3-Methylindole-Based Tripodal Tetraphosphine Ruthenium Complexes in N2 Coordination and Reduction and Formic Acid Dehydrogenation


 , and
, and
Abstract
: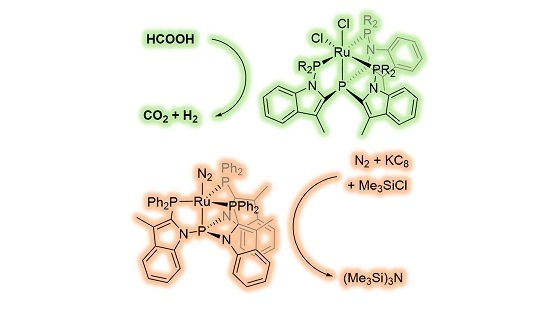
1. Introduction
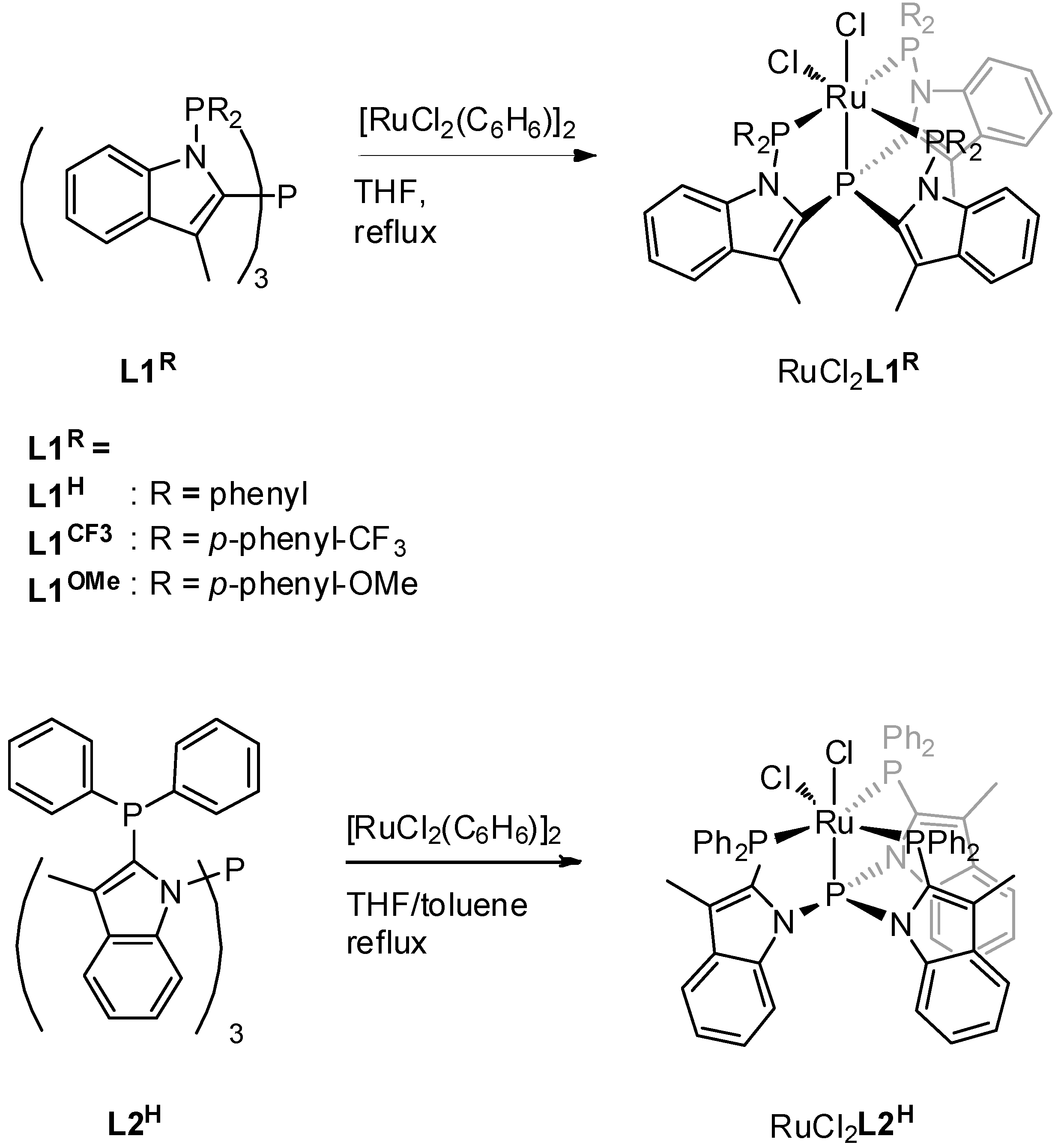
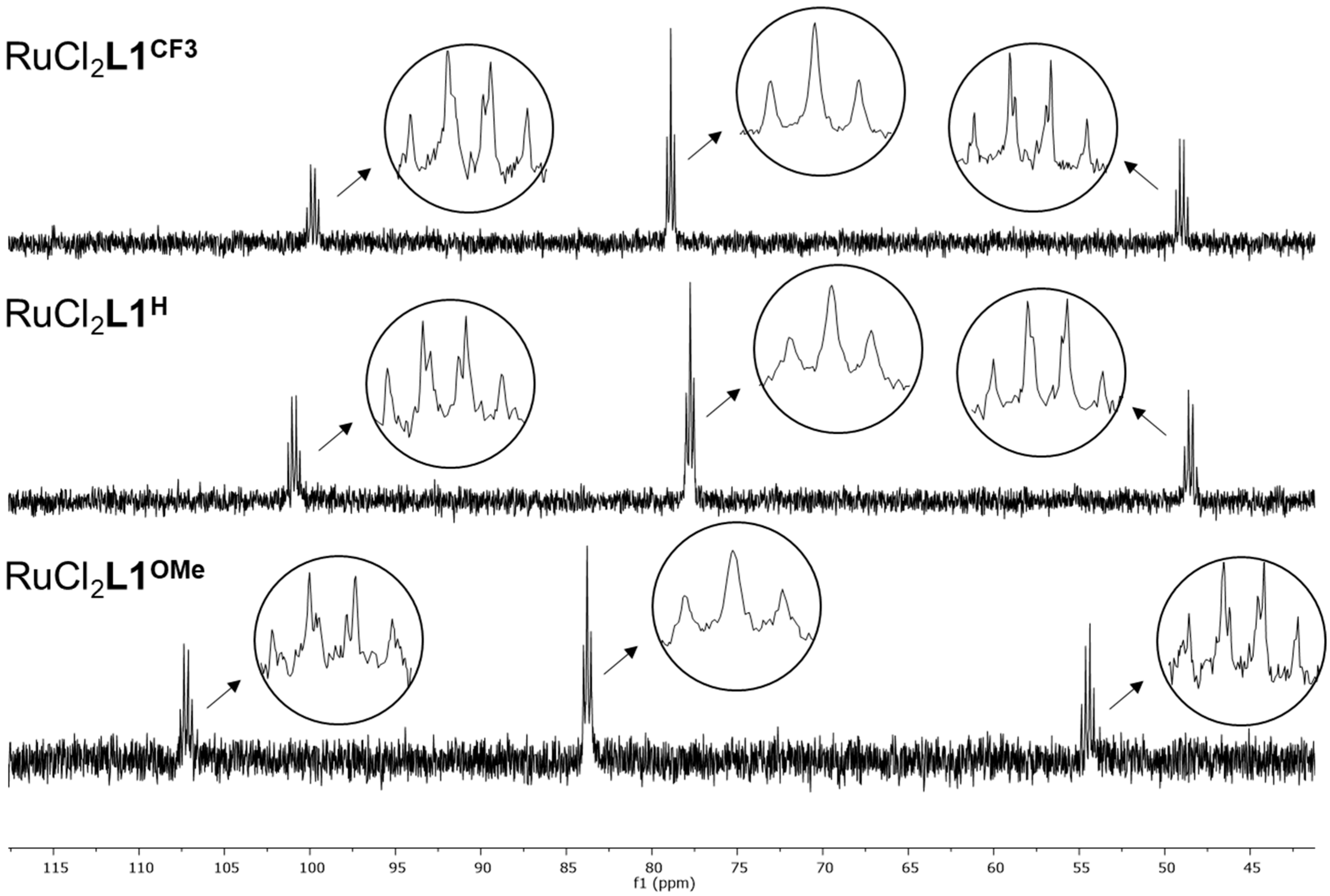
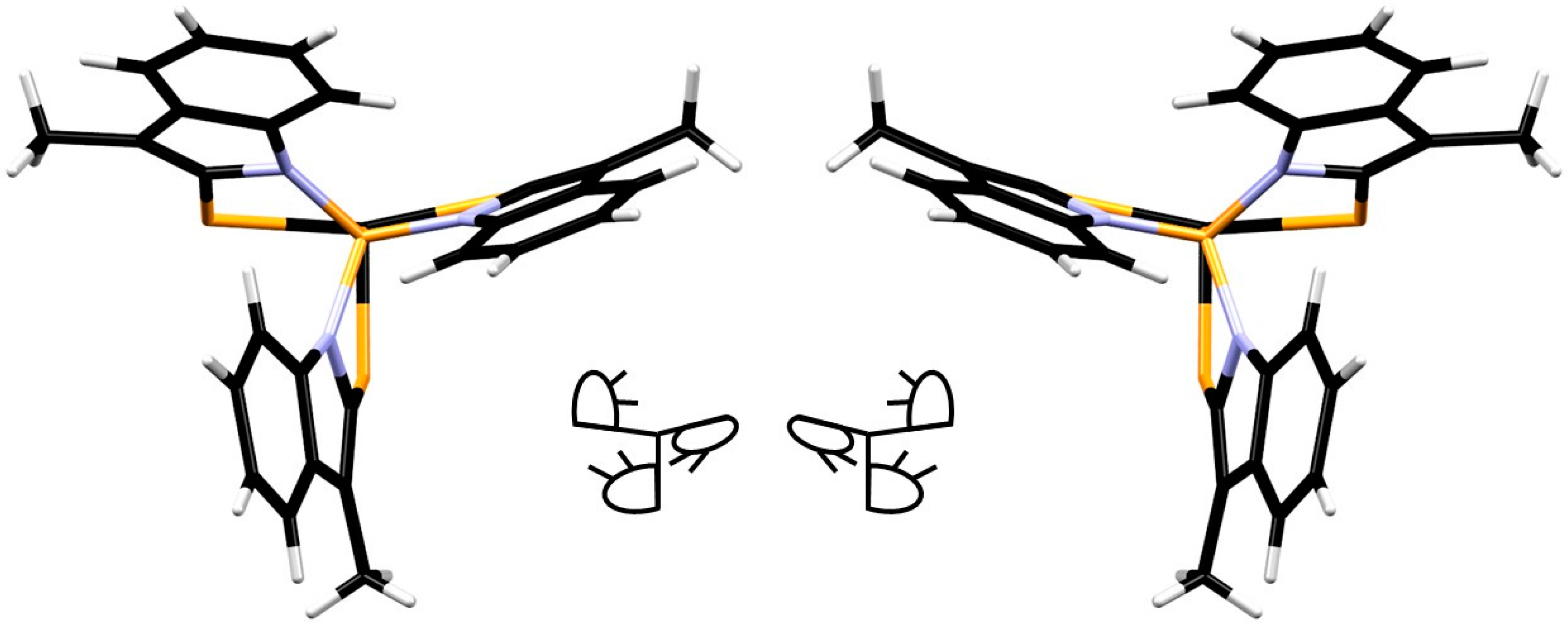
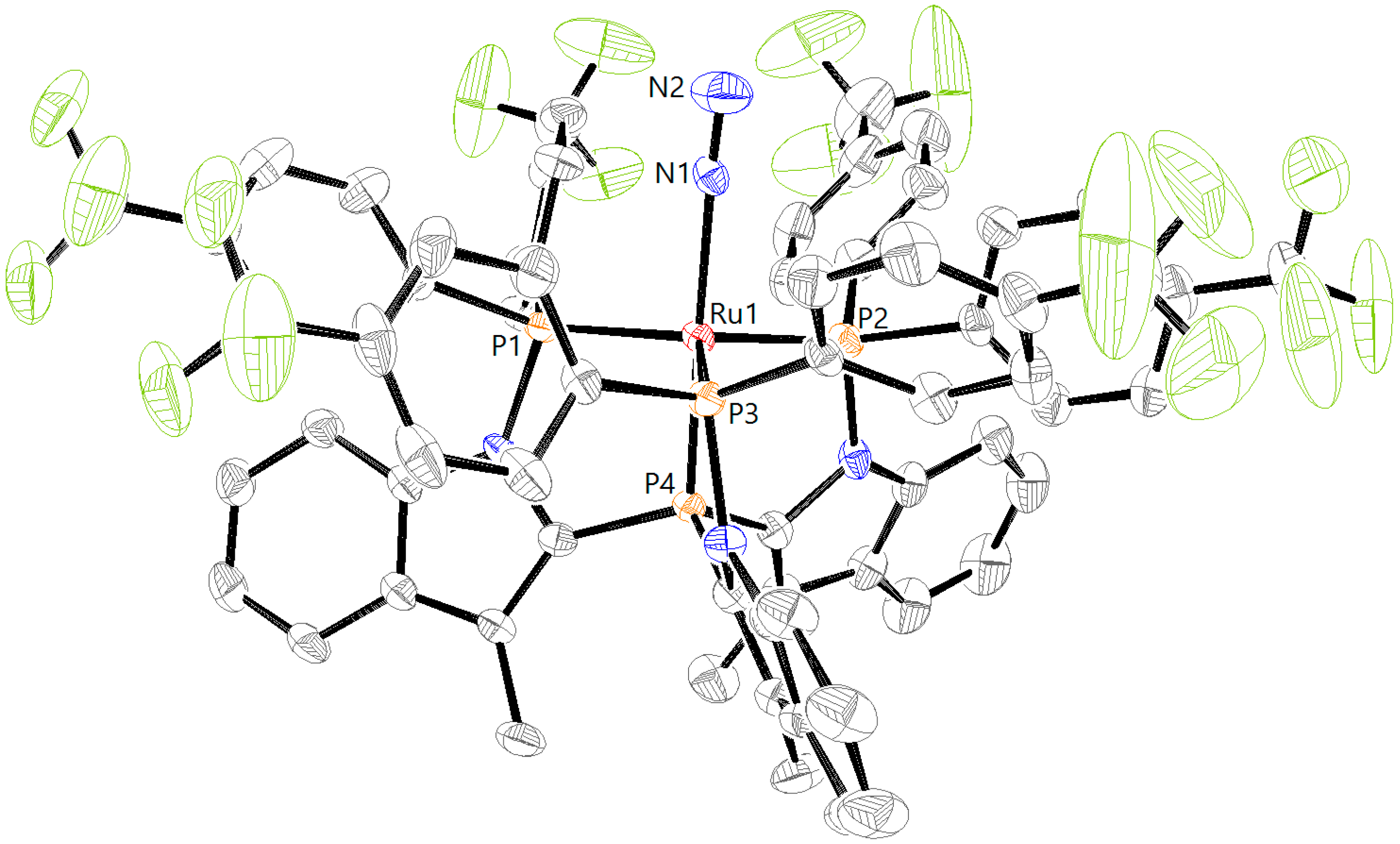
2. Results
3. Materials and Methods
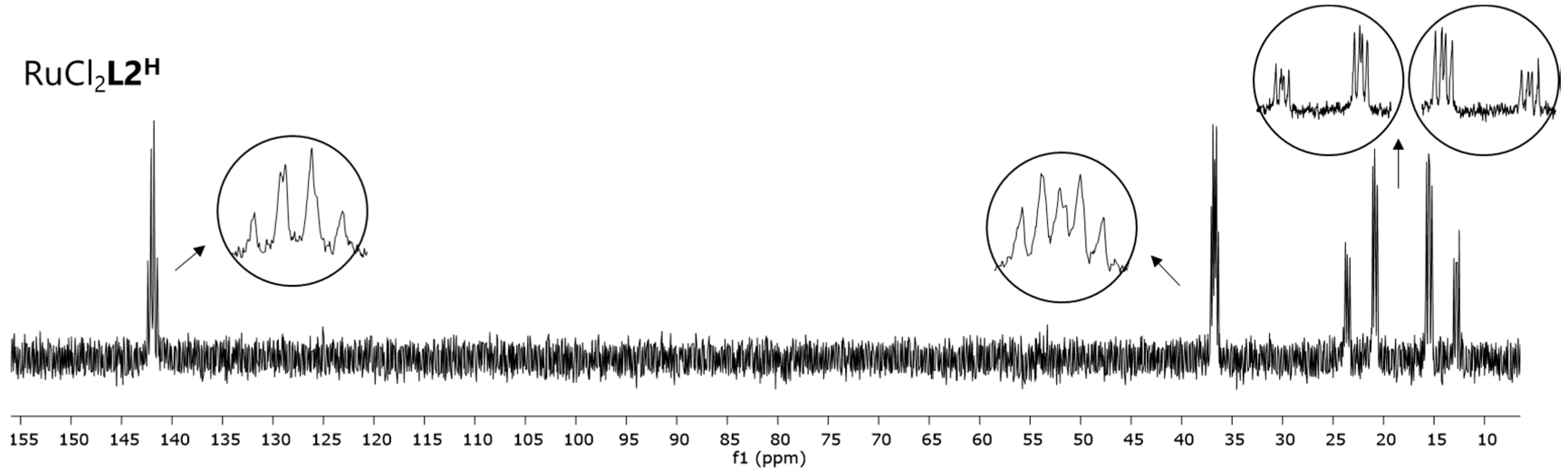
3.1. Reduction of RuCl2L to RuN2L
3.2. Catalysis: Dinitrogen Reduction
3.3. Catalysis in Time: Dinitrogen Reduction
3.4. Catalysis: Formic Acid Dehydrogenation
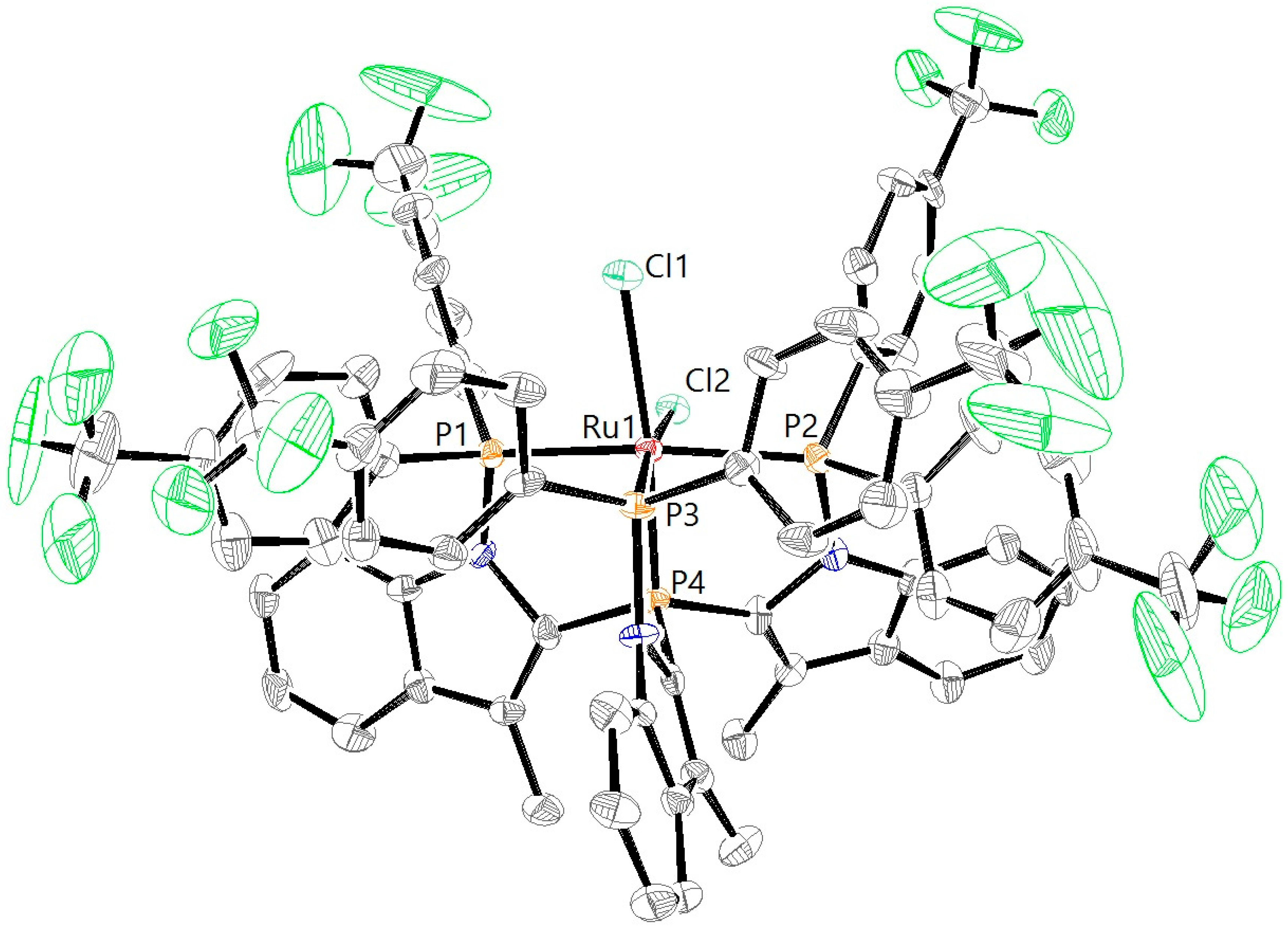
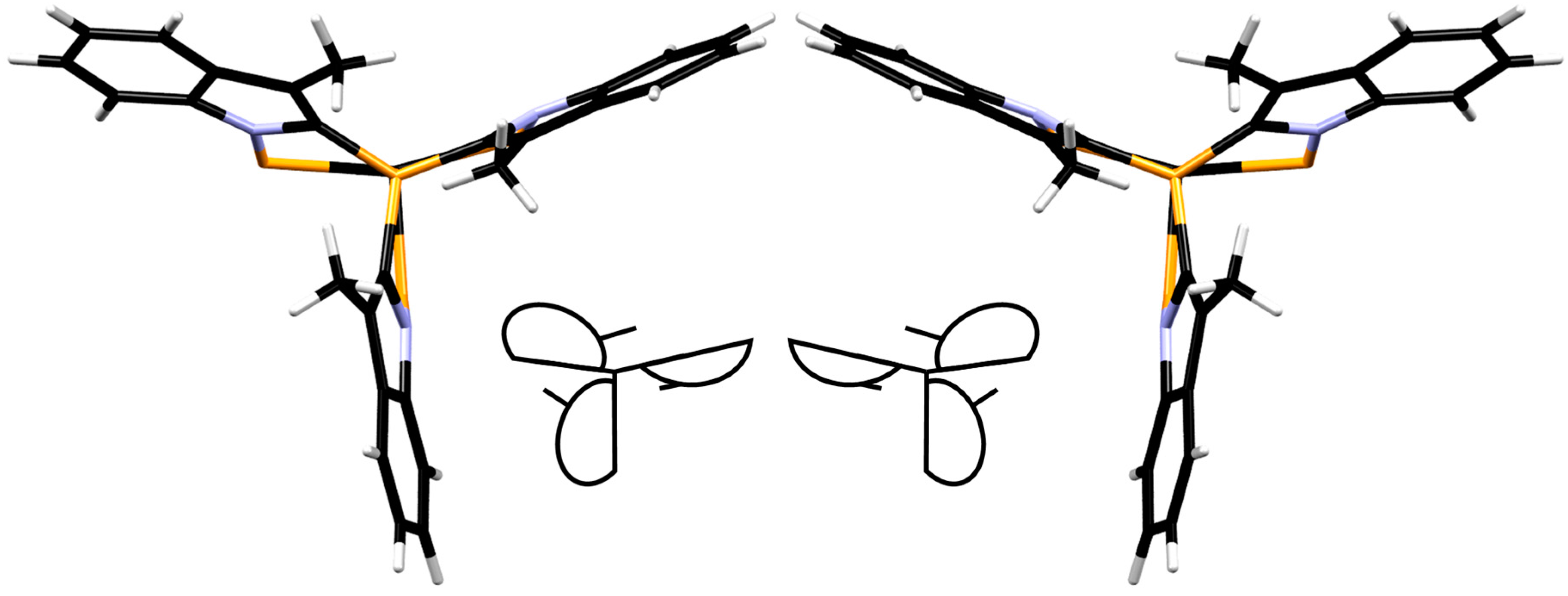
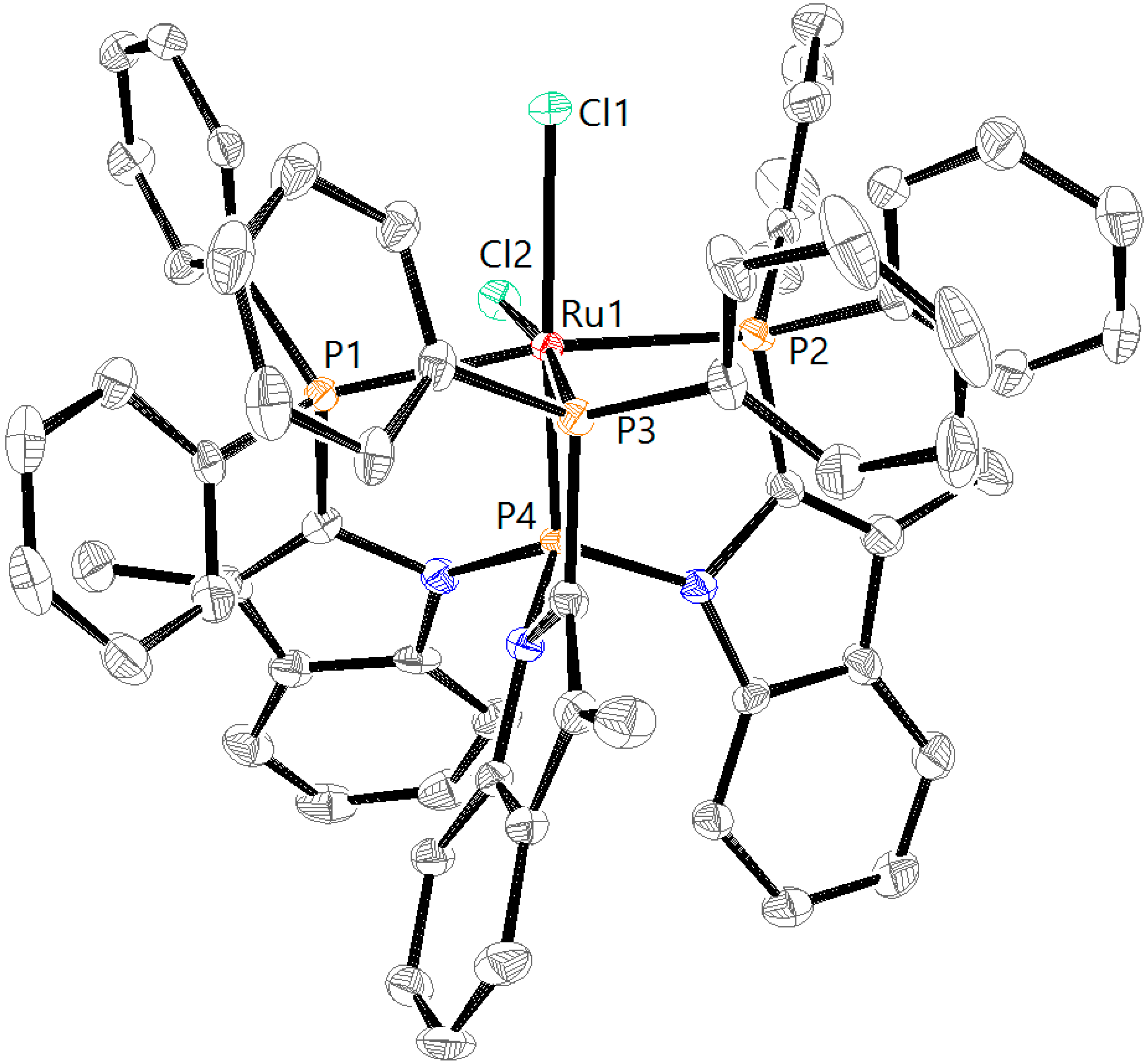
3.5. X-ray Diffraction Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Field, L.D.; Messerle, B.A.; Smernik, R.J. Synthesis and Properties of Iron(II) Hydride Complexes Containing the Tripodal Tetraphosphine Ligand P(CH2CH2PMe2)3. Inorg. Chem. 1997, 36, 5984–5990. [Google Scholar] [CrossRef] [PubMed]
- Bruneau, C.; Dixneuf, P.H. Ruthenium in Catalysis; Bruneau, C., Dixneuf, P.H., Eds.; Springer: Rennes, France, 2014; Volume 48. [Google Scholar]
- Bianchini, C. Stoichiometric and catalytic functionalization reactions of alkynes at transition metal complexes stabilized by tripodal polyphosphine ligands. Pure Appl. Chem. 1991, 63, 829–834. [Google Scholar] [CrossRef]
- Yandulov, D.V.; Schrock, R.R.; Rheingold, A.L.; Ceccarelli, C.; Davis, W.M. Synthesis and Reactions of Molybdenum Triamidoamine Complexes Containing Hexaisopropylterphenyl Substituents. Inorg. Chem. 2003, 42, 796–813. [Google Scholar] [CrossRef] [PubMed]
- Mankad, N.P.; Whited, M.T.; Peters, J.C. Terminal FeI–N2 and FeII···H–C Interactions Supported by Tris(phosphino)silyl Ligands. Angew. Chem. Int. Ed. 2007, 46, 5768–5771. [Google Scholar] [CrossRef] [PubMed]
- Moret, M.E.; Peters, J.C. Terminal Iron Dinitrogen and Iron Imide Complexes Supported by a Tris(phosphino)borane Ligand. Angew. Chem. Int. Ed. 2011, 50, 2063–2067. [Google Scholar] [CrossRef] [PubMed]
- Creutz, S.E.; Peters, J.C. Catalytic Reduction of N2 to NH3 by an Fe–N2 Complex Featuring a C–Atom Anchor. J. Am. Chem. Soc. 2014, 136, 1105–1115. [Google Scholar] [CrossRef] [PubMed]
- Siedschlag, R.B.; Bernales, V.; Vogiatzis, K.D.; Planas, N.; Clouston, L.J.; Bill, E.; Gagliardi, L.; Lu, C.C. Catalytic Silylation of Dinitrogen with a Dicobalt Complex. J. Am. Chem. Soc. 2015, 137, 4638–4641. [Google Scholar] [CrossRef] [PubMed]
- Del Castillo, T.J.; Thompson, N.B.; Peters, J.C. A Synthetic Single-Site Fe Nitrogenase: High Turnover, Freeze-Quench 57Fe Mössbauer Data, and a Hydride Resting State. J. Am. Chem. Soc. 2016, 138, 5341–5350. [Google Scholar] [CrossRef] [PubMed]
- Yandulov, D.V.; Schrock, R.R. Catalytic reduction of dinitrogen to ammonia at a single molybdenum center. Science 2003, 301, 76–78. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-N.; Ma, R.; He, L.-N.; Diao, Z.-F. Homogeneous hydrogenation of carbon dioxide to methanol. Catal. Sci. Technol. 2014, 4, 1498–1512. [Google Scholar] [CrossRef]
- Wang, W.H.; Himeda, Y.; Muckerman, J.T.; Manbeck, G.F.; Fujita, E. CO2 Hydrogenation to Formate and Methanol as an Alternative to Photo- and Electrochemical CO2 Reduction. Chem. Rev. 2015, 115, 12936–12973. [Google Scholar] [CrossRef] [PubMed]
- Federsel, C.; Jackstell, R.; Boddien, A.; Laurenczy, G.; Beller, M. Ruthenium-Catalyzed Hydrogenation of Bicarbonate in Water. ChemSusChem 2010, 3, 1048–1050. [Google Scholar] [CrossRef] [PubMed]
- Wassenaar, J.; de Bruin, B.; Siegler, M.A.; Spek, A.L.; Reek, J.N.H.; van der Vlugt, J.I. Activation of H2 by a highly distorted RhII complex with a new C3-symmetric tripodal tetraphosphine ligand. Chem. Commun. 2010, 46, 1232–1234. [Google Scholar] [CrossRef] [PubMed]
- Wassenaar, J.; Siegler, M.A.; Spek, A.L.; De Bruin, B.; Reek, J.N.H.; van der Vlugt, J.I. Versatile New C3-Symmetric Tripodal Tetraphosphine Ligands; Structural Flexibility to Stabilize CuI and RhI Species and Tune Their Reactivity. Inorg. Chem. 2010, 49, 6495–6508. [Google Scholar] [CrossRef] [PubMed]
- van de Watering, F.F.; van der Vlugt, J.I.; Dzik, W.I.; de Bruin, B.; Reek, J.N.H. Metalloradical Reactivity of RuI and Ru0 Stabilized by an Indole-Based Tripodal Tetraphosphine Ligand. Chem. Eur. J. 2017, 23, 12709–12713. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.O.; Browning, C.S.; Farrar, D.H. Tris-2-(3-methylindolyl)phosphine as an anion receptor. Chem. Commun. 2008, 1020–1022. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.O.; Lam, E.; Sereda, J.L.; Rampersad, N.C.; Lough, A.J.; Browning, C.S.; Farrar, D.H. 2-Indolylphosphines, a New Class of Tunable Ligands: Their Synthesis, Facile Derivatization, and Coordination to Palladium(II). Organometallics 2005, 24, 37–47. [Google Scholar] [CrossRef]
- Yu, J.O. An Exploration of the Structural, Electronic, and Anion Binding Properties of 2-Indolylphosphines. Ph.D. Thesis, University of Toronto, Toronto, ON, Canada, 2008. [Google Scholar]
- Ciclosi, M.; Lloret, J.; Estevan, F.; Lahuerta, P.; Sanaú, M.; Pérez-Prieto, J. A C3-Symmetric Palladium Catalyst with a Phosphorus-Based Tripodal Ligand. Angew. Chem. Int. Ed. 2006, 45, 6741–6744. [Google Scholar] [CrossRef] [PubMed]
- Ciclosi, M.; Estevan, F.; Lahuerta, P.; Passarelli, V.; Perez-Prieto, J.; Sanaú, M. Synthesis and reactivity of the novel hydride derivative RhHCl(TIMP3) (HTIMP3 = tris[1-(diphenylphosphino)-3-methyl-1H-indol-2-yl]methane). Dalton Trans. 2009, 2290–2297. [Google Scholar] [CrossRef] [PubMed]
- Penno, D.; Koshevoy, I.O.; Estevan, F.; Sanaú, M.; Ubeda, M.A.; Pérez-Prieto, J. Synthesis of a New C3-Symmetric Tripodal P4-Tetradentate Ligand and Its Application to the Formation of Chiral Metal Complexes. Organometallics 2010, 29, 703–706. [Google Scholar] [CrossRef]
- Kuo, YY.; Haddow, M.F.; Jamieson, A.L.; Owen, G.R. Synthesis, structural characterisation and catalytic application of dichloro(η6-p-cymene){diphenyl(3-methyl-2-indolyl)phosphine}-ruthenium(II) in the transfer hydrogenation of ketones. Transit. Met. Chem. 2013, 38, 641–648. [Google Scholar] [CrossRef]
- Imayoshi, R.; Tanaka, H.; Matsuo, Y.; Yuki, M.; Nakajima, K.; Yoshizawa, K.; Nishibayashi, Y. Cobalt-Catalyzed Transformation of Molecular Dinitrogen into Silylamine under Ambient Reaction Conditions. Chem. Eur. J. 2015, 21, 8905–8909. [Google Scholar] [CrossRef] [PubMed]
- Yuki, M.; Tanaka, H.; Sasaki, K.; Miyake, Y.; Yoshizawa, K.; Nishibayashi, Y. Iron-catalysed transformation of molecular dinitrogen into silylamine under ambient conditions. Nat. Commun. 2012, 3, 1254. [Google Scholar] [CrossRef] [PubMed]
- Dzik, W.I. Silylation of Dinitrogen Catalyzed by Hydridodinitrogentris(Triphenylphosphine)Cobalt(I). Inorganics 2016, 4, 21. [Google Scholar] [CrossRef]
- Oldenhof, S.; de Bruin, B.; Lutz, M.; Siegler, M.A.; Patureau, F.W.; van der Vlugt, J.I.; Reek, J.N.H. Base-Free Production of H2 by Dehydrogenation of Formic Acid Using an Iridium–bisMETAMORPhos Complex. Chem. Eur. J. 2013, 19, 11507–11511. [Google Scholar] [CrossRef] [PubMed]
- Oldenhof, S.; Lutz, M.; de Bruin, B.; van der Vlugt, J.I.; Reek, J.N.H. Dehydrogenation of formic acid by Ir–bisMETAMORPhos complexes: Experimental and computational insight into the role of a cooperative ligand. Chem. Sci. 2015, 6, 1027–1034. [Google Scholar] [CrossRef]
- Oldenhof, S.; van der Vlugt, J.I.; Reek, J.N.H. Hydrogenation of CO2 to formic acid with iridiumIII(bisMETAMORPhos)(hydride): The role of a dormant fac-IrIII(trihydride) and an active trans-IrIII(dihydride) species. Catal. Sci. Technol. 2016, 6, 404–408. [Google Scholar] [CrossRef]
- Jongbloed, L.S.; de Bruin, B.; Reek, J.N.H.; Lutz, M.; van der Vlugt, J.I. Reversible cyclometalation at RhI as a motif for metal–ligand bifunctional bond activation and base-free formic acid dehydrogenation. Catal. Sci. Technol. 2016, 6, 1320–1327. [Google Scholar] [CrossRef]
- Van de Watering, F.F.; Lutz, M.; Dzik, W.I.; de Bruin, B.; Reek, J.N.H. Reactivity of a Ruthenium–Carbonyl Complex in the Methanol Dehydrogenation Reaction. ChemCatChem 2016, 8, 2752–2756. [Google Scholar] [CrossRef] [PubMed]
- De Boer, S.Y.; Korstanje, T.J.; La Rooij, S.R.; Kox, R.; Reek, J.N.H.; van der Vlugt, J.I. Ruthenium PNN(O) Complexes: Cooperative Reactivity and Application as Catalysts for Acceptorless Dehydrogenative Coupling Reactions. Organometallics 2017, 36, 1541–1549. [Google Scholar] [CrossRef]
- Trincado, M.; Sinha, V.; Rodriguez-Lugo, R.E.; Pribanic, B.; de Bruin, B.; Grützmacher, H. Homogeneously catalysed conversion of aqueous formaldehyde to H2 and carbonate. Nat. Commun. 2017, 8, 14990. [Google Scholar] [CrossRef] [PubMed]
- Himeda, Y.; Miyazawa, S.; Hirose, T. Interconversion between formic acid and H2/CO2 using rhodium and ruthenium catalysts for CO2 fixation and H2 storage. ChemSusChem 2011, 4, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Scholten, J.D.; Prechtl, M.H.G.; Dupont, J. Decomposition of Formic Acid Catalyzed by a Phosphine-Free Ruthenium Complex in a Task-Specific Ionic Liquid. ChemCatChem 2010, 2, 1265–1270. [Google Scholar] [CrossRef]
- Wang, Z.; Lu, S.-M.; Li, J.; Wang, J.; Li, C. Unprecedentedly High Formic Acid Dehydrogenation Activity on an Iridium Complex with an N,N′-Diimine Ligand in Water. Chem. Eur. J. 2015, 21, 12592–12595. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Lugo, R.E.; Trincado, M.; Vogt, M.; Tewes, F.; Santiso-Quinones, G.; Grützmacher, H. A homogeneous transition metal complex for clean hydrogen production from methanol-water mixtures. Nat. Chem. 2013, 5, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Mellone, I.; Bertini, F.; Peruzzini, M.; Gonsalvi, L. An active, stable and recyclable Ru(II) tetraphosphine-based catalytic system for hydrogen production by selective formic acid dehydrogenation. Catal. Sci. Technol. 2016, 6, 6504–6512. [Google Scholar] [CrossRef]
- Weitz, I.S.; Rabinovitz, M. The application of C8K for organic synthesis: Reduction of substituted naphthalenes. J. Chem. Soc. Perkin Trans. 1993, 1, 117–120. [Google Scholar] [CrossRef]
- Nielsen, M.; Alberico, E.; Baumann, W.; Drexler, H.-J.; Junge, H.; Gladiali, S.; Beller, M. Catalysis: A step closer to a methanol economy. Nature 2013, 495, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Bruker. SAINT-Plus; Bruker AXS Inc.: Madison, WI, USA, 2001. [Google Scholar]
- Sheldrick, G.M. SADABS and TWINABS; Universität Göttingen: Göttingen, Germany, 2008. [Google Scholar]
- Sheldrick, G.M. SHELXT; Universität Göttingen: Göttingen, Germany, 2012. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. Structure validation in chemical crystallography. Acta Cryst. 2009, D65, 148–155. [Google Scholar] [CrossRef] [PubMed]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
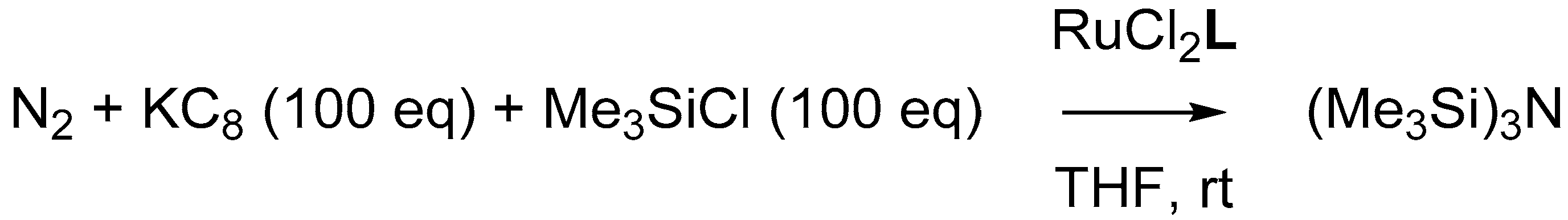{kind=link}
{kind=link}
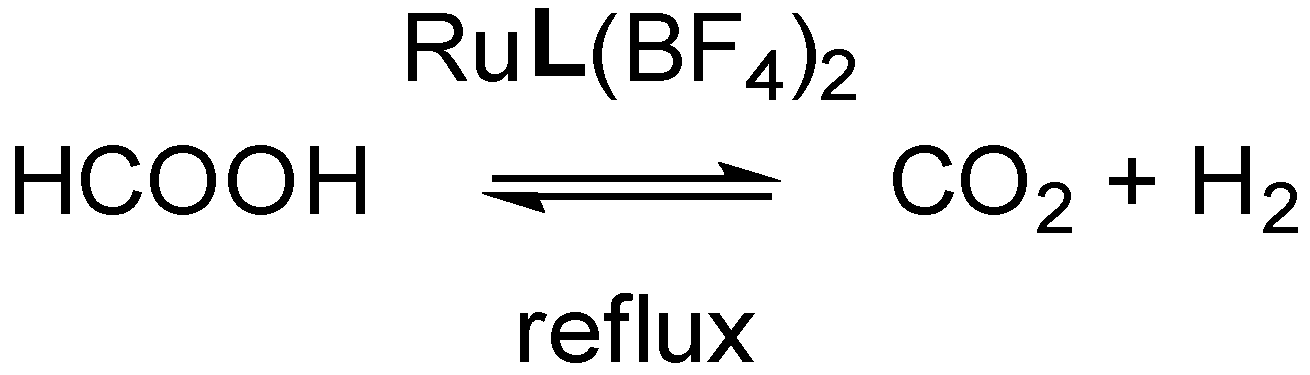{kind=link}
{kind=link}
{kind=link}
| RuCl2L1H [a] | RuCl2L1CF3 | RuCl2L2H | RuN2L1H | RuN2L1CF3 | |
|---|---|---|---|---|---|
| Ru1–P1 | 2.3189(9) | 2.3097(19) | 2.3295(8) | 2.2747(12) | 2.2613(10) |
| Ru1–P2 | 2.3727(9) | 2.3445(19) | 2.4079(8) | 2.2752(11) | 2.2702(10) |
| Ru1–P3 | 2.2671(9) | 2.2383(17) | 2.2913(8) | 2.2774(11) | 2.2554(10) |
| Ru1–P4 | 2.1932(9) | 2.2023(17) | 2.1363(8) | 2.2133(11) | 2.2193(9) |
| Ru1–Cl1 | 2.4869(9) | 2.4869(16) | 2.4829(7) | ||
| Ru1–Cl2 | 2.4471(9) | 2.4487(16) | 2.4705(7) | ||
| Ru1–N1 | 2.011(4) | 2.066(3) | |||
| N1–N2 | 1.085(5) | 1.064(5) | |||
| P1–Ru1–P2 | 160.04(3) | 157.78(6) | 159.24(3) | 122.85(4) | 119.79(4) |
| P1–Ru1–P3 | 102.23(3) | 101.39(7) | 101.30(3) | 115.80(4) | 119.28(4) |
| P2–Ru1–P3 | 94.27(3) | 97.42(7) | 93.60(3) | 118.33(4) | 118.16(4) |
| P1–Ru1–P4 | 87.06(3) | 86.13(6) | 86.13(3) | 84.14(4) | 84.54(4) |
| P2–Ru1–P4 | 82.67(3) | 83.12(6) | 80.63(3) | 83.91(4) | 84.13(4) |
| P3–Ru1–P4 | 86.85(3) | 87.02(6) | 85.62(3) | 84.63(4) | 84.73(3) |
| Complex | νN2 (cm−1) |
|---|---|
| RuN2L1CF3 | 2136 |
| RuN2L1H | 2125 |
| RuN2L1OMe | 2113 |
| RuN2L2H | 2136 |
| Complex | Equiv (Me3Si)3N [a] |
|---|---|
| RuCl2L1CF3 | 0.92 [b] |
| RuCl2L1H | 1.83 [b] |
| RuCl2L1OMe | 1.74 [b] |
| RuCl2L2H | 1.40 [b] |
| [Ru(η6-benzene)Cl(μ-Cl)]2 | 1.29 [c] |
| - | trace |
| Complex | TOF (h−1) [a] |
|---|---|
| RuCl2L1CF3 | 124 |
| RuCl2L1H | 76 |
| RuCl2L1OMe | 121 |
| RuCl2L2H | 33 |
| – | – [b] |
| [{RuCl2(p-cymene)}2] | 1540 [c] |
| Complex | TOF (h−1) #1 | TOF (h−1) #2 | TOF (h−1) Average |
|---|---|---|---|
| RuCl2L1CF3 | 120.8 | 127.2 | 124 |
| RuCl2L1H | 79.3 | 71.9 | 75.6 |
| RuCl2L1OMe | 123.5 | 117.4 | 120.5 |
| RuCl2L2H | 22.4 | 42.7 | 32.6 |
| Complex | RuCl2L1CF3 | RuCl2L2H | RuN2L1CF3 |
|---|---|---|---|
| Empirical formula | C69H45Cl2F18N3P4Ru + solvent | C63H51Cl2N3P4Ru, 2(CH2Cl2) + solvent | C69H45F18N5P4Ru + solvent |
| FW | 1553.93 a | 1315.77 a | 1511.05 a |
| Temperature [K] | 150 | 150 | 150 |
| Radiation | Mo Kα | Mo Kα | Mo Kα |
| Wavelength [Å] | 0.71073 | 0.71073 | 0.71073 |
| Cryst syst. | monoclinic | monoclinic | monoclinic |
| Space group | C 2/c | P 21/n | P 21/c |
| a [Å] | 21.594(3) | 19.1879(9) | 14.2851(6) |
| b [Å] | 27.753(4) | 18.1604(8) | 18.9090(8) |
| c [Å] | 23.938(3) | 19.7879(9) | 29.8721(12) |
| α [deg.] | 90 | 90 | 90 |
| β [deg.] | 93.631(3) | 91.012(2) | 97.011(2) |
| γ [deg.] | 90 | 90 | 90 |
| Volume [Å3] | 14317(3) | 6894.2(5) | 8008.6(6) |
| Z | 8 | 4 | 4 |
| Color | pale yellow | yellow | dark red |
| θ-max | 25.135 | 25.030 | 26.450 |
| Density [kg·m−3] | 1.442 a | 1.268 a | 1.253 a |
| Absorp. Coeff. [mm−1] | 0.472 a | 0.591 a | 0.356 a |
| F(000) | 6240 a | 2688.0 | 3040 a |
| R1/wR2/S | 0.0715/0.1975/1.053 | 0.0412/0.1435/1.134 | 0.0666/0.2002/1.436 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van de Watering, F.F.; Heijtbrink, N.; Van der Vlugt, J.I.; Dzik, W.I.; De Bruin, B.; Reek, J.N.H. 3-Methylindole-Based Tripodal Tetraphosphine Ruthenium Complexes in N2 Coordination and Reduction and Formic Acid Dehydrogenation. Inorganics 2017, 5, 73. https://doi.org/10.3390/inorganics5040073
Van de Watering FF, Heijtbrink N, Van der Vlugt JI, Dzik WI, De Bruin B, Reek JNH. 3-Methylindole-Based Tripodal Tetraphosphine Ruthenium Complexes in N2 Coordination and Reduction and Formic Acid Dehydrogenation. Inorganics. 2017; 5(4):73. https://doi.org/10.3390/inorganics5040073
Chicago/Turabian StyleVan de Watering, Fenna F., Nicol Heijtbrink, Jarl Ivar Van der Vlugt, Wojciech I. Dzik, Bas De Bruin, and Joost N. H. Reek. 2017. "3-Methylindole-Based Tripodal Tetraphosphine Ruthenium Complexes in N2 Coordination and Reduction and Formic Acid Dehydrogenation" Inorganics 5, no. 4: 73. https://doi.org/10.3390/inorganics5040073
APA StyleVan de Watering, F. F., Heijtbrink, N., Van der Vlugt, J. I., Dzik, W. I., De Bruin, B., & Reek, J. N. H. (2017). 3-Methylindole-Based Tripodal Tetraphosphine Ruthenium Complexes in N2 Coordination and Reduction and Formic Acid Dehydrogenation. Inorganics, 5(4), 73. https://doi.org/10.3390/inorganics5040073