

New Iron(III)-Containing Composite of Salinomycinic Acid with Antitumor Activity—Synthesis and Characterization
 , , , , , ,
, , , , , ,  , , , ,
, , , ,
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Elemental Analysis
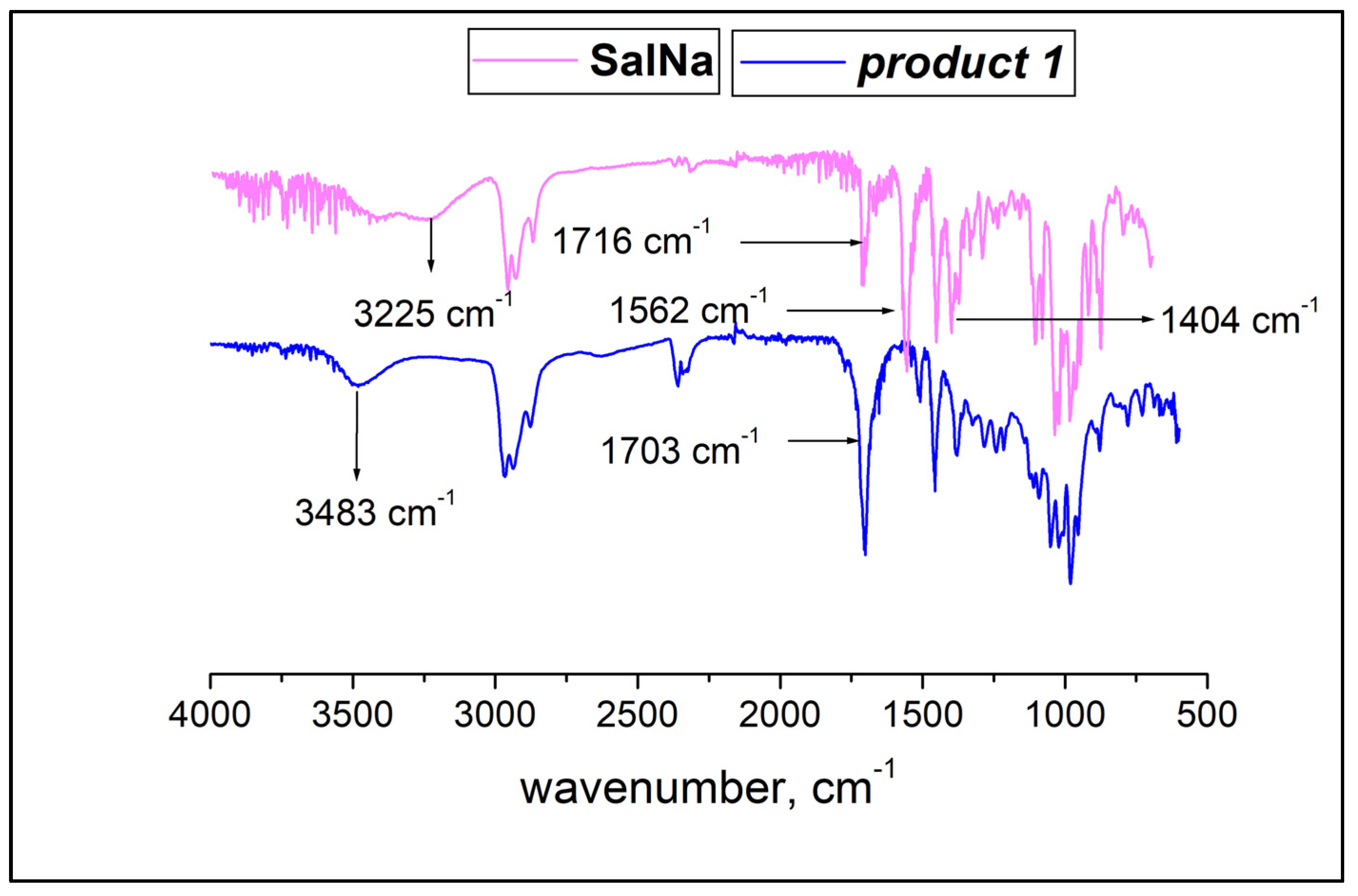
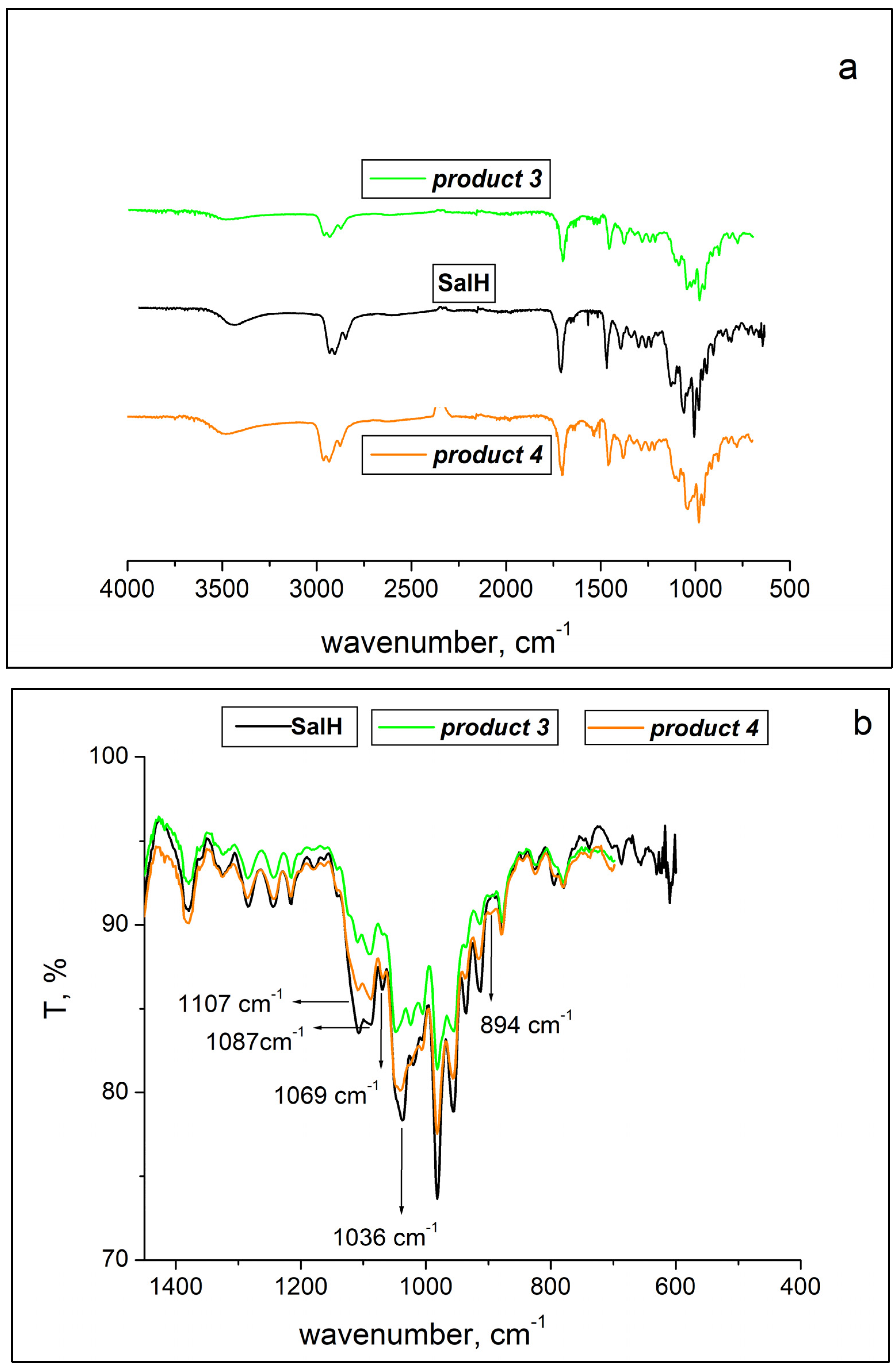
2.2. ATR-FTIR Spectroscopy
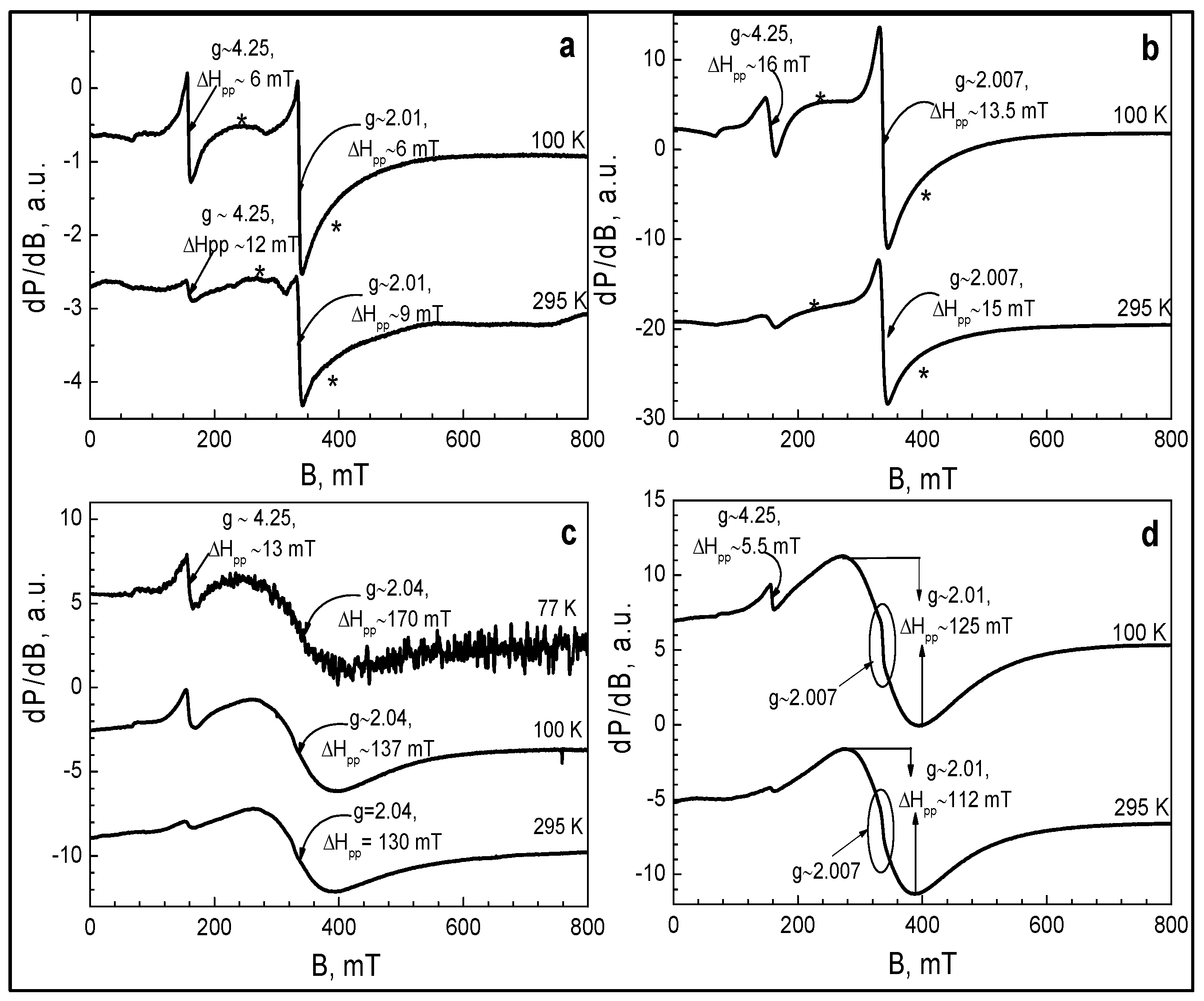
2.3. Electron Paramagnetic Resonance Spectroscopy (EPR)
2.4. Thermogravimetric Studies
2.5. Electrospray-Ionization Mass Spectrometry
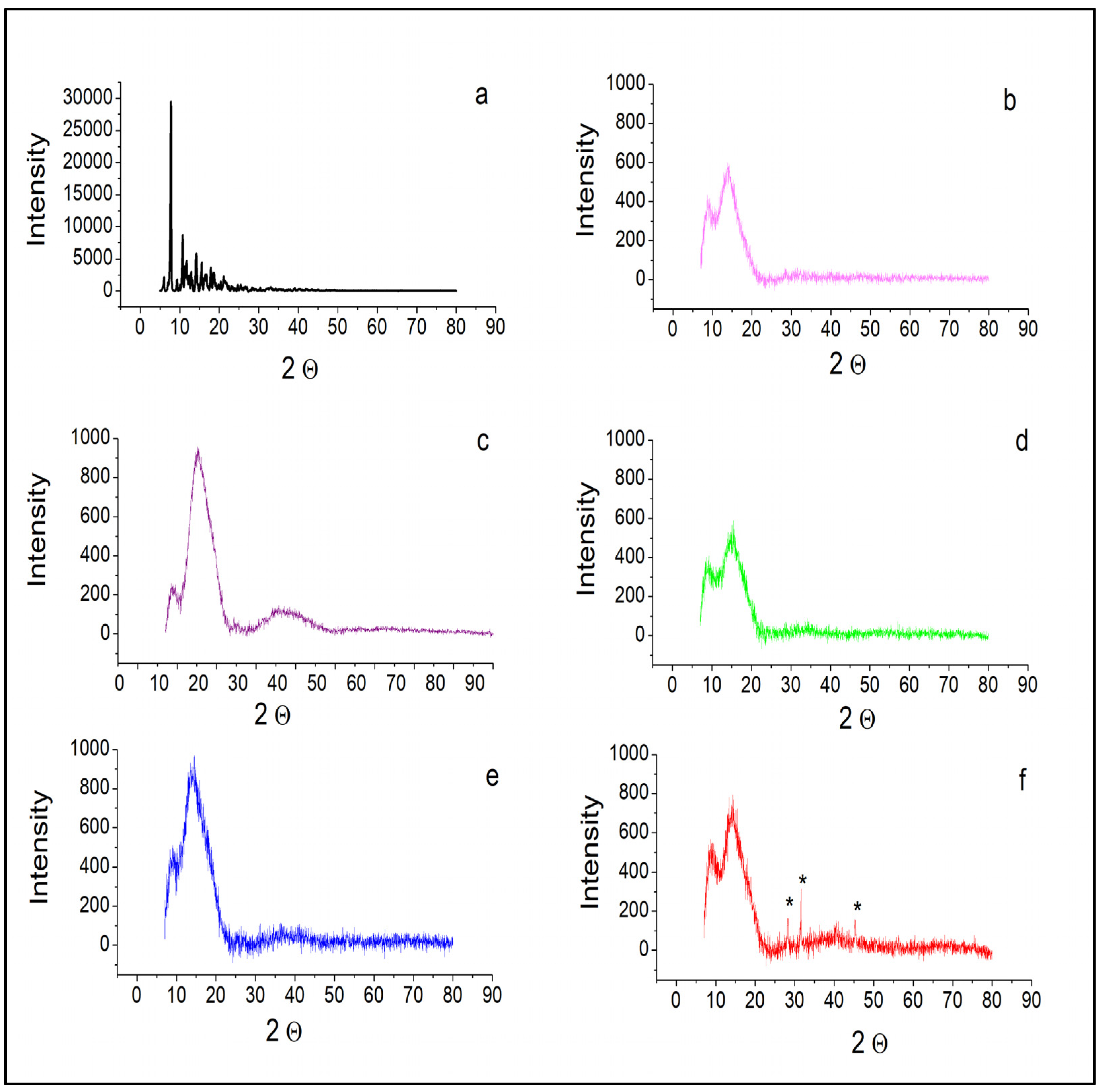
2.6. Powder XRD Analysis
2.7. Proposed Compositions of the Isolated Products
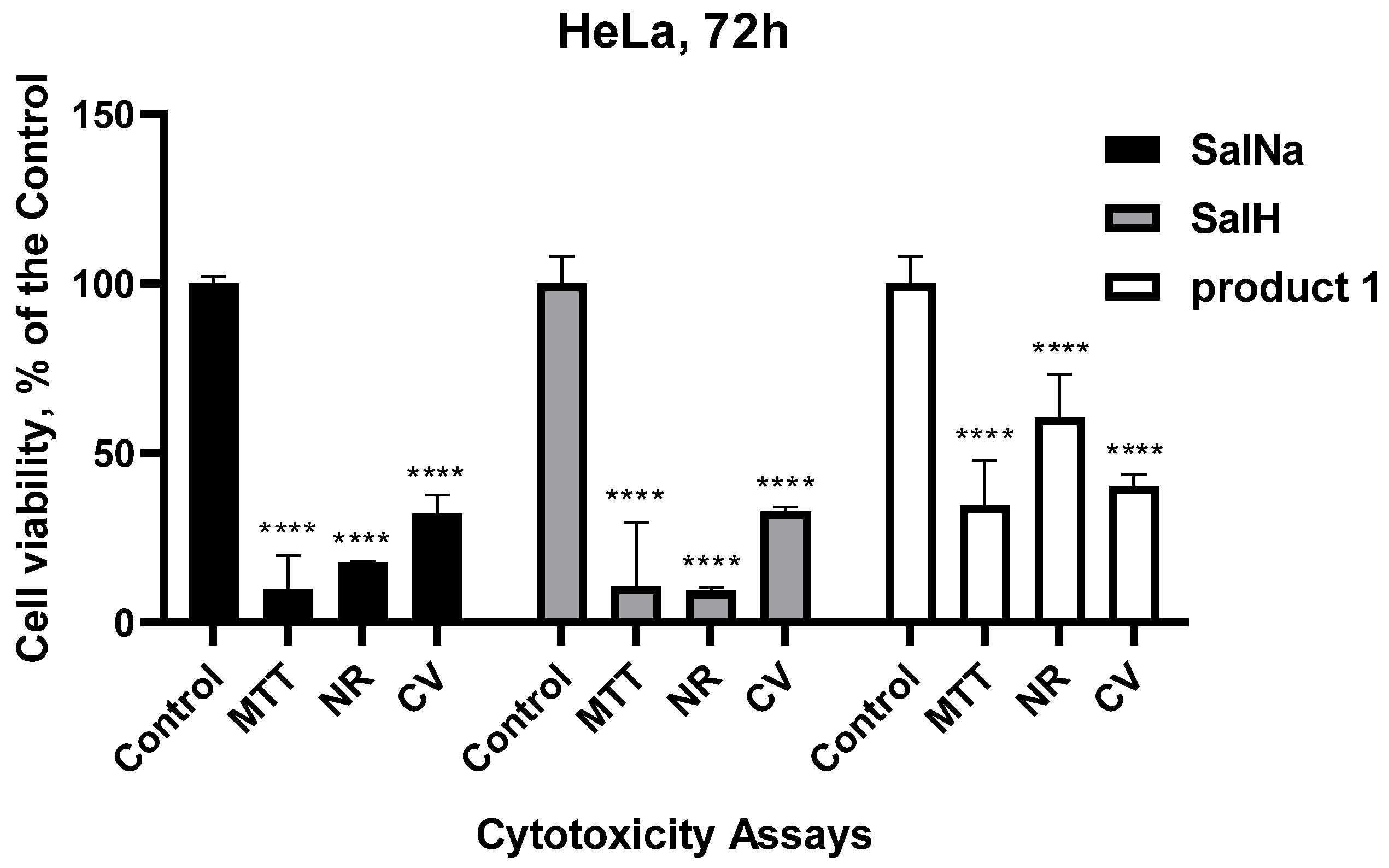
2.8. Biological Evaluation
3. Materials and Methods
3.1. Chemicals
3.2. Synthesis
3.3. Elemental Analysis
3.4. Attenuated Total Reflectance–Fourier-Transform Spectroscopy (ATR-FTIR)
3.5. Electron Paramagnetic Resonance Spectroscopy (EPR)
3.6. Powder X-ray Diffraction (XRD)
3.7. Electrospray-Ionization Mass Spectrometry (ESI-MS)
3.8. Thermogravimetric Analysis with Differential Thermal Analysis (DTA) and Mass Spectrometry
3.9. Biological Activity Evaluation
Cytotoxicity Tests
- Cell culture models
- MTT Assay
- NR test
- CV staining
- Double staining with acridine orange (AO) and propidium iodide (PI)
- Three-dimensional colony-forming assay
3.10. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huczyński, A.; Janczak, J.; Antoszczak, M.; Wietrzyk, J.; Maj, E.; Brzezinski, B. Antiproliferative activity of salinomycin and its derivatives. Bioorganic Med. Chem. Lett. 2012, 22, 7146–7150. [Google Scholar] [CrossRef]
- Antoszczak, M.; Huczyński, A. Salinomycin and its derivatives—A new class of multiple-targeted “magic bullets”. Eur. J. Med. Chem. 2019, 176, 208–227. [Google Scholar] [CrossRef]
- Mai, T.T.; Hamaï, A.; Hienzsch, A.; Cañeque, T.; Müller, S.; Wicinski, J.; Cabaud, O.; Leroy, C.; David, A.; Acevedo, V.; et al. Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nat. Chem. 2017, 9, 1025–1033. [Google Scholar] [CrossRef]
- Dewangan, J.; Srivastava, S.; Rath, S.K. Salinomycin: A new paradigm in cancer therapy. Tumor Biol. 2017, 39, 1010428317695035. [Google Scholar] [CrossRef]
- Ebokaiwe, A.P.; Njoya, E.M.; Sheng, Y.; Zhang, Z.; Li, S.; Zhou, Z.; Qiang, Z.; Peng, T.; Hussein, A.A.; Zhang, G.; et al. Salinomycin promotes T-cell proliferation by inhibiting the expression and enzymatic activity of immunosuppressive indoleamine-2,3-dioxygenase in human breast cancer cells. Toxicol. Appl. Pharmacol. 2020, 404, 115203. [Google Scholar] [CrossRef]
- Yuan, C.; Huang, X.; Zhai, R.; Ma, Y.; Xu, A.; Zhang, P.; Yang, Q. In Vitro Antiviral Activities of Salinomycin on Porcine Epidemic Diarrhea Virus. Viruses 2021, 13, 580. [Google Scholar] [CrossRef]
- Jang, Y.; Shin, J.S.; Yoon, Y.-S.; Go, Y.Y.; Lee, H.W.; Kwon, O.S.; Park, S.; Park, M.-S.; Kim, M. Salinomycin Inhibits Influenza Virus Infection by Disrupting Endosomal Acidification and Viral Matrix Protein 2 Function. J. Virol. 2018, 92, e01441-18. [Google Scholar] [CrossRef]
- Qayed, W.S.; Ferreira, R.S.; Silva, J.R.A. In Silico Study towards Repositioning of FDA-Approved Drug Candidates for Anticoronaviral Therapy: Molecular Docking, Molecular Dynamics and Binding Free Energy Calculations. Molecules 2022, 27, 5988. [Google Scholar] [CrossRef]
- Gurukkalot, K.; Rajendran, V. Repurposing Polyether Ionophores as a New-Class of Anti-SARS-CoV-2 Agents as Adjunct Therapy. Curr. Microbiol. 2023, 80, 273. [Google Scholar] [CrossRef]
- Wang, Z.; Xiong, Y.; Yu, S.; Li, H.; Fan, J.; Li, F.; Su, Z.; Song, J.; Sun, Q.; Liu, S.; et al. Salinomycin exerts anti-colorectal cancer activity by targeting the β-catenin/T-cell factor complex. Br. J. Pharmacol. 2019, 176, 3390–3406. [Google Scholar] [CrossRef]
- Klose, J.; Trefz, S.; Wagner, T.; Steffen, L.; Charrier, A.P.; Radhakrishnan, P.; Volz, C.; Schmidt, T.; Ulrich, A.; Dieter, S.M.; et al. Salinomycin: Anti-tumor activity in a pre-clinical colorectal cancer model. PLoS ONE 2019, 14, e0211916. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhuo, J.; Tao, Y.; Xu, S.; Chen, Z.; Yang, F.; Ke, Q.; Xie, H.; Zheng, S.; Wang, H.; et al. Salinomycin-Loaded Small-Molecule Nanoprodrugs Enhance Anticancer Activity in Hepatocellular Carcinoma. Int. J. Nanomed. 2020, 15, 6839–6854. [Google Scholar] [CrossRef] [PubMed]
- Naujokat, C.; Steinhart, R. Salinomycin as a Drug for Targeting Human Cancer Stem Cells. J. Biomed. Biotechnol. 2012, 2012, 950658. [Google Scholar] [CrossRef] [PubMed]
- Pashkunova-Martic, I.; Kukeva, R.; Stoyanova, R.; Pantcheva, I.; Dorkov, P.; Friske, J.; Hejl, M.; Jakupec, M.; Hohagen, M.; Legin, A.; et al. Novel Salinomycin-Based Paramagnetic Complexes—First Evaluation of Their Potential Theranostic Properties. Pharmaceutics 2022, 14, 2319. [Google Scholar] [CrossRef] [PubMed]
- Momekova, D.; Momekov, G.; Ivanova, J.; Pantcheva, I.; Drakalska, E.; Stoyanov, N.; Guenova, M.; Michova, A.; Balashev, K.; Arpadjan, S.; et al. Sterically stabilized liposomes as a platform for salinomycin metal coordination compounds: Physicochemical characterization and in vitro evaluation. J. Drug Deliv. Sci. Technol. 2013, 23, 215–223. [Google Scholar] [CrossRef]
- Ivanova, J.; Pantcheva, I.N.; Zhorova, R.; Momekov, G.; Simova, S.; Stoyanova, R.; Zhecheva, E.; Ivanova, S.; Mitewa, M. Synthesis, spectral properties, antibacterial and antitumor activity of salinomycin complexes with Co(II), Ni(II), Cu(II) and Zn(II) transition metal ions. J. Chem Chem Eng. 2012, 6, 551–562. [Google Scholar]
- Vaidya, S.P.; Gadre, S.; Kamisetti, R.T.; Patra, M. Challenges and opportunities in the development of metal-based anticancer theranostic agents. Biosci. Rep. 2022, 42, BSR20212160. [Google Scholar] [CrossRef] [PubMed]
- Snyder, E.M.; Asik, D.; Abozeid, S.M.; Burgio, A.; Bateman, G.; Turowski, S.G.; Spernyak, J.A.; Morrow, J.R. A Class of FeIII Macrocyclic Complexes with Alcohol Donor Groups as Effective T1 MRI Contrast Agents. Angew. Chem. Int. Ed. 2020, 59, 2414–2419. [Google Scholar] [CrossRef]
- Palagi, L.; Di Gregorio, E.; Costanzo, D.; Stefania, R.; Cavallotti, C.; Capozza, M.; Aime, S.; Gianolio, E. Fe(deferasirox)2: An Iron(III)-Based Magnetic Resonance Imaging T1 Contrast Agent Endowed with Remarkable Molecular and Functional Characteristics. J. Am. Chem. Soc. 2021, 143, 14178–14188. [Google Scholar] [CrossRef]
- Baranyai, Z.; Carniato, F.; Nucera, A.; Horváth, D.; Tei, L.; Platas-Iglesias, C.; Botta, M. Defining the conditions for the development of the emerging class of FeIII-based MRI contrast agents. Chem. Sci. 2021, 12, 11138–11145. [Google Scholar] [CrossRef]
- Asik, D.; Abozeid, S.M.; Turowski, S.G.; Spernyak, J.A.; Morrow, J.R. Dinuclear Fe(III) Hydroxypropyl-Appended Macrocyclic Complexes as MRI Probes. Inorg. Chem. 2021, 60, 8651–8664. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; An, L.; Yang, S. Low-Molecular-Weight Fe(III) Complexes for MRI Contrast Agents. Molecules 2022, 27, 4573. [Google Scholar] [CrossRef] [PubMed]
- Kras, E.A.; Snyder, E.M.; Sokolow, G.E.; Morrow, J.R. Distinct Coordination Chemistry of Fe(III)-Based MRI Probes. Accounts Chem. Res. 2022, 55, 1435–1444. [Google Scholar] [CrossRef] [PubMed]
- Sahiner, N.; Umut, E.; Suner, S.S.; Sahiner, M.; Culha, M.; Ayyala, R.S. Hyaluronic acid (HA)-Gd(III) and HA-Fe(III) microgels as MRI contrast enhancing agents. Carbohydr. Polym. 2022, 277, 118873. [Google Scholar] [CrossRef] [PubMed]
- Botta, M.; Geraldes, C.F.; Tei, L. High spin Fe(III)-doped nanostructures as T1 MR imaging probes. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2023, 15, e1858. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wong, A.; Lewis, L.C.; Nemeth, G.R.; Jordan, V.C.; Bacon, J.W.; Caravan, P.; Shafaat, H.S.; Gale, E.M. Rational Ligand Design Enables pH Control over Aqueous Iron Magnetostructural Dynamics and Relaxometric Properties. Inorg. Chem. 2020, 59, 17712–17721. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, J.; Pantcheva, I.N.; Mitewa, M.; Simova, S.; Tanabe, M.; Osakada, K. Cd(II) and Pb(II) complexes of the polyether ionophorous antibiotic salinomycin. Chem. Central J. 2011, 5, 52. [Google Scholar] [CrossRef] [PubMed]
- Pantcheva, I.; Petkov, N.; Simova, S.; Zhorova, R.; Dorkov, P. Alkaline-earth metal(II) complexes of salinomycin—Spectral properties and antibacterial activity. Phys. Sci. Rev. 2022, 8, 3799–3811. [Google Scholar] [CrossRef]
- Sorokin, A.B. Recent progress on exploring µ-oxo bridged binuclear porphyrinoid complexes in catalysis and material science. Coord. Chem. Rev. 2019, 389, 141–160. [Google Scholar] [CrossRef]
- Wang, H.-L.; Cui, J.-Y.; Jiang, W.-F. Synthesis, characterization and flocculation activity of novel Fe(OH)3–polyacrylamide hybrid polymer. Mater. Chem. Phys. 2011, 130, 993–999. [Google Scholar] [CrossRef]
- Muralidhara, R.; Kesavulu, C.; Rao, J.; Anavekar, R.; Chakradhar, R. EPR and optical absorption studies of Fe3+ ions in sodium borophosphate glasses. J. Phys. Chem. Solids 2010, 71, 1651–1655. [Google Scholar] [CrossRef]
- Vercamer, V.; Lelong, G.; Hijiya, H.; Kondo, Y.; Galoisy, L.; Calas, G. Diluted Fe3+ in silicate glasses: Structural effects of Fe-redox state and matrix composition. An optical absorption and X-band/Q-band EPR study. J. Non-Crystalline Solids 2015, 428, 138–145. [Google Scholar] [CrossRef]
- Ardelean, I.; Peteanu, M.; Simon, V.; Ciorcas, F.; Ioncu, V. Structural investigation of Fe2O3-TeO2-B2O3-SrO glasses by EPR. J. Mater. Sci. Lett. 2001, 20, 947–949. [Google Scholar] [CrossRef]
- Castner, T.; Newell, G.S.; Holton, W.C.; Slichter, C.P. Note on the Paramagnetic Resonance of Iron in Glass. J. Chem. Phys. 1960, 32, 668–673. [Google Scholar] [CrossRef]
- Dzieniszewska, A.; Kyziol-Komosinska, J.; Pająk, M. Adsorption and bonding strength of chromium species by ferrihydrite from acidic aqueous solutions. PeerJ 2020, 8, e9324. [Google Scholar] [CrossRef] [PubMed]
- Pouran, H.M.; Banwart, S.A.; Romero-Gonzalez, M. Coating a polystyrene well-plate surface with synthetic hematite, goethite and aluminium hydroxide for cell mineral adhesion studies in a controlled environment. Appl. Geochem. 2014, 42, 60–68. [Google Scholar] [CrossRef]
- Perton, F.; Cotin, G.; Kiefer, C.; Strub, J.-M.; Cianferani, S.; Greneche, J.-M.; Parizel, N.; Heinrich, B.; Pichon, B.; Mertz, D.; et al. Iron Stearate Structures: An Original Tool for Nanoparticles Design. Inorg. Chem. 2021, 60, 12445–12456. [Google Scholar] [CrossRef] [PubMed]
- Hellman, H.; Laitinen, R.S.; Kaila, L.; Jalonen, J.; Hietapelto, V.; Jokela, J.; Sarpola, A.; Rämö, J. Identification of hydrolysis products of FeCl3·6H2O by ESI-MS. J. Mass Spectrom. 2006, 41, 1421–1429. [Google Scholar] [CrossRef] [PubMed]
- Hosny, W.M. Formation of Fe(III) Ternary Complexes with Related Bio-relevant Ligands. In Descriptive Inorganic Chemistry Researches of Metal Compounds; IntechOpen: Rijeka, Croatia, 2017. [Google Scholar] [CrossRef]
- Nurwahdah, N.; Rodiansono, R.; Kamari, A.; Abdullah, R.; Sunardi, S. The effects of FeCl3 concentration on hydrothermal pretreatment of oil palm fronds to enhance reducing sugar production. IOP Conf. Series Earth Environ. Sci. 2020, 591, 012024. [Google Scholar] [CrossRef]
- Jolivet, J.P.; Chanéac, C.; Tronc, E. Iron oxide chemistry. From molecular clusters to extended solid networks. Chem Communi-cations 2004, 5, 481–487. [Google Scholar]
- Zhu, M.; Frandsen, C.; Wallace, A.F.; Legg, B.; Khalid, S.; Zhang, H.; Mørup, S.; Banfield, J.F.; Waychunas, G.A. Precipitation pathways for ferrihydrite formation in acidic solutions. Geochim. Cosmochim. Acta 2016, 172, 247–264. [Google Scholar] [CrossRef]
- Gimsing, A.L.; Borggaard, O.K. Phosphate and glyphosate adsorption by hematite and ferrihydrite and comparison with other variable-charge minerals. Clays Clay Miner. 2007, 55, 108–114. [Google Scholar] [CrossRef]
- Hiemstra, T.; Van Riemsdijk, W.H. A surface structural model for ferrihydrite I: Sites related to primary charge, molar mass, and mass density. Geochim. Cosmochim. Acta 2009, 73, 4423–4436. [Google Scholar] [CrossRef]
- Kandioller, W.; Theiner, J.; Keppler, B.K.; Kowol, C.R. Elemental analysis: An important purity control but prone to manipulations. Inorg. Chem. Front. 2021, 9, 412–416. [Google Scholar] [CrossRef]
- Michel, F.M.; Ehm, L.; Antao, S.M.; Lee, P.L.; Chupas, P.J.; Liu, G.; Strongin, D.R.; Schoonen, M.A.A.; Phillips, B.L.; Parise, J.B. The Structure of Ferrihydrite, a Nanocrystalline Material. Science 2007, 316, 1726–1729. [Google Scholar] [CrossRef] [PubMed]
- Hiemstra, T. Surface and mineral structure of ferrihydrite. Geochim. Cosmochim. Acta 2012, 105, 316–325. [Google Scholar] [CrossRef]
- Ma, B.; Fernandez-Martinez, A.; Mancini, A.; Lothenbach, B. Spectroscopic investigations on structural incorporation pathways of FeIII into zeolite frameworks in cement-relevant environments. Cem. Concr. Res. 2020, 140, 106304. [Google Scholar] [CrossRef]
- Koksharov, Y.A.; Pankratov, D.A.; Gubin, S.P.; Kosobudsky, I.D.; Beltran, M.; Khodorkovsky, Y.; Tishin, A.M. Electron paramagnetic resonance of ferrite nanoparticles. J. Appl. Phys. 2001, 89, 2293–2298. [Google Scholar] [CrossRef]
- Jahagirdar, A.; Dhananjaya, N.; Monika, D.; Kesavulu, C.; Nagabhushana, H.; Sharma, S.; Nagabhushana, B.; Shivakumara, C.; Rao, J.; Chakradhar, R. Structural, EPR, optical and magnetic properties of α-Fe2O3 nanoparticles. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2012, 104, 512–518. [Google Scholar] [CrossRef]
- Naik, R.; Prashantha, S.; Nagabhushana, H.; Girish, K. Electrochemical, photoluminescence and EPR studies of Fe3+ doped nano Forsterite: Effect of doping on tetra and octahedral sites. J. Lumin. 2018, 197, 233–241. [Google Scholar] [CrossRef]
- Vallina, B.; Rodriguez-Blanco, J.D.; Brown, A.P.; Benning, L.G.; Blanco, J.A. Enhanced magnetic coercivity of α-Fe2O3 obtained from carbonated 2-line ferrihydrite. J. Nanoparticle Res. 2014, 16, 2322. [Google Scholar] [CrossRef]
- Boily, J.-F.; Song, X. Direct identification of reaction sites on ferrihydrite. Commun. Chem. 2020, 3, 79. [Google Scholar] [CrossRef]
- Varbanov, H.P.; Göschl, S.; Heffeter, P.; Theiner, S.; Roller, A.; Jensen, F.; Jakupec, M.A.; Berger, W.; Galanski, M.S.; Keppler, B.K. ANovel Class of Bis- and Tris-Chelate Diam(m)inebis(dicarboxylato)platinum(IV) Complexes as Potential Anticancer Prodrugs. J. Med. Chem. 2014, 57, 6751–6764. [Google Scholar] [CrossRef] [PubMed]
- Varbanov, H.P.; Valiahdi, S.M.; Kowol, C.R.; Jakupec, M.A.; Galanski, M.S.; Keppler, B.K. Novel tetracarboxylatoplati-num(iv)complexes as carboplatin prodrugs. Dalton Trans. 2012, 41, 14404–14415. [Google Scholar] [CrossRef] [PubMed]
- Śliwka, L.; Wiktorska, K.; Suchocki, P.; Milczarek, M.; Mielczarek, S.; Lubelska, K.; Cierpiał, T.; Łyżwa, P.; Kiełbasiński, P.; Jaromin, A.; et al. The Comparison of MTT and CVS Assays for the Assessment of Anticancer Agent Interactions. PLoS ONE 2016, 11, e0155772. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Borenfreund, E.; Puerner, J. Toxicity determination in vitro by morphological alterations and neural red absorption. Toxicol. Lett. 1985, 24, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Saotome, K.; Morita, H.; Umeda, M. Cytotoxicity test with simplified crystal violet staining method using microtitre plates and its application to injection drugs. Toxicol. Vitr. 1989, 3, 317–321. [Google Scholar] [CrossRef]
- Abdel Wahab, S.I.; Abdul, A.B.; Alzubairi, A.S.; Mohamed Elhassan, M.; Mohan, S. In Vitro Ultramorphological Assessment of Apoptosis Induced by Zerumbone on (HeLa). J. Biomed. Biotechnol. 2009, 2009, 769568. [Google Scholar] [CrossRef]
- Abudalleh, A.M.; Alexandrov, M.; Alexandrova, R. Ammonium vanadate decreases viability and proliferation activity of cul-tured virus-transformed rat sarcoma cells. Compt. Rend Acad. Bulg. Sci. 2013, 66, 61–66. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cexp., % | Ctheor., % | Hexp., % | Htheor., % | Oexp., % | Otheor., % | Feexp., % | Fe theor., % | |
|---|---|---|---|---|---|---|---|---|
| product 1, [(FeOOH)(C42H70O11)3], MW = 2341.88 g/mol | 64.65 ± 0.79 | 64.62 | 9.28 ± 0.09 | 9.08 | 24.67 ± 0.52 | 23.91 | 0.39 ± 0.01 | 2.38 |
| product 2, [(FeOOH)(C42H70O11)3]·4H2O, MW = 2413.944 g/mol | 60.95 ± 0.13 | 62.69 | 8.76 ± 0.03 | 9.14 | 25.20 ± 0.95 | 25.85 | 2.42 ± 0.06 | 2.32 |
| product 3, [(FeOOH)(C42H70O11)3]·4H2O, MW = 2413.944 g/mol | 62.58 ± 0.18 | 62.69 | 9.09 ± 0.09 | 9.14 | 27.55 ± 2.71 | 25.85 | 2.17 ± 0.04 | 2.32 |
| product 4, [(FeOOH)(C42H70O11)3]·4H2O, MW = 2413.944 g/mol | 61.89 ± 0.09 | 62.69 | 8.98 ± 0.03 | 9.14 | 25.85 ± 0.89 | 25.85 | 2.03 ± 0.27 | 2.32 |
| Cell Line | |||
|---|---|---|---|
| Compound | A549 | SW480 | CH1/PA-1 |
| SalH | 0.23 ± 0.06 | 1.1 ± 0.6 | 0.32 ± 0.12 |
| SalNa | 0.27 ± 0.02 | 0.88 ± 0.44 | 0.43 ± 0.11 |
| Product 1 | 0.43 ± 0.08 | 3.5 ± 0.1 | 0.27 ± 0.05 |
| Method | |||
|---|---|---|---|
| Compound | MTT | NR | CV |
| SalH | 2.5 | 5.7 | 3.9 |
| SalNa | 1.2 | 10.7 | 4.1 |
| Product 1 | 0.98 | 6.8 | 0.47 |
| Compound | Effective Inhibitory Concentration, μM |
|---|---|
| SalH | ≥1.33 |
| SalNa | ≥1.29 |
| Product 1 | ≥0.21 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ivanova, J.; Kukeva, R.; Stoyanova, R.; Zhivkova, T.; Abudalleh, A.; Dyakova, L.; Alexandrova, R.; Pashkunova-Martic, I.; Theiner, J.; Dorkov, P.; et al. New Iron(III)-Containing Composite of Salinomycinic Acid with Antitumor Activity—Synthesis and Characterization. Inorganics 2024, 12, 206. https://doi.org/10.3390/inorganics12080206
Ivanova J, Kukeva R, Stoyanova R, Zhivkova T, Abudalleh A, Dyakova L, Alexandrova R, Pashkunova-Martic I, Theiner J, Dorkov P, et al. New Iron(III)-Containing Composite of Salinomycinic Acid with Antitumor Activity—Synthesis and Characterization. Inorganics. 2024; 12(8):206. https://doi.org/10.3390/inorganics12080206
Chicago/Turabian StyleIvanova, Juliana, Rositsa Kukeva, Radostina Stoyanova, Tanya Zhivkova, Abedulkadir Abudalleh, Lora Dyakova, Radostina Alexandrova, Irena Pashkunova-Martic, Johannes Theiner, Peter Dorkov, and et al. 2024. "New Iron(III)-Containing Composite of Salinomycinic Acid with Antitumor Activity—Synthesis and Characterization" Inorganics 12, no. 8: 206. https://doi.org/10.3390/inorganics12080206