaldoximes. Org. Lett. 2014, 16, 6140–6143. [Google Scholar] [CrossRef]
- Chang, X.-Y.; Low, K.-H.; Wang, J.-Y.; Huang, J.-S.; Che, C.-M. From Cluster to Polymer: Ligand Cone Angle Controlled Syntheses and Structures of Copper(I) Alkynyl Complexes. Angew. Chem. Int. Ed. 2016, 55, 10312–10316. [Google Scholar] [CrossRef]
- Weng, Y.; Min, L.; Zeng, X.; Shan, L.; Wang, X.; Hu, Y. General Synthesis of α-Alkyl Ynones from Morpholine Amides and 1-Copper(I) Alkynes Promoted by Triflic Anhydride. Org. Lett. 2020, 22, 8296–8301. [Google Scholar] [CrossRef]
- Davis, R.B.; Scheiber, D.H. The Preparation of Acetylenic Ketones Using Soluble Silver Acetylides. J. Am. Chem. Soc. 1956, 78, 1675–1678. [Google Scholar] [CrossRef]
- CIBALtd. Silver Propiolic Acid Esters and Process for Their Manufacture. Patent: BE 620022A; GB 977227A; GB 977228A. 1963. Available online: https://worldwide.espacenet.com/patent/search/family/025701225/publication/BE620022A?q=BE%20620022A (accessed on 12 June 2024).
- De Las Heras, F.G.; Tam, S.Y.K.; Klein, R.S.; Fox, J.J. Nucleosides. XCIV. Synthesis of Some C Nucleosides by 1,3-Dipolar Cycloadditions to 3-(Ribofuranosyl) Propiolates. J. Org. Chem. 1976, 41, 84–90. [Google Scholar] [CrossRef]
- Yerino, L.V.; Osborn, M.E.; Mariano, P.S. The hydroazocine route to highly functionalized pyrrolizidines. Tetrahedron 1982, 38, 1579–1591. [Google Scholar] [CrossRef]
- Schubert, T. Chemoenzymatische Synthese α,β-propargylischer Alkohole In Berichte des Forschungszentrums Jülich; Forschungszentrum Jülich, Jülich, Germany, 2002; Jül-3983.
- Gorin, D.J.; Toste, F.D. Relativistic effects in homogeneous gold catalysis. Nature 2007, 446, 395–403. [Google Scholar] [CrossRef]
- Ghosh, T.; Chatterjee, J.; Bhakta, S. Gold-catalyzed hydroarylation reactions: A comprehensive overview. Org. Biomol. Chem. 2022, 20, 7151–7187. [Google Scholar] [CrossRef]
- Lo, V.K.-Y.; Chan, A.O.-Y.; Che, C.-M. Gold and silver catalysis: From organic transformation to bioconjugation. Org. Biomol. Chem. 2015, 13, 6667–6680. [Google Scholar] [CrossRef]
- Carlos Lima, J.; Rodríguez, L. Applications of gold(i) alkynyl systems: A growing field to explore. Chem. Soc. Rev. 2011, 40, 5442–5456. [Google Scholar] [CrossRef]
- Widenhoefer, R.A. Recent Developments in Enantioselective Gold(I) Catalysis. Chem. Eur. J. 2008, 14, 5382–5391. [Google Scholar] [CrossRef]
- Arcadi, A. Alternative Synthetic Methods through New Developments in Catalysis by Gold. Chem. Rev. 2008, 108, 3266–3325. [Google Scholar] [CrossRef]
- Hobosyan, N.G.; Balyan, K.V.; Pogosyan, A.R.; Sargsyan, A.B. Alkylacetylenes in Mercuration-Demercuration Reactions with 1,3-Dicarbonyl Compounds. Russ. J. Gen. Chem. 2021, 91, 2123–2128. [Google Scholar] [CrossRef]
- Nishizawa, M.; Imagawa, H.; Yamamoto, H. A new catalyst for organic synthesis: Mercuric triflate. Org. Biomol. Chem. 2010, 8, 511–521. [Google Scholar] [CrossRef]
- Meyer, R.J. Gmelins Handbuch der Anorganischen Chemie, Kupfer Teil B—Lieferung 1, 8th ed.; Verlag Chemie GmbH: Weinheim/Bergstraße, Germany, 1958. [Google Scholar]
- Weidlein, J.; Müller, U.; Dehnicke, K. Schwingungsfrequenzen/Hautgruppenelemente; Georg Thieme Verlag: Stuttgar, Germany, 1981. [Google Scholar]
- Kester, J.G.; Keller, D.; Huffman, J.C.; Benefiel, M.A.; Geiger, W.E., Jr.; Atwood, C.; Siedle, A.R.; Korba, G.A.; Todd, L.J. Synthesis and Properties of Copper and Nickel Complexes of the General Formula (B11H11)2Mn−. Crystal Structure of [(n-Bu)4N]3[Cu(B11H11)2]. Inorg. Chem. 1994, 33, 5438–5442. [Google Scholar] [CrossRef]
- Bartl, H.; Falbe, J. Makromolekulare Stoffe, II. In Methoden der Organischen Chemie (Houben-Weyl); Georg Thieme Verlag: Stuttgart, Germany, 1987; Volume E20. [Google Scholar]
- Matnishyan, A.A.; Kobryanskii, V.M. Characteristic Features of the Polymerisation of Acetylene and Its Derivatives. Russ. Chem. Rev. 1983, 52, 751–756. [Google Scholar] [CrossRef]
- Almarzoqi, B.; George, A.V.; Isaacs, N.S. The quaternization of tertiary amines with dihalomethane. Tetrahedron 1986, 42, 601–607. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, X.; Chen, T.; Zhou, Y.; Yin, S.-F.; Han, L.-B. Catalyst-Free and Selective C-N Bond Functionalization: Stereospecific Three-Component Coupling of Amines, Dichloromethane, and >P(O)H Species Affording α-Aminophosphorus Compounds. J. Org. Chem. 2015, 80, 62–69. [Google Scholar] [CrossRef]
- Wright, D.A.; Wulff, C.A. Chloromethyltriethylammonium Chloride. A Serendipitous Preparation. J. Org. Chem. 1970, 35, 4252. [Google Scholar] [CrossRef]
- Larsen, A.S.; Holbrey, J.D.; Tham, F.S.; Reed, C.A. Designing Ionic Liquids: Imidazolium Melts with Inert Carborane Anions. J. Am. Chem. Soc. 2000, 122, 7264–7272. [Google Scholar] [CrossRef]
- Novikov, A.P.; Bezdomnikov, A.A.; Grigoriev, M.S.; German, K.E. Synthesis, crystal structure and Hirshfeld surface analysis of 2-(perfluorophenyl)acetamide in comparison with some related compounds. Acta Crystallogr. Sect. E Struct. Rep. Online 2022, 78, 80–83. [Google Scholar] [CrossRef]
- Skelton, B.W.; Pakawatchai, C.; White, A.H. CCDC 642663: Experimental Crystal Structure Determination; The Cambridge Crystallographic Data Centre (CCDC): Cambridge, UK, 2017. [Google Scholar]
- Ganesan, M.; Nagaraaj, P. Recent developments in dehydration of primary amides to nitriles. Org. Chem. Front. 2020, 7, 3792–3814. [Google Scholar] [CrossRef]
- Rigaku. CrysAlisPRO; Oxford Diffraction/Agilent Technologies U.K. Ltd.: Yarnton, UK, 2016. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar]
- Brandenburg, K. Diamond, v.3.2f; Crystal Impact GbR: Bonn, Germany, 2001. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond | [NEt3CH2Cl] [NH2C(O)CH2-CB11H11] a | K[NH2C(O)CH2-CB11H11] a | NH2C(O)CH2-C6F5 b | NH2C(O)CH2-C6H5 c,d |
|---|---|---|---|---|
| C1-C2 | 155.0(12) | 153.43(12) | 150.43(17) | 150.2 |
| C2-C3 | 150.5(12) | 152.10(13) | 151.92(18) | 151.5 |
| C3-O1 | 124.8(9) | 123.74(12) | 124.30(15) | 123.9 |
| C3-N1 | 133.6(9) | 133.09(12) | 132,72(17) | 131.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barra, V.C.; Bernhardt, E.; Fellinger, S.; Jenne, C.; Langenbach, S.S. Direct Synthesis of C-Substituted [RC(O)CH2-CB11H11]− Carborate Anions. Inorganics 2024, 12, 173. https://doi.org/10.3390/inorganics12060173
Barra VC, Bernhardt E, Fellinger S, Jenne C, Langenbach SS. Direct Synthesis of C-Substituted [RC(O)CH2-CB11H11]− Carborate Anions. Inorganics. 2024; 12(6):173. https://doi.org/10.3390/inorganics12060173
Chicago/Turabian StyleBarra, Vanessa C., Eduard Bernhardt, Sarah Fellinger, Carsten Jenne, and Shiomi S. Langenbach. 2024. "Direct Synthesis of C-Substituted [RC(O)CH2-CB11H11]− Carborate Anions" Inorganics 12, no. 6: 173. https://doi.org/10.3390/inorganics12060173
APA StyleBarra, V. C., Bernhardt, E., Fellinger, S., Jenne, C., & Langenbach, S. S. (2024). Direct Synthesis of C-Substituted [RC(O)CH2-CB11H11]− Carborate Anions. Inorganics, 12(6), 173. https://doi.org/10.3390/inorganics12060173