Abstract
In this study, mono- and bimetallic platinum (Pt), palladium (Pd) and Pt-Pd nanoparticles were synthesized using the wet sol–gel method, employing a carbon-based XC72R as catalytic carrier. The overall metal content was set at 40 wt.% at varying Pt:Pd ratios. Characterization of the morphology and surface structure was conducted through scanning electron microscopy (SEM), energy-dispersive X-ray spectroscopy (EDX), Brunauer–Emmett–Teller (BET) and X-ray diffraction (XRD) analyses. The electrochemical performance and catalytic activity against the hydrogen evolution reaction (HER) were assessed in a three-electrode cell for screening purposes, as well as in a prototype cell of an electrochemical hydrogen pump/compressor (EHP/C) where the catalysts served as cathodes, while the anode was Pt/XC72 40% wt. with 0.38 mgPt·cm−2 within a membrane electrode assembly (MEA) with a 180 µm thick Nafion 117 proton-conductive membrane. The results obtained indicated superior catalytic activity of the bimetallic catalysts in comparison to the pure metal samples. Further electrochemical tests in an EHP/C cell at varying differential pressures in the range of 0–3 bar revealed stable behavior and high current density, reaching approximately 0.7 A cm−2 at 60 °C. The accelerated durability tests performed demonstrated excellent stability of the synthesized composite catalysts. These findings underscore the potential of Pt-Pd nanoparticles as efficient catalysts with sustainable performance for electrochemical hydrogen pumping/compressing applications.
1. Introduction
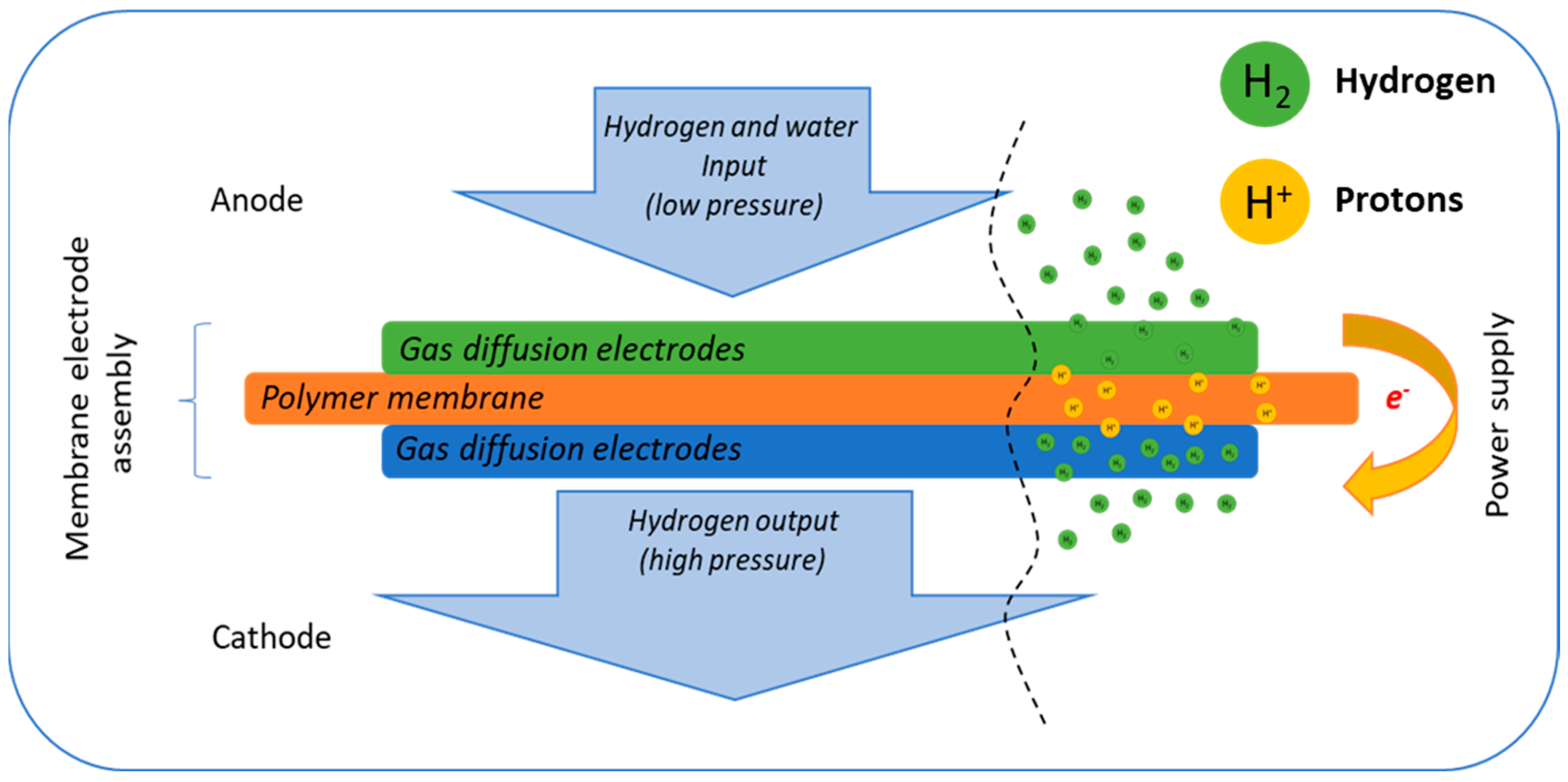
Hydrogen and the hydrogen economy are poised to play a crucial role in the global transition toward sustainable energy systems, providing a pathway to achieving zero carbon emissions [1,2]. However, a major challenge in the widespread adoption of hydrogen as a primary energy carrier lies in its efficient storage and transportation [3,4,5]. Current commercially available compression methods demand substantial energy inputs, significantly increasing the cost of green hydrogen and hindering its industrial scalability. To address these limitations, electrochemical hydrogen pumps/compressors (EHP/C) have emerged as a promising alternative to conventional piston compressors. EHP/C technology demonstrates high efficiency and superior capabilities for both hydrogen pressurization and purification, positioning it as a viable solution for integrating hydrogen into modern energy infrastructures [6,7,8]. The operational principles of the electrochemical hydrogen pump/compressor are depicted schematically in Figure 1.
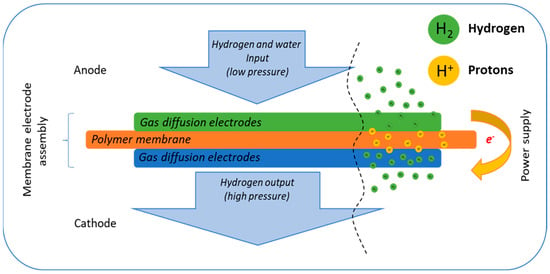
Figure 1.
Principle scheme of the electrochemical compression of hydrogen.
Hydrogen compression in electrochemical hydrogen pumps/compressors (EHP/C) is achieved through an efficient electrochemical process. At the anode, hydrogen molecules undergo dissociation into protons and electrons. The protons migrate across the membrane to the cathode, where they recombine with the electrons supplied through an external electric circuit to reform the hydrogen molecules [9]. The hydrogen recombination rate is directly governed by the cell voltage, following the Faraday law, enabling precise control over the process. By maintaining a constant cell voltage, the pressure within the cathode chamber increases, while the pressure in the anode chamber remains constant. As a result, differential pressure between the anode and cathode departments of the cell is generated, which is a key operation parameter for the compressors. The EHP/C is a highly efficient and reliable technology for hydrogen storage and transportation. The overall reaction governing the EHP/C process is represented by Equation (1).
H2(anode) → H2(cathode)
The partial electrode reactions proceeding on the anode (hydrogen oxidation reaction, HOR) and the cathode (hydrogen reduction reaction, HRR) are described by Equation (2) and Equation (3), respectively.
H2 → 2H+ + 2e− (anode)
2H+ + 2e− → H2 (cathode)
The membrane electrode assembly (MEA) is the core of the electrochemical hydrogen pump/compressor (EHP/C) systems, comprising two gas diffusion electrodes (GDE) where electrochemical reactions occur [10,11]. The polymer electrolyte membrane, commonly known as Nafion or modified perfluorosulfonic acid (PFSA), provides high ionic conductivity, low hydrogen permeability and mechanical stability at operating temperatures of up to 80 °C [12,13]. For hydrogen purification applications, the membrane may be substituted with polybenzimidazole (PBI) doped with phosphoric acid, allowing operation at elevated temperatures of up to 160 °C [14]. This configuration minimizes the contamination of the platinum catalyst but limits the operating differential pressure due to the high back-diffusion forces [15]. The phosphoric acid is not chemically bound to the polymer. And when the differential pressure increases, it moves toward the anode department of the cell, thus breaking the electrolytic contact between both electrodes. For this reason, the PBI membranes are usually used at differential pressure up to 5 bar, e.g., in an operation mode suitable only for hydrogen purification (such as an electrochemical hydrogen pump, EHP). On the other hand, the usage of PFSA or Nafion membranes in hydrogen a compression mode sustaining high differential pressure necessitates adequate hydration of the membrane to ensure optimal proton conductivity. In addition, at elevated current densities, phenomena such as electro-osmotic drag and hydrogen back-diffusion may also occur and negatively impact the system efficiency [16]. The gas diffusion electrodes also play a pivotal role in maintaining stable EHP/C operation. These electrodes consist of multiple sublayers, each with a distinct function: the diffusion layer (DL) ensures mechanical integrity and electrical conductivity; the microporous layer (MPL), composed of carbon-based materials mixed with polytetrafluoroethylene (PTFE), facilitates uniform current distribution and efficient hydrogen transport; and the catalyst layer (CL), containing nanostructured catalysts dispersed on a conductive support, accelerates the electrochemical reactions [17,18,19]. The overall resistance of each electrode is the sum of the resistances of its individual components, as defined in Equation (4.)
REL = RDL + RM PL + RCL
The catalytic activity of the integrated anode and cathode catalysts is a critical factor influencing the energy efficiency and overall performance of the electrochemical hydrogen pump/compressor (EHP/C) systems. The activation energy required for the partial electrode reactions defined in Equation (5) is fundamental to enhancing the system efficiency and achieving high differential pressure
Eactivation = Eact. Anode + Eact. Cathode
As detailed in prior studies, the anodic reaction limits the process at low differential pressures, whereas at high pressures, the cathodic reaction becomes the rate-determining step [20]. Platinum (Pt) and palladium (Pd) are widely utilized in EHP/C systems due to their superior catalytic activity, as demonstrated in the well-known Volcano plot [21]. These catalysts exhibit low activation energy, contributing to high energy efficiency. However, Pt nanoparticles are prone to aggregation during electrochemical reactions, which diminishes their performance. To mitigate this issue, support materials such as carbon black, carbon nanotubes and graphene are employed, which stabilize the nanoparticles and enhance the catalyst utilization. The advanced nanocarbon supports effectively decrease the required Pt loading while maintaining high catalytic efficiency. Additionally, alloying Pt with other metals such as Ru or Ni has a proven effect in improving the catalytic performance and reducing the costs. Alloying prevents particle agglomeration and enhances catalytic activity through synergetic interatomic and/or interelectronic interactions [22,23]. According to [24], carbon-supported catalysts (Ir/C) have also shown superior performance against HOR compared to pure Pt catalysts. It has been shown that Pd/C catalysts, when applied to MEA with a Nafion 115 membrane, demonstrate an 80% increase in energy efficiency for the purification of hydrogen from reformate mixtures compared to Pt/C catalysts [25,26,27]. These findings emphasize the critical role of optimizing electrocatalysts and their support materials for EHP/C applications.
In the present study, mono- and bimetallic catalysts comprising Pt and Pd supported on carbon XC72R were synthesized and evaluated as cathodes in a custom-designed electrochemical pump/compressor. Various Pt and Pd ratios were tested to determine the impact of both metals’ content on hydrogen oxidation reaction (HOR) overpotential, which directly affects the EHP/C cell voltage and the overall system efficiency. For comparison, the performance of pure Pt/C and Pd/C catalysts was also assessed under identical operating conditions. The total metal content in all catalyst samples was fixed at 40 wt.%.
2. Results
The surface morphology of the synthesized mono- and bimetallic catalysts studied by means of scanning electron microscopy (SEM) is shown on the images presented in Figure 2. The morphology of the mono- and bimetallic samples appears visually identical to that of the catalytic support, suggesting a potentially well-developed active surface.

Figure 2.
SEM images of the synthesized carbon-supported mono- and bimetallic catalysts: (a) Pt/XC72R; (b) Pd/XC72R; (c) 3Pt-1Pd/XC72R; (d) 1Pt-1Pd/XC72R; (e) 1Pt3Pd/XC72R; (f) the carbon support XC72R.
To determine the amount of platinum (Pt) and palladium (Pd) in the synthesized materials, Energy-dispersive X-ray spectroscopy (EDX) analysis was performed. The results are summarized in Table 1.

Table 1.
The composition of the synthesized samples determined by EDX analysis.
The carbon content dominates in all samples, since the carbon is the supporting material. The amounts of palladium (Pd) and platinum (Pt) in samples 2 to 6 align well with the metallic content introduced by the Pt- and Pd-acethylacetonates used as precursors for their synthesis, although some losses obviously occur during the synthesis.
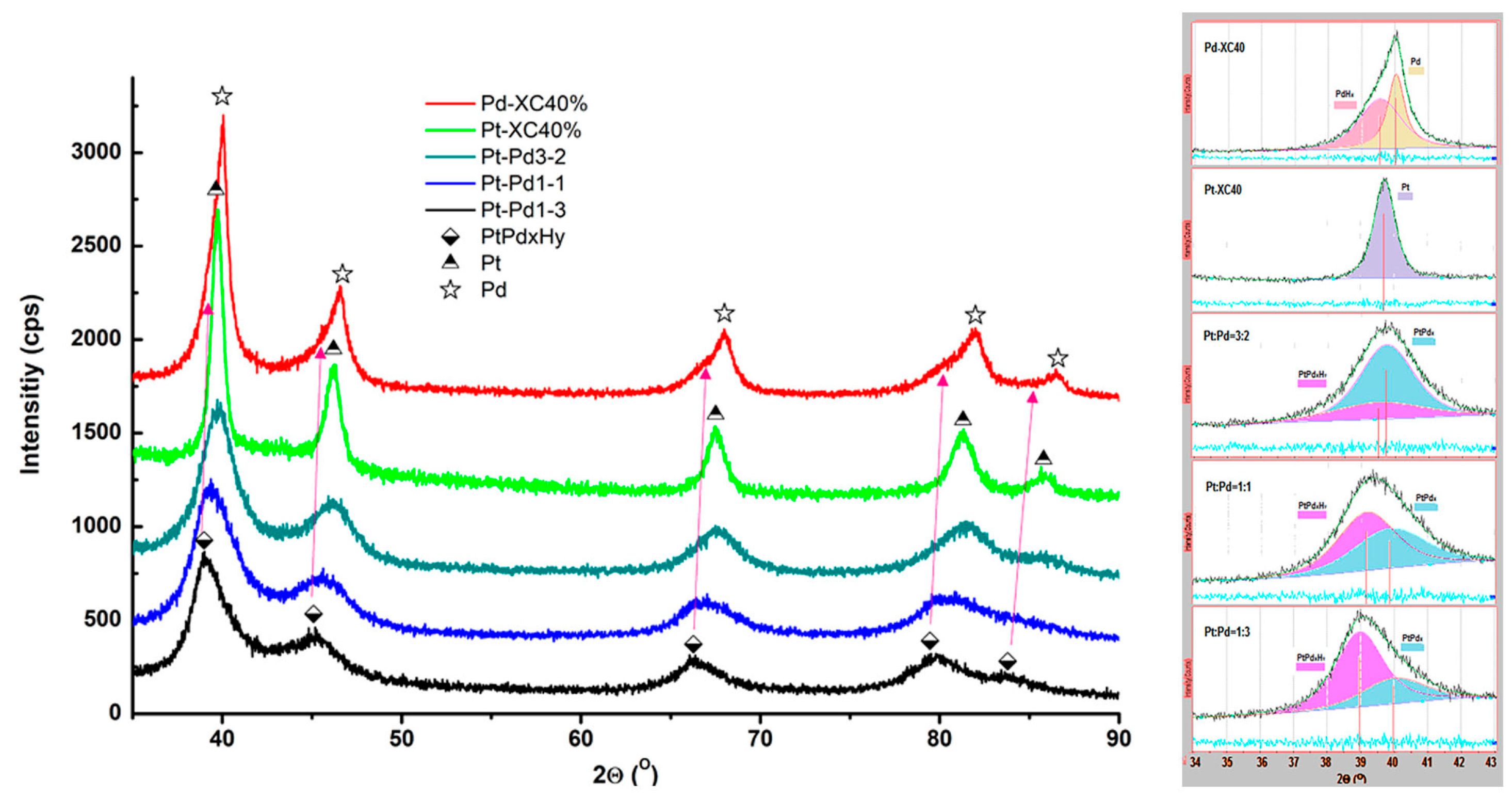
To investigate the crystallinity of the prepared samples, X-ray diffraction (XRD) measurements were conducted. The results obtained are shown in Figure 3.
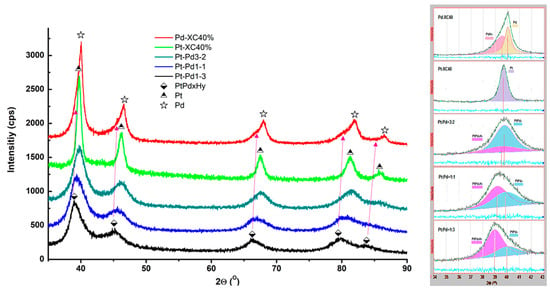
Figure 3.
XRD spectra of the synthesized carbon-supported mono- and bimetallic catalysts.
The diffraction peaks in the diffractogram of Pd/XC72R are asymmetric, broadening on the side of smaller 2θ angles. This is explained by the registered presence of the hydride PdHx. In the PtPd samples, the peaks are broader compared to those of the monometallic Pt and Pd. They shift towards smaller 2θ angles with increasing content of palladium and exhibit asymmetry—additional broadening on the side of larger 2θ. The decomposition of the (111) diffraction peak confirms the presence of hydride in the Pd-containing catalysts (the component at the smaller angles) and the PtPdx alloy. The calculated crystallographic data for the catalysts under study are summarized in Table 2.
Table 2.
The lattice parameters and the crystallite sizes of the synthesized samples.
The monometallic Pt/XC72R shows a crystallite size of 11 nm, while Pd/XC72R has slightly larger crystallites with a size of 14 nm. In contrast, the crystallite sizes of the bimetallic and hydride phases are significantly smaller. For instance, PtPdx alloys exhibit crystallite sizes ranging from 2 to 4 nm, indicating much finer particles compared to the monometallic samples. The presence of PdHx and β-PtPdHx phases contributes to variations in the cell parameters and crystallite sizes, as the hydrides generally show smaller crystallite sizes.
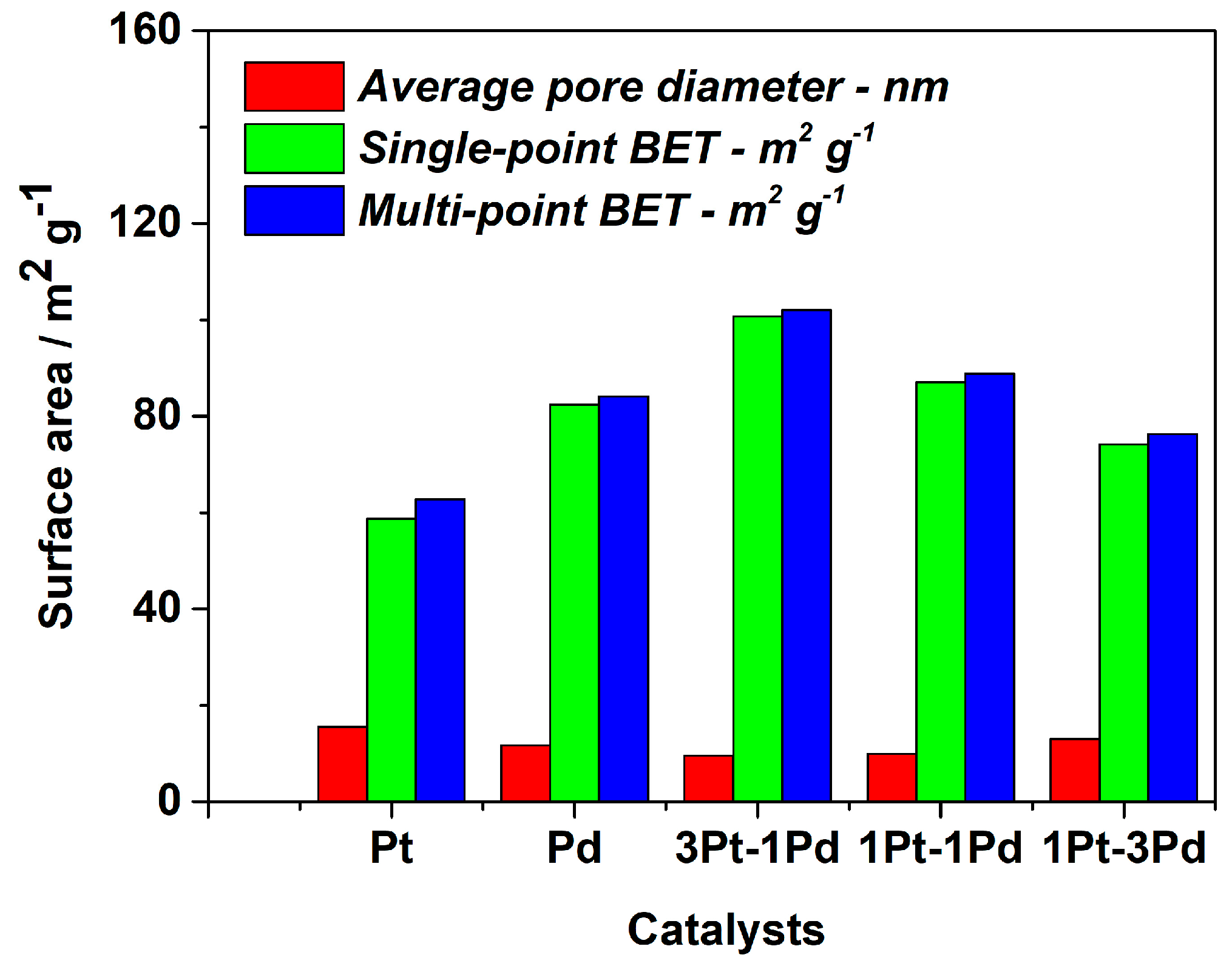
Brunauer–Emmett–Teller (BET) analysis was conducted on all samples to evaluate the surface area of the synthesized composites. The results are illustrated in Figure 4. The samples containing Pt and Pd dispersed on XC72R exhibit surface areas of around 60 m2·g−1 and 80 m2·g−1, respectively, with a nearly identical pore diameter. The surface area of the bimetallic samples is higher than that of the monometallic phases, and it could be concluded that the formation of bimetallic phases enhances the surface area of the synthesized catalysts. Indeed, incorporating 25% wt. palladium increases the surface area to approximately 100 m2·g−1 and concurrently reduces the pore diameter. However, a further increase in the amount of palladium in the platinum catalyst results in a decrease in the active surface area and an increase in the average pore diameter.
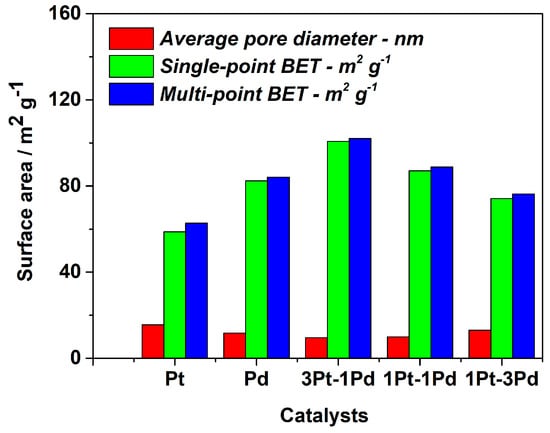
Figure 4.
BET analysis on mono- and bimetallic Pt and Pd catalyst supported on XC72R.
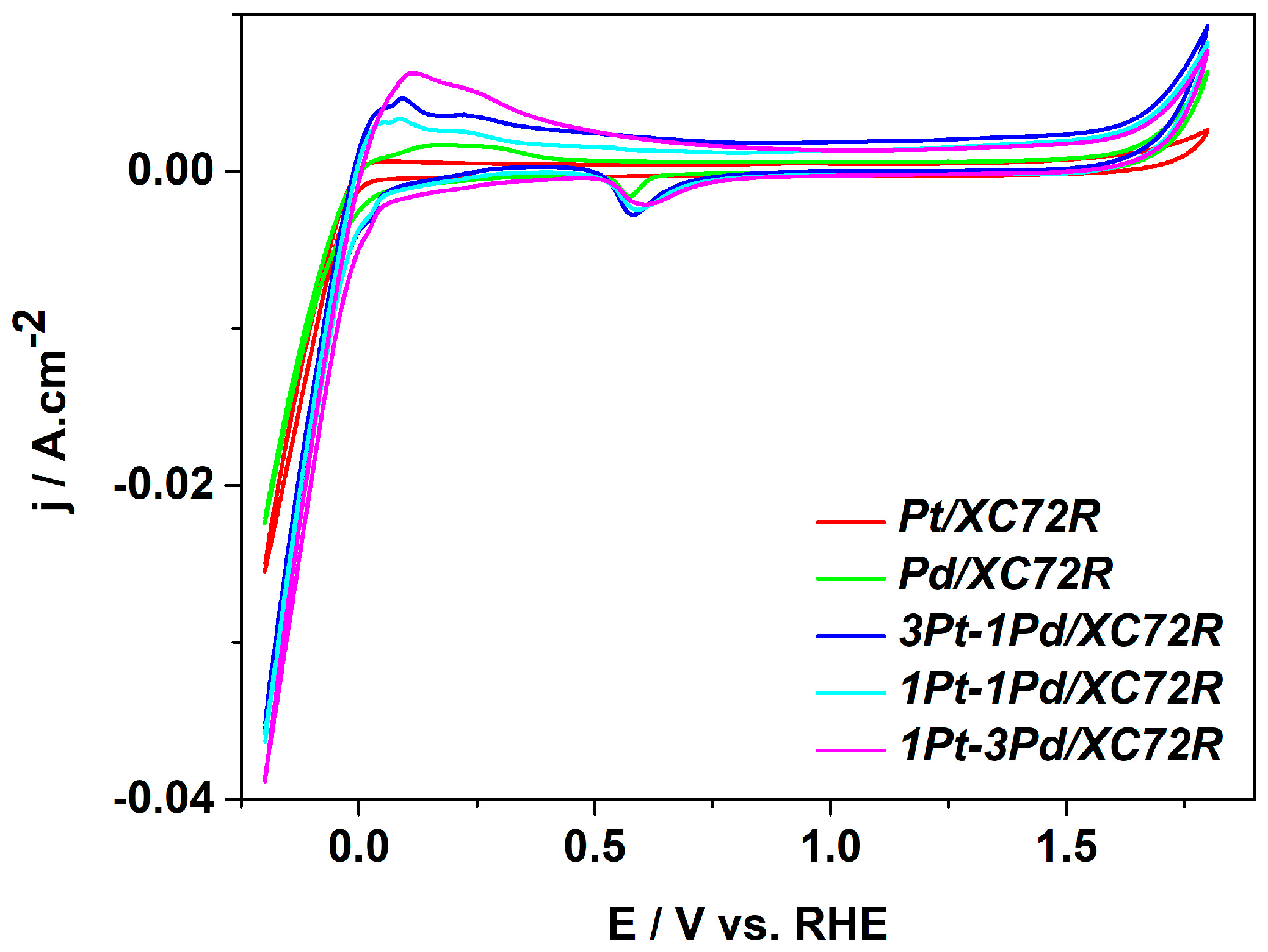
To investigate the electrochemical activity of the synthesized samples, the catalysts were deposited onto flat graphite-based electrodes using a spray technique, achieving a catalyst loading of 0.25 mg cm−2 for each sample. The electrochemical performance of the thus-prepared electrodes was evaluated in 0.5 M H2SO4 electrolyte using a standard three-electrode electrochemical cell at room temperature. The recorded cyclic voltammograms (CV) are presented in Figure 5.
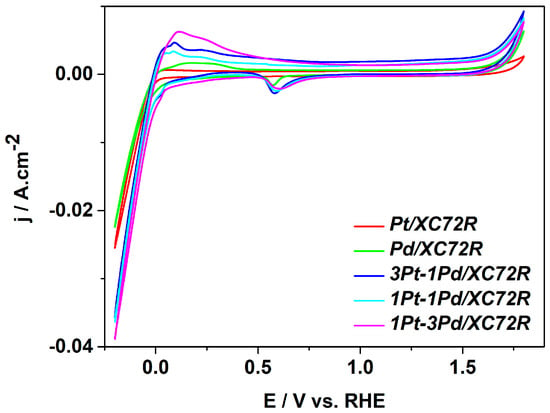
Figure 5.
Cyclic voltammograms of the catalysts under study obtained in 0.5 M H2SO4 at room temperature, recorded at 100 mV·s−1.
The cyclic voltammograms exhibit characteristic peaks for Pt and Pd catalysts. During potential scanning in the positive direction (from −0.25 V to 1.8 V), hydrogen oxidation occurs in the potential range up to around 0.45 V, as, again, the current density of the process is higher on the bimetallic samples. The formation of oxygen coverage on the catalyst surface proceeds in the range 0.95–1.55 V, followed by oxygen evolution at more positive potentials.
During reverse negative potential scanning (from 1.8 V to −1.25 V), the oxygen reduction reaction occurs in the potential range of 0.68 V to 0.55 V, and the adsorption of hydrogen starts at around 0.15 V, followed by the commencement of hydrogen evolution at around −0.005 V. The HER is visibly more intensive on the bimetallic catalysts than on pure Pt and Pd ones. The HER occurs most actively over the sample 1Pt-3Pd/XC72R as the reaction rate, resp. the current density, decreases in the order 1Pt-3Pd/XC72R > 3Pt-1Pd/XC72R > 1Pt-1Pd/XC72R > Pd > Pt. It is evident that the hydrogen evolution is more pronounced on the bimetallic samples compared to the pure Pt and Pd catalysts. This phenomenon can be explained by the nature of the Pd, which builds hydrates, as described in [28,29].
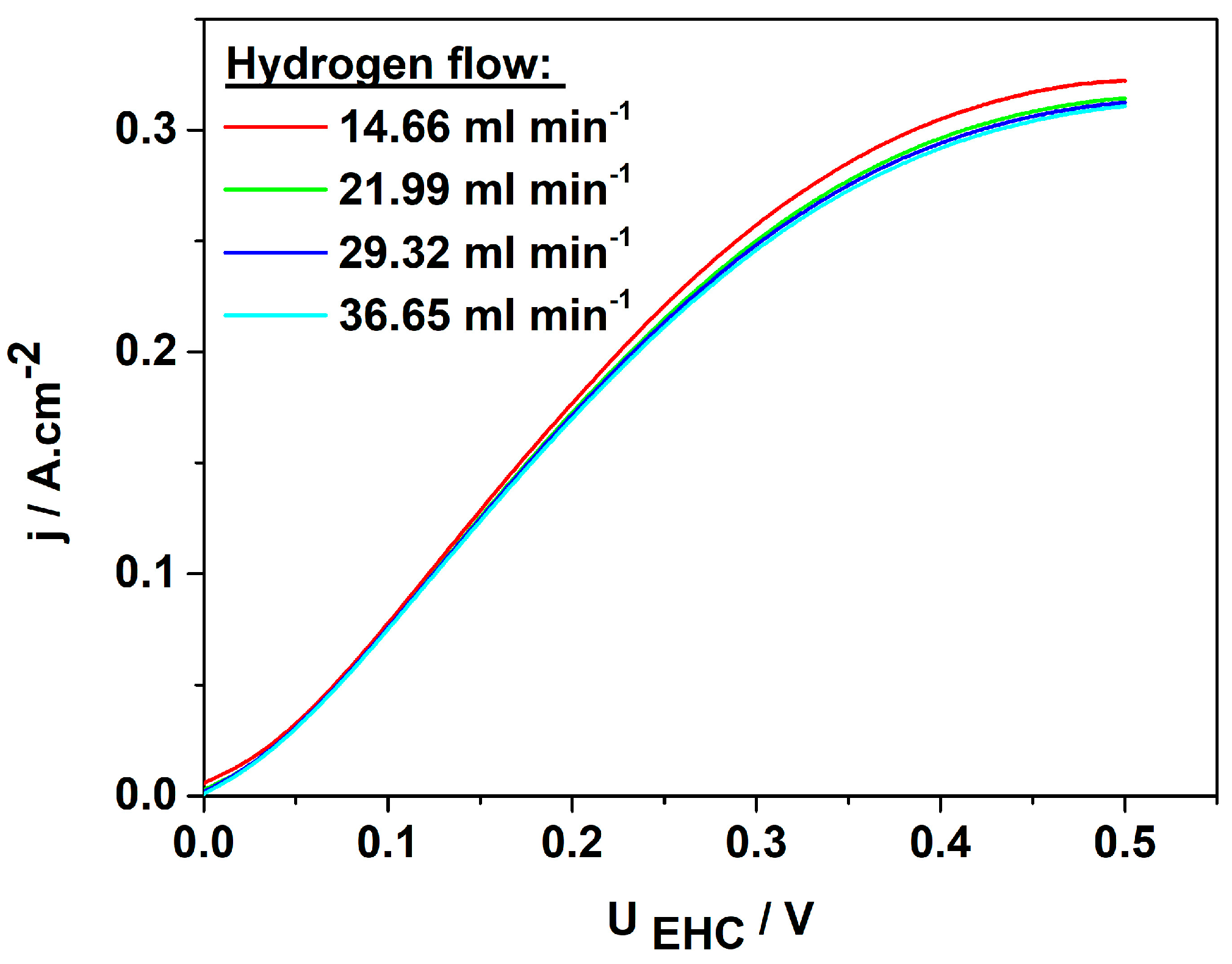
Following the preliminary screening of the synthesized catalysts carried out in 0.5 M H2SO4, the best-performing catalyst, 3Pt-1Pd/XC72R, was tested in a custom-made laboratory PEM cell, enabling us to simulate the operating conditions in a real electrochemical hydrogen pump/compressor (EHP/C). Membrane electrode assemblies with a cathode containing 0.25 mg cm−2 3PtPd/XC72R dispersed over the H2315C2 gas diffusion electrode and a commercially available anode with 0.38 mg cm−2 Pt/XC72 attached on both sides of the polymer electrolyte membrane Nafion 117 (thickness of 180 µm in dry state) were prepared by applying a standard assembly procedure [30]. The MEA’s performance was first investigated at room temperature by varying the hydrogen inflow rate. The obtained voltampere characteristics (j/U curves) are shown in Figure 6.
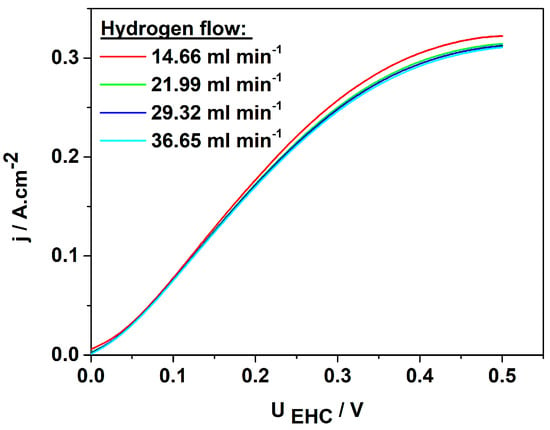
Figure 6.
Voltampere characteristics of MEA with 3Pt−1Pd/XC72R as cathode and 0.38 mg·cm−2 Pt−XC72 as anode at different hydrogen flow rates; potential scan rate: 1 mV s−1.
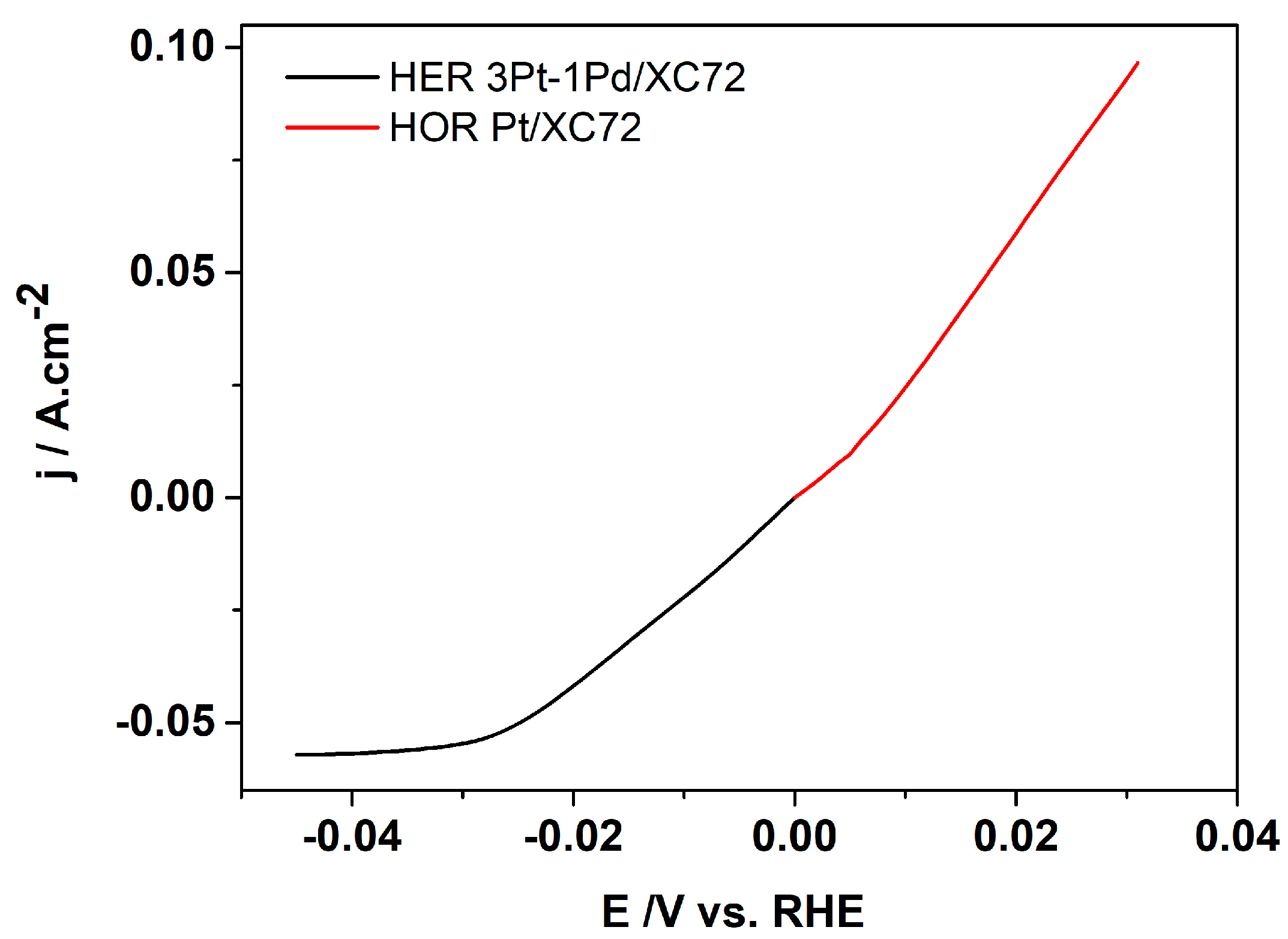
It is seen that the hydrogen flow rate does not affect the rate of the process in the kinetic (0–25 mV) and ohmic regions (25 mV to 300 mV) of the curve (Figure 6). However, in the diffusion-limited region, the flow rate influences both the slope of the j/U curves and the rate of the process. The current density reaches a maximum of approximately 0.3 A cm−2 at a cell voltage of 0.35 V, beyond which the diffusion limitations become apparent. To investigate the efficiency of both partial reactions (anode and cathode), linear sweep voltammetry was performed at a hydrogen inflow rate of 14.66 mL min−1. The results obtained are presented in Figure 7. Both curves were recorded in the potential range close to the open-circuit potential in order to investigate the operation of the EHC at low overvoltage, thereby achieving high energy efficiency.
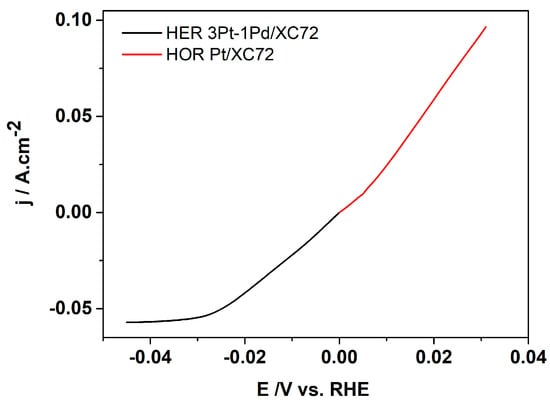
Figure 7.
Polarization curves obtained at hydrogen inflow rate of 14.66 mL min−1, room temperature and scan rate of 1 mV s−1; HOR (red), HER (black).
On the cathode, where the hydrogen evolution reaction (HER) takes place, the current density reaches about 0.06 A cm−2 at −0.03 V vs. RHE, after which the diffusion limitations become apparent. In comparison, on the anode gas diffusion electrode, where the hydrogen oxidation reaction (HOR) occurs, the current density reaches approximately 0.1 A cm−2 at a potential of 0.03 V vs. RHE.
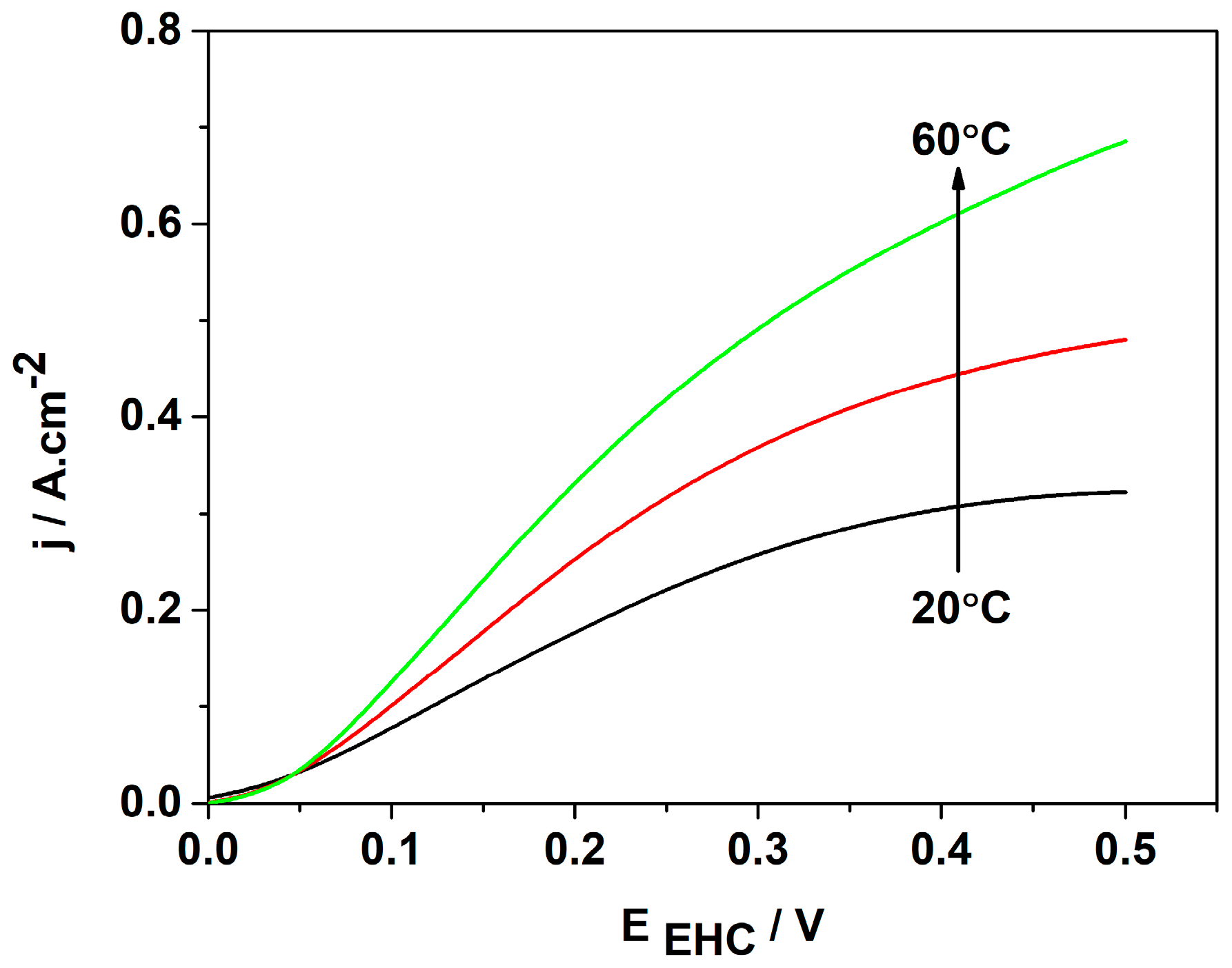
It is well known that an increase in the temperature enhances the ionic conductivity of a polymer membrane, thus accelerating both partial reactions. To investigate the electrochemical behavior of the selected best catalyst, the temperature of the EHP/C test cell was varied in the range of 20 °C to 60 °C. The results obtained are presented in Figure 8. They show that with increasing temperature, the cell performance improves considerably—the diffusion limitations shift to higher voltages and the current density rises up to 0.7 A cm−2 at a cell voltage of 0.5 V. These effects are related to the decrease in the activation energy and volume changes in the hydrogen reagent gas.
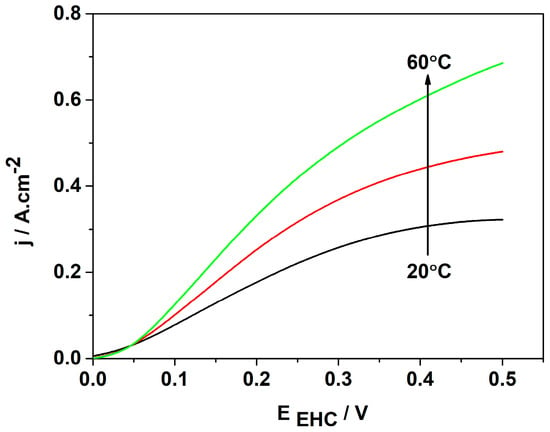
Figure 8.
The influence of the operation temperature on the voltampere characteristics of MEA with the 3Pt-1Pd/XC72R cathode and Pt/XC72.
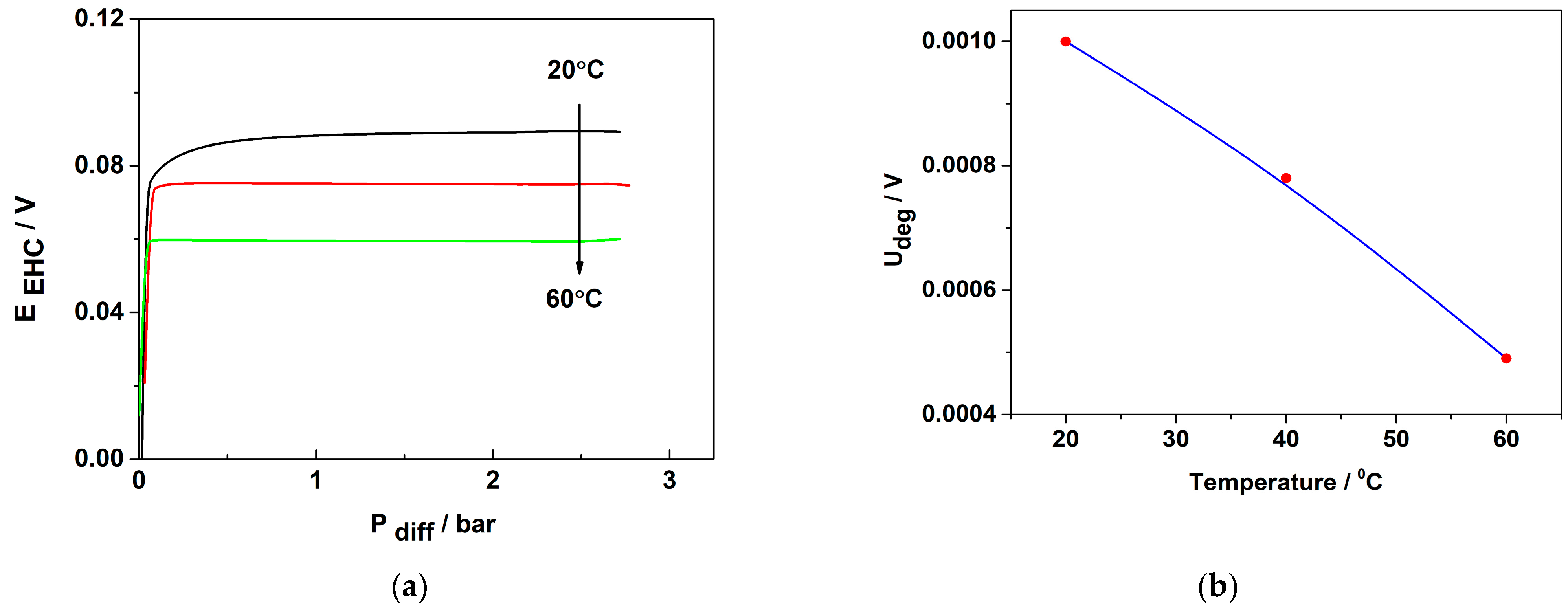
The EHP/C operation under pressure was evaluated using steady-state polarization curves at a constant current density of 60 mA cm−2 and operating temperatures ranging from 20 to 60 °C. The differential pressure of the cell was varied in the range of 0–3 bar, corresponding to a force of 3 kg f−1 cm−2. The obtained results are presented in Figure 9a,b.
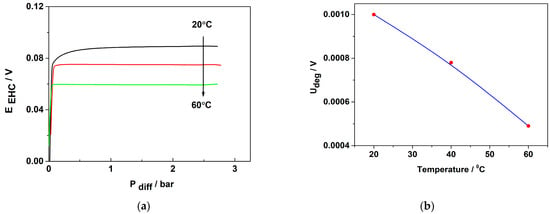
Figure 9.
Influence of differential pressure on EHC cell performance at constant current density of 0.06 A cm−2: (a) change in Ucell with changing differential pressure; (b) change in ΔUcell with temperature.
Figure 9a shows that at a constant current density, resp. the constant rate of the process, at the beginning of the test, the differential pressure between the anode and cathode departments of the cell sharply increases, then stabilizes and does not change until the final test pressure of 3 bar is achieved. These results confirm that 3Pt-1Pd/XC72 catalyst demonstrates stable electrochemical behavior under differential pressure of 3 bar and temperatures up to 60 °C. In order to have a quantitative measure of the stability of the catalyst’s performance with increasing pressure, as shown in Figure 9b, the parameter Udeg/V (the change in Ucell with each 1 bar increase in the differential pressure) was calculated and presented as a function of the temperature, as shown in Figure 9b. It is seen that increasing temperature facilitates the process of hydrogen pumping/compression, which could be related to a decrease in activation energy as well as to an improvement in membrane proton conductivity.
3. Materials and Methods
Catalyst synthesis: The catalysts were synthesized via the sol–gel method, employing platinum and palladium acetylacetonate precursors with the common formula M((C5H7O2)n)m. The catalytic support was a commercial XC72R provided by the Cabot Corporation, with an average particle size of 30 nm. The experimental procedure included pretreatment of both the catalytic support and the precursors in isopropanol, homogenization with a magnetic stirrer and treatment in an ultrasonic bath. The subsequent steps involved their meticulous mixing and heating at a temperature of 60 °C until a fine gel was achieved. Then, a gradual thermal treatment in a hydrogen atmosphere was conducted with a heating rate of 2 °C per minute until a temperature of 240 °C was reached. This temperature was maintained for 2 h and then gradually decreased to room temperature. The details of the synthesis are provided in [30].
Physical characterization: The structural and phase composition of the thus-prepared materials were examined using a Malvern PANalytical B.V. (Malvern, UK) X-ray diffraction (XRD) machine in a compact size. The diffraction data were collected using a Philips ADP15 (Eindhoven, The Netherlands) X-ray diffractometer with Cu-Kα radiation λ = 1.54178 Å) at a constant rate of 0.02 s−1 over an angle range of 2θ = 4° ÷ 80°. The metal content in the composite catalysts was determined using energy-dispersive X-ray analysis, while the morphology and surface structure were investigated via scanning electron microscopy. The SEM analysis was carried out using a Zeiss Evo 10 (Carl Zeiss Microscopy, Oberkochen, Germany) with SmartSEM (version 7.05) software upgraded with an EDS probe: Oxford Ultim Max 40 (Oxford Instruments, Abingdon, UK).
Test cell: The cell used in this study was constructed from stainless steel (AISI 316) and equipped with reference electrodes (Pt/XC72 ETEK, 40 wt.% Pt) in both the anode and cathode compartments. This design enabled simultaneous monitoring of the polarization behavior of each electrode, as well as the tracking of changes in the overall cell voltage. These features allowed for the recording of Ucell/j and Ucell/pdiff characteristics.
Both the anode and cathode current collectors were also fabricated from AISI 316 stainless steel, ensuring durability and compatibility with the system. Copper foam was utilized for gas distribution and to facilitate uniform current density throughout the cell, enhancing its overall performance.
Electrochemical tests: The electrochemical tests were conducted using a three-electrode cell containing 200 mL of a liquid electrolyte (0.5 M H2SO4) with an electrode size of 1 cm2. Additionally, membrane electrode assemblies (MEAs) were employed, incorporating a polymer proton-conductive membrane, Nafion 117 (Alfa Aesar, Heysham, UK). The investigated catalysts, namely Pt, Pd and their alloy, were integrated into the working electrode (the cathode). The electrode structure included a multi-layered configuration consisting of a hydrophobic gas diffusion layer H2315C2 from Freudenberg, Germany, featuring a graphite microporous layer and an active catalytic layer applied using a spray technique with a catalytic ink. Each electrode had a catalytic loading of 1 mg cm−2, with a metal content of 40% by weight. The membrane electrode assembly was prepared using the hot-pressing procedure outlined in [31]. The performance characteristics of the MEAs were evaluated in a custom-made electrochemical hydrogen pump/compression cell, registered as a utility model (4303U1) under patent 67663B1 with the Bulgarian patent association. The electrochemical behavior and catalytic activity were examined through linear sweep voltammetry and quasi-steady-state polarization curves at both room temperature and an elevated temperature up to 60 °C, at varying differential pressures of up to 3 bar. The measurements were carried out using a Galvanostat/Potentiostat (Gamry Reference 3000, Gamry Instruments, Warminster, PA, USA) and DAQ recorder NI6008 from National Instruments (Austin, TX, USA).
4. Conclusions
All of the samples used in this study exhibit a morphology similar to that of the catalytic support, indicating a well-developed active surface. The newly synthesized bimetallic catalysts exhibit high catalytic activity for the hydrogen evolution reaction (HRR) in EHP/C. Both monometallic catalysts, Pt and Pd, show similar catalytic behavior for HRR in EHP/C. Alloying platinum (Pt) with palladium (Pd) reduces particle size, thus increasing the catalyst’s active surface area. However, higher Pd content leads to a reduction in surface area and hydride formation, which absorbs hydrogen and diminishes catalytic activity. Additionally, an increase in Pd content results in a decrease in the average pore diameter. The most efficient catalytic activity is observed in the 3Pt-1Pd/XC72R sample with a 34.8:11.7wt.% Pt:Pd ratio, which successfully operates at elevated temperatures in the range of 20 °C to 60 °C. Increasing the operating temperature of PEM cells increases the current density approximately 3 times and stabilizes the catalyst’s performance.
Author Contributions
Conceptualization, G.B. and E.S.; methodology, B.M.; software, N.B.; validation, G.B., B.M. and N.B.; formal analysis, G.B. and E.S.; investigation, G.B. and E.S.; resources, E.S.; data curation, B.M.; writing—original draft preparation, G.B., E.S. and B.M.; writing—review and editing, E.S.; visualization, N.B.; supervision, E.S.; project administration, E.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The data that support the findings of this study are available within the article.
Acknowledgments
The authors acknowledge the financial support of project No. BG05M2OP001-1.002-0014 “Center of competence HITMOBIL—Technologies and systems for generation, storage and consumption of clean energy” funded by the Operational Program “Science and Education for Smart Growth” 2014–2020, co-funded by the EU from the European Regional Development Fund.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Wei, X.; Qiu, R.; Liang, Y.; Liao, Q.; Klemeš, J.J.; Xue, J.; Zhang, H. Roadmap to carbon emissions neutral industrial parks: Energy, economic and environmental analysis. Energy 2022, 238, 121732. [Google Scholar] [CrossRef]
- Yue, M.; Lambert, H.; Pahon, E.; Roche, R.; Jemei, S.; Hissel, D. Hydrogen energy systems: A critical review of technologies, applications, trends and challenges. Renew. Sustain. Energy Rev. 2021, 146, 111180. [Google Scholar] [CrossRef]
- Züttel, A. Materials for hydrogen storage. Mater. Today 2003, 6, 24–33. [Google Scholar] [CrossRef]
- Kumar, N.; Lee, S.-Y.; Park, S.-J. Advancements in hydrogen storage technologies: A comprehensive review of materials, methods, and economic policy. Nano Today 2024, 56, 102302. [Google Scholar] [CrossRef]
- Tarkowski, R.; Uliasz-Misiak, B. Towards underground hydrogen storage: A review of barriers. Renew. Sustain. Energy Rev. 2022, 162, 112451. [Google Scholar] [CrossRef]
- Casati, C.; Longhi, P.; Zanderighi, L.; Bianchi, F. Some fundamental aspects in electrochemical hydrogen purifica-tion/compression. J. Power Sources 2008, 180, 103–113. [Google Scholar] [CrossRef]
- Sdanghi, G.; Maranzana, G.; Celzard, A.; Fierro, V. Review of the current technologies and performances of hydrogen com-pression for stationary and automotive applications. Renew. Sustain. Energy Rev. 2019, 102, 150–170. [Google Scholar] [CrossRef]
- Ströbel, R.; Oszcipok, M.; Fasil, M.; Rohland, B.; Jörissen, L.; Garche, J. The compression of hydrogen in an electrochemical cell based on a PE fuel cell design. J. Power Sources 2002, 105, 208–215. [Google Scholar] [CrossRef]
- Bampaou, M.; Panopoulos, K.; Papadopoulos, A.; Seferlis, P.; Voutetakis, S. An Electrochemical Hydrogen Compression Model. Chem. Eng. Trans. 2018, 70, 1213–1218. [Google Scholar]
- Perry, K.A.; Eisman, G.A.; Benicewicz, B.C. Electrochemical hydrogen pumping using a high-temperature polybenzimidazole (PBI) membrane. J. Power Sources 2008, 177, 478–484. [Google Scholar] [CrossRef]
- Paidar, M.; Fateev, V.; Bouzek, K. Membrane electrolysis—History, current status and perspective. Electrochim. Acta 2016, 209, 737–756. [Google Scholar] [CrossRef]
- Saito, M.; Arimura, N.; Hayamizu, K.; Okada, T. Mechanisms of ion and water transport in perfluorosulfonated ionomer membranes for fuel cells. J. Phys. Chem. C 2004, 108, 16064–16070. [Google Scholar] [CrossRef]
- Mauritz, K.A.; Moore, R.B. State of understanding of nafion. Chem. Rev. 2004, 104, 4535–4586. [Google Scholar] [CrossRef]
- Huang, F.; Pingitore, A.T.; Benicewicz, B.C. Electrochemical hydrogen separation from reformate using high-temperature polybenzimidazole (PBI) membranes: The role of chemistry. ACS Sustain. Chem. Eng. 2020, 8, 6234–6242. [Google Scholar] [CrossRef]
- Maricle, J.; Benicewicz, C. Electrochemical hydrogen separation and compression using polybenzimidazole (PBI) fuel cell technology. J. Power Sources 2013, 243, 1–8. [Google Scholar]
- Pineda-Delgado, J.L.; Menchaca-Rivera, J.A.; Pérez-Robles, J.F.; Aviles-Arellano, L.M.; Chávez-Ramirez, A.U.; de Jesús Hernández-Cortes, R.; Rivera, J.G.; Rivas, S. Energetic evaluations of an electrochemical hydrogen compressor. J. Energy Storage 2022, 55, 105675. [Google Scholar] [CrossRef]
- Durmus, G.N.B.; Colpan, C.O.; Devrim, Y. A review on the development of the electrochemical hydrogen compressors. J. Power Sources 2021, 494, 229743. [Google Scholar] [CrossRef]
- Li, Q.; Aili, D.; Hjuler, H.; Jensen, J. High Temperature Polymer Electrolyte Membrane Fuel Cells; Springer International Publishing: Cham, Switzerland, 2016; Volume 44, pp. 527–540. [Google Scholar]
- Park, J.; Lim, J.; Lim, M.; Kim, S.; Kim, O.; Lee, D.; Lee, J.; Cho, Y.; Sung, Y. Gas diffusion layer/flow-field unified mem-brane-electrode assembly in fuel cell using graphene foam. Electrochim. Acta 2019, 323, 134808. [Google Scholar] [CrossRef]
- Nordioa, M.; Rizzia, F.; Manzolinib, G.; Mulder, M.; Raymakers, L.; Annalanda, M.; Galluccia, F. Experimental and modelling study of an electrochemical hydrogen compressor. Chem. Eng. J. 2019, 369, 432–442. [Google Scholar] [CrossRef]
- Ďurovič, M.; Hnát, J.; Bouzek, K. Electrocatalysts for the hydrogen evolution reaction in alkaline and neutral media. A comparative review. J. Power Sources 2021, 493, 229708. [Google Scholar] [CrossRef]
- Wei, C.; Rao, R.R.; Peng, J.; Huang, B.; Stephens, I.; Risch, M. Recommended practices and benchmark activity for hydrogen and oxygen electrocatalysis in water splitting and fuel cells. Adv. Mater. 2019, 31, 1806296. [Google Scholar] [CrossRef]
- Chen, W.X.; Lee, J.Y.; Liu, Z. Microwave-assisted synthesis of carbon supported Pt nanoparticles for fuel cell applications. Chem. Commun. 2002, 2588–2589. [Google Scholar] [CrossRef]
- Kim, S.; Park, H.; Ahn, S.; Lee, B.; Kim, H.; Gho, E.; Henkensmeier, D.; Nam, S.; Kim, S.; Yoo, S.; et al. Highly active and CO2 tolerant Ir nanocatalysts for H2/CO2 separation in electrochemical hydrogen pumps. Appl. Catal. B Environ. 2014, 158–159, 348–354. [Google Scholar] [CrossRef]
- Wu, M.; Shen, P.; Wei, Z.; Song, S.; Nie, M. High activity PtPd-WC/C electrocatalyst for hydrogen evolution reaction. J. Power Sources 2007, 166, 310–316. [Google Scholar] [CrossRef]
- Wu, X.; Benziger, J.; He, G. Comparison of Pt and Pd catalysts for hydrogen pump separation from reformate. J. Power Sources 2012, 218, 424–434. [Google Scholar] [CrossRef]
- Bharti, A.; Cheruvally, G. Surfactant assisted synthesis of Pt-Pd/MWCNT and evaluation as cathode catalyst for proton ex-change membrane fuel cell. Int. J. Hydrogen Energy 2018, 43, 14729–14741. [Google Scholar] [CrossRef]
- Boasiako, C.A.; Zhou, Z.; Huo, X.; Ye, T. Development of Pd-based catalysts for hydrogenation of nitrite and nitrate in water: A review. J. Hazard. Mater. 2023, 446, 130661. [Google Scholar] [CrossRef]
- Chen, A.; Ostrom, C. Palladium-Based Nanomaterials: Synthesis and Electrochemical Applications. Chem. Rev. 2015, 115, 11999–12044. [Google Scholar] [CrossRef]
- Slavcheva, E.; Borisov, G.; Lefterova, E.; Petkucheva, E.; Boshnakova, I. Ebonex supported iridium as anode catalyst for PEM water electrolysis. Int. J. Hydrogen Energy 2015, 40, 11356–11361. [Google Scholar] [CrossRef]
- Borisov, G.; Stoyanova, A.; Lefterova, E.; Slavcheva, E. Binary catalysts for PEM water electrolysis. Proc. Bulg. Acad. Sci. 2012, 65, 457812. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).