Redox-Induced Aromatic C–H Bond Functionalization in Metal Complex Catalysis from the Electrochemical Point of View



Abstract
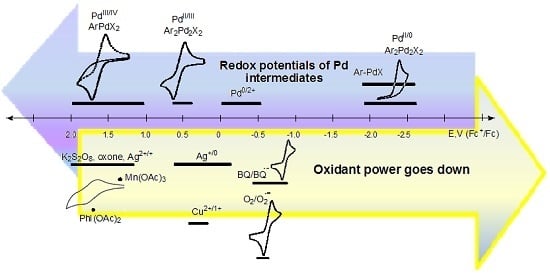
:
1. Introduction
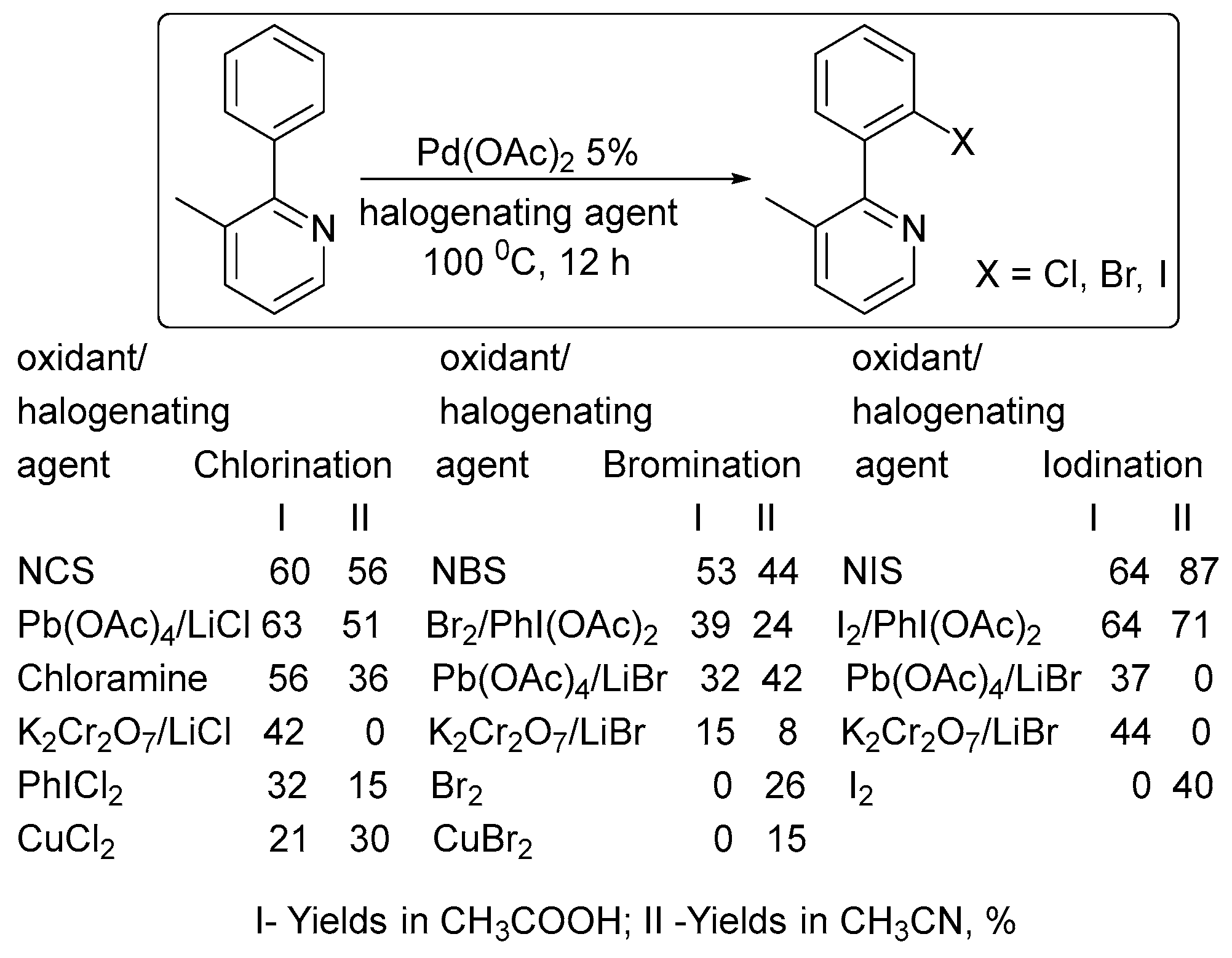
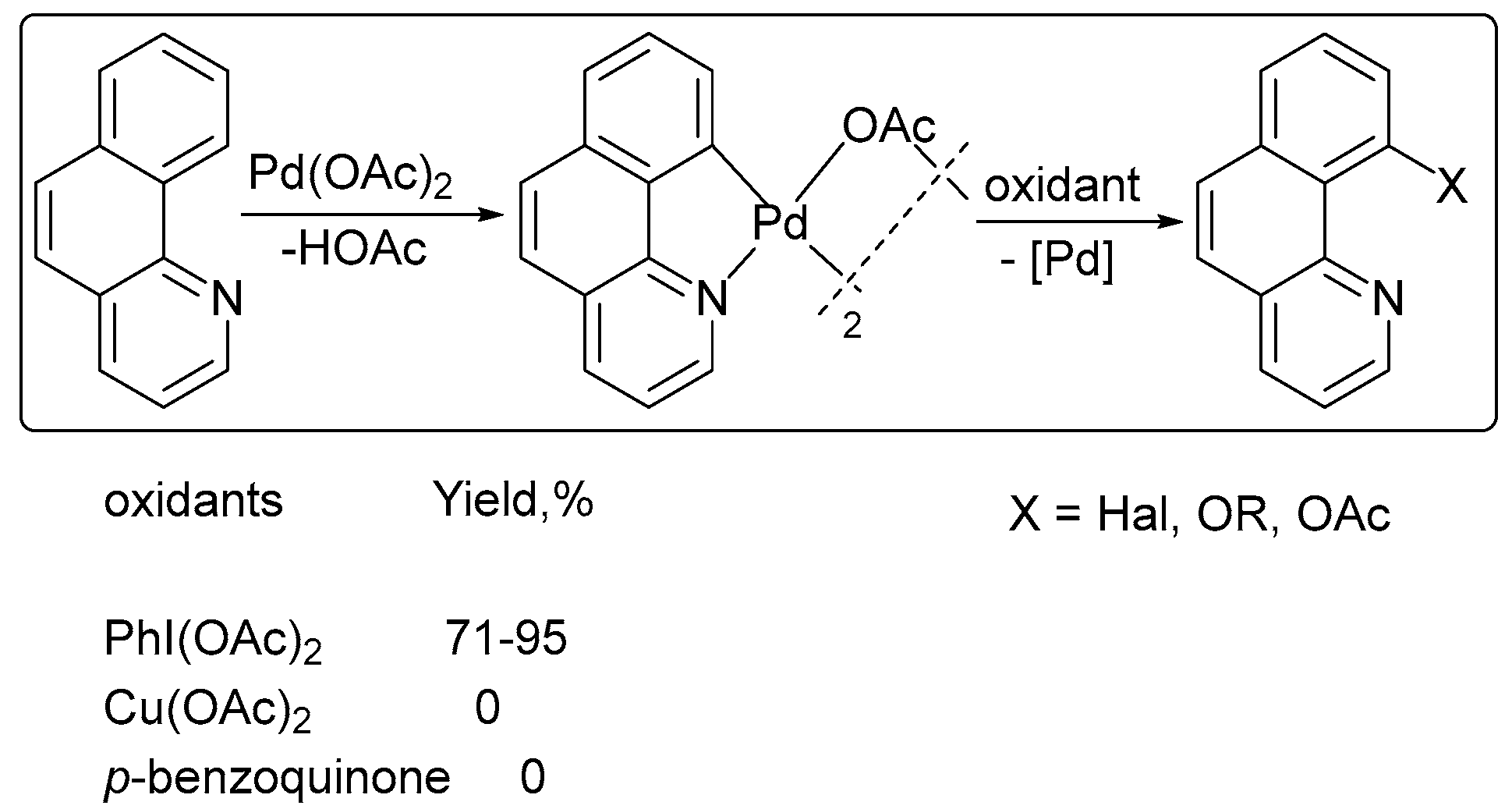
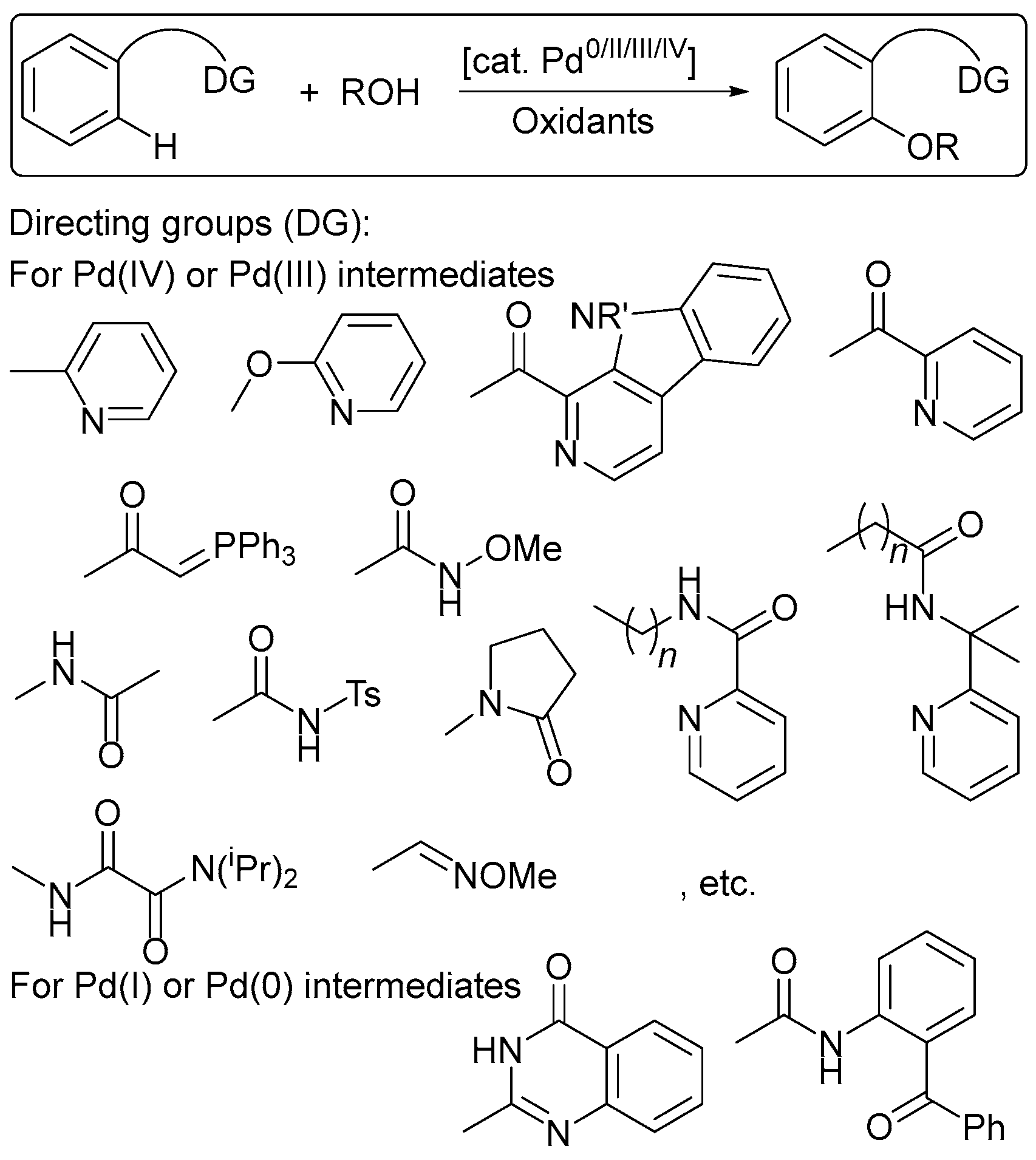
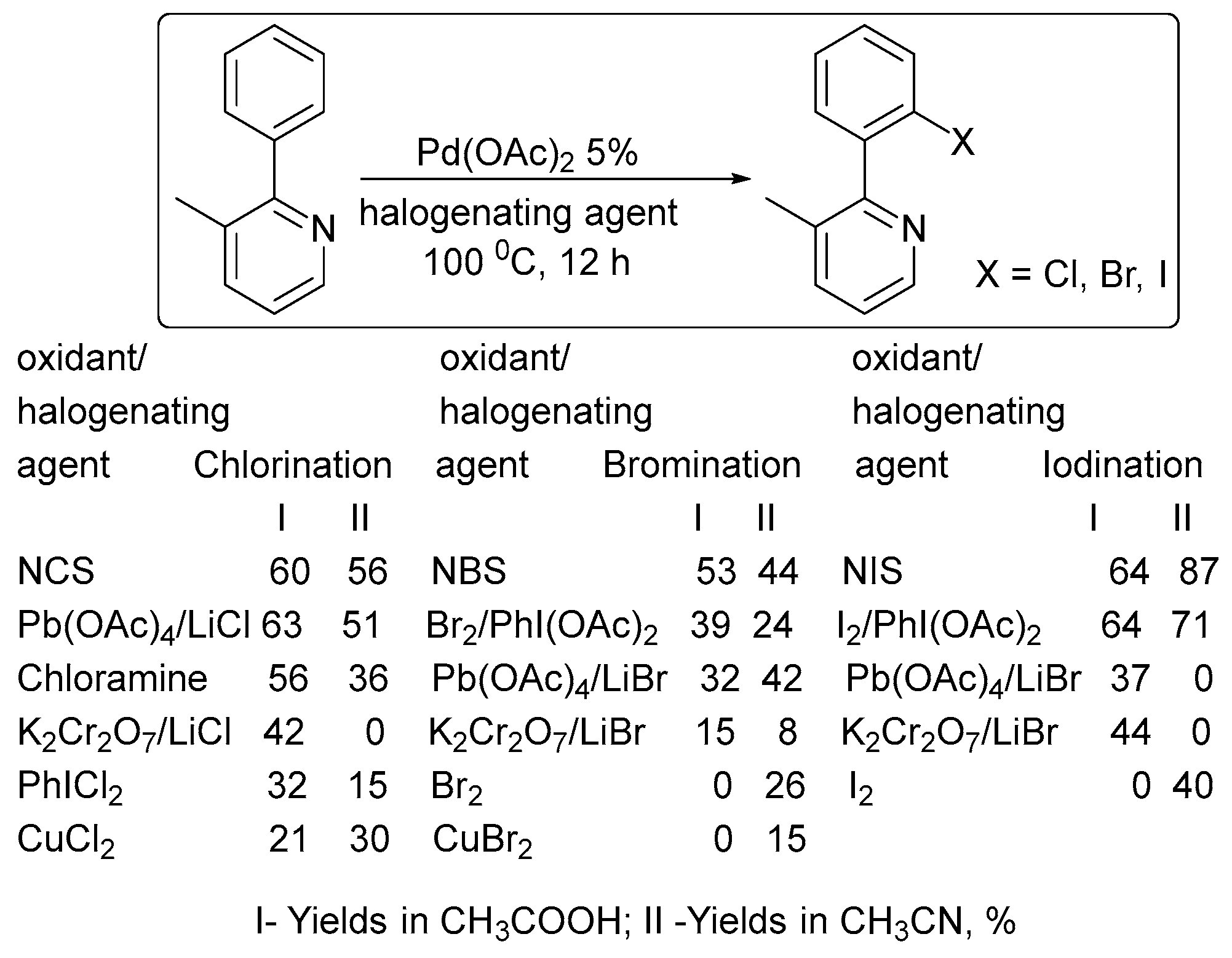
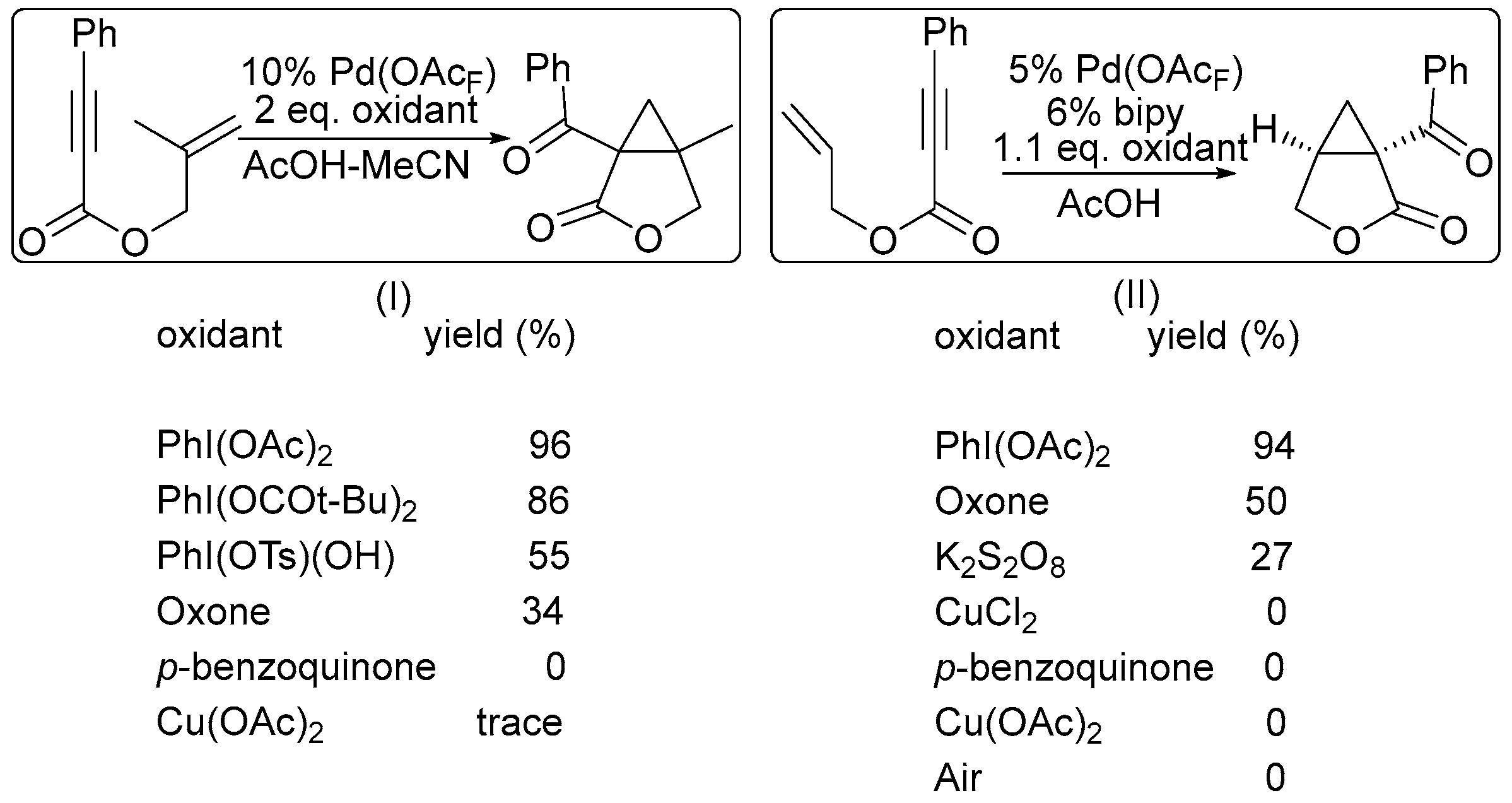
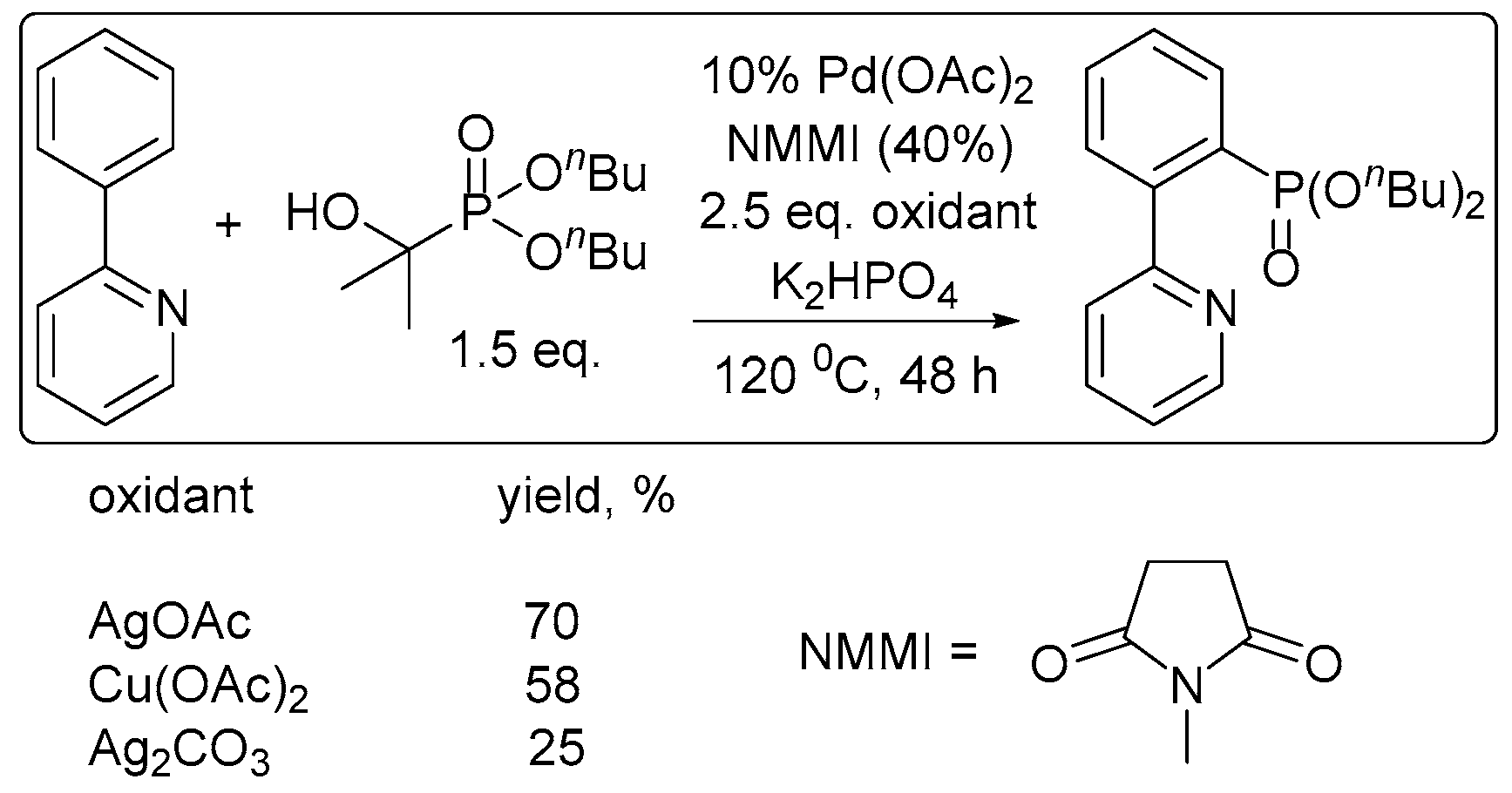
2. Oxidant Screening in Palladium Catalysis
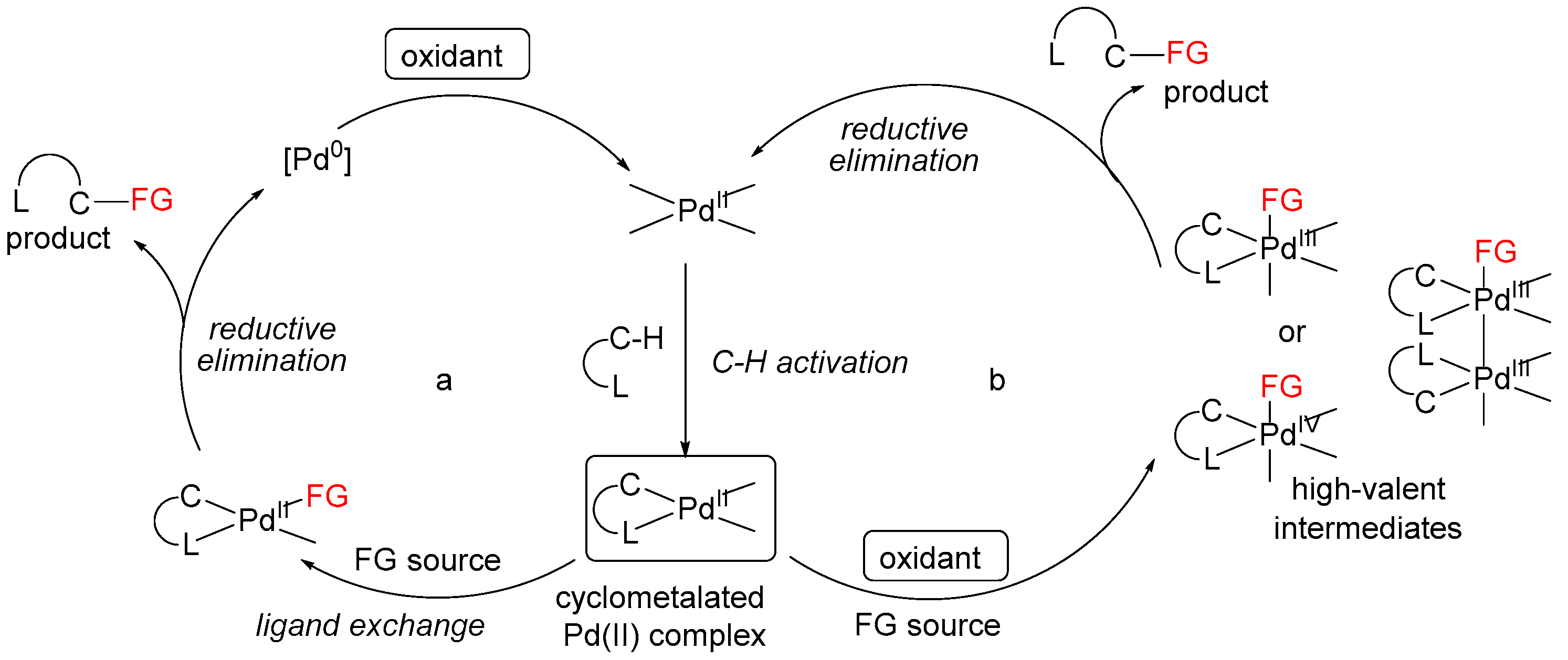
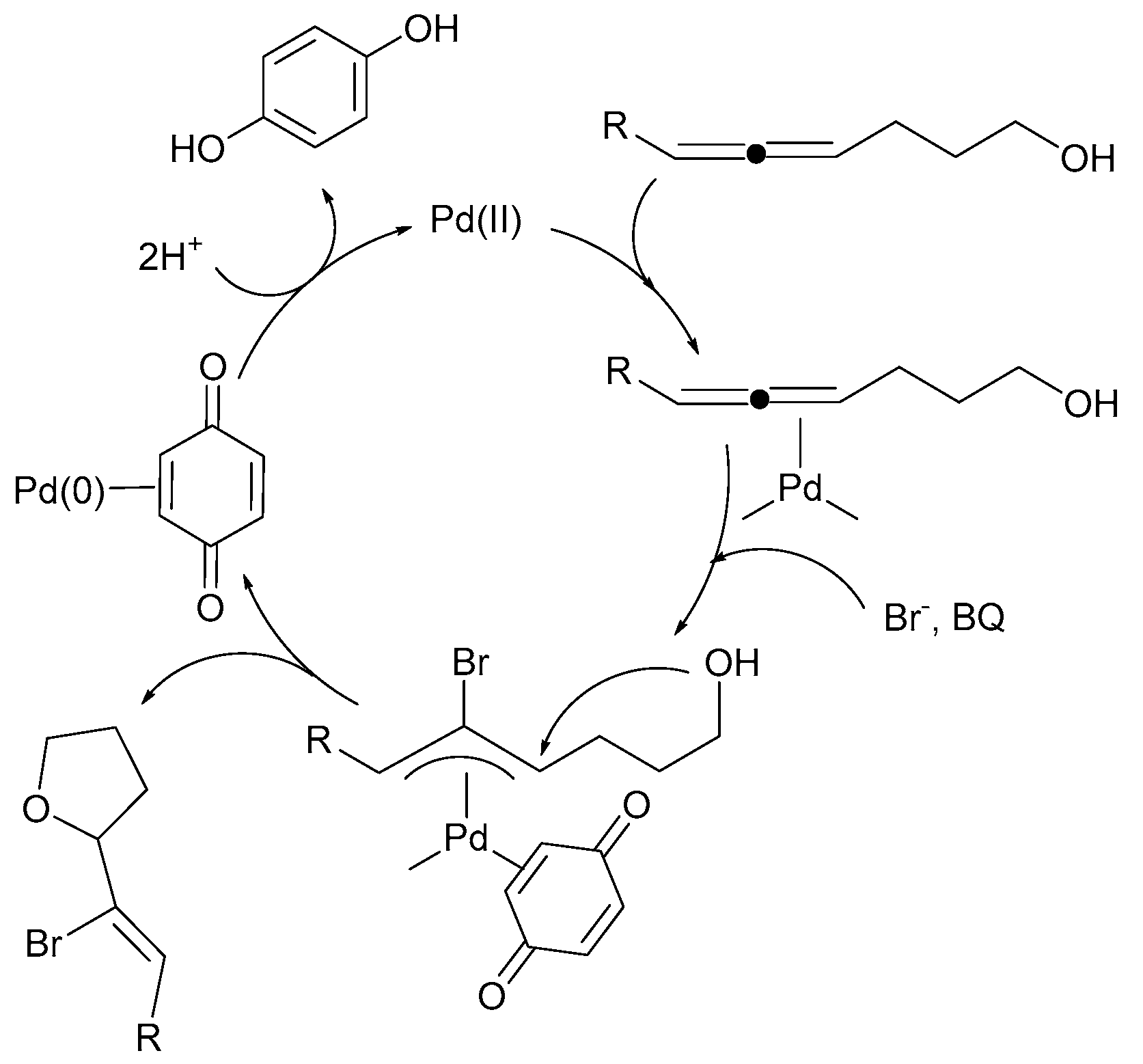
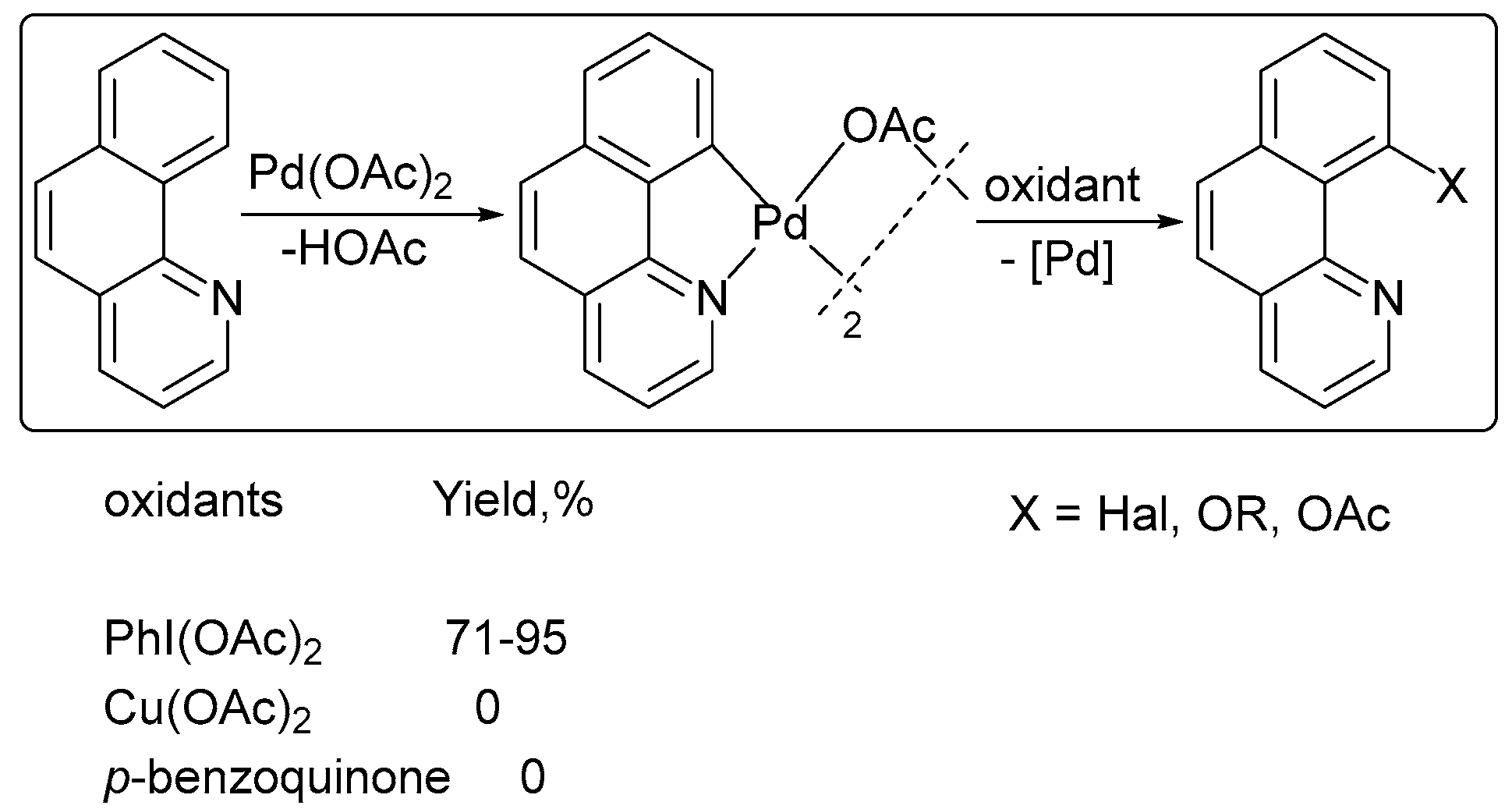
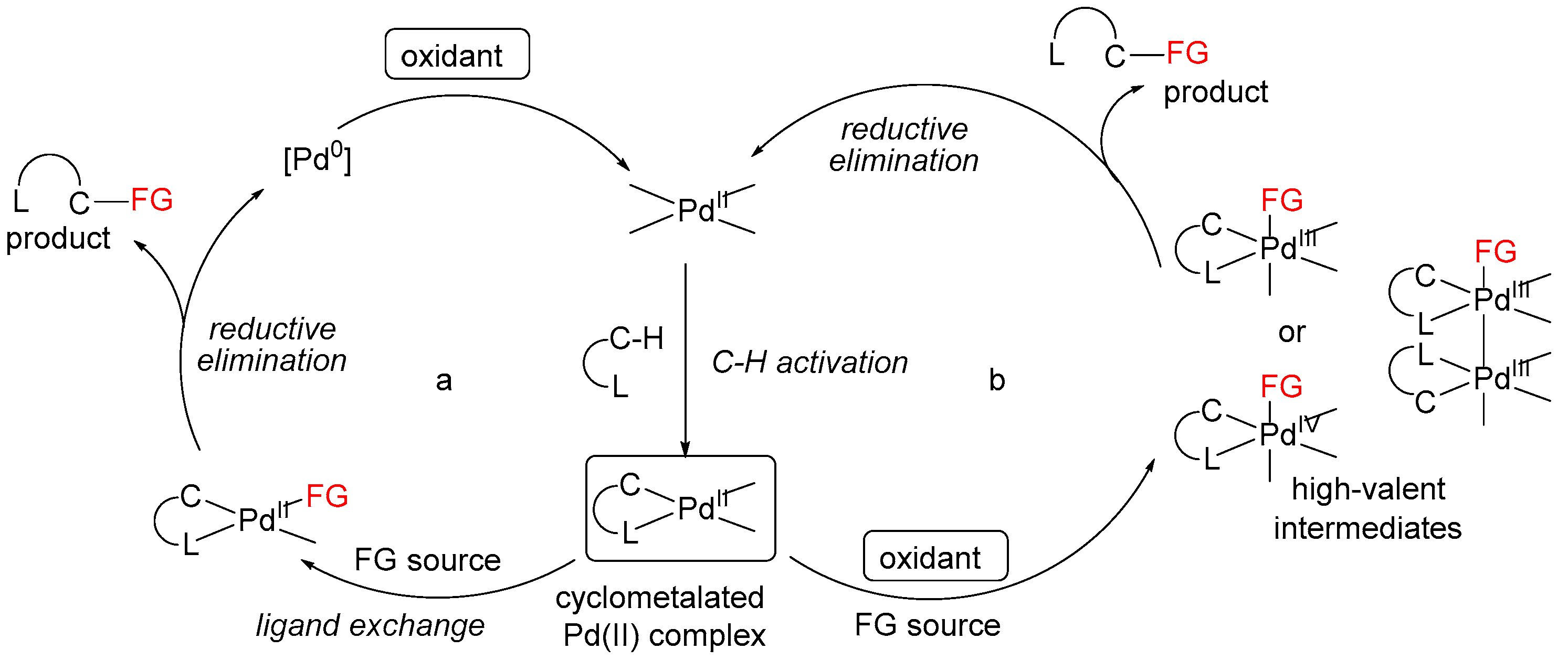
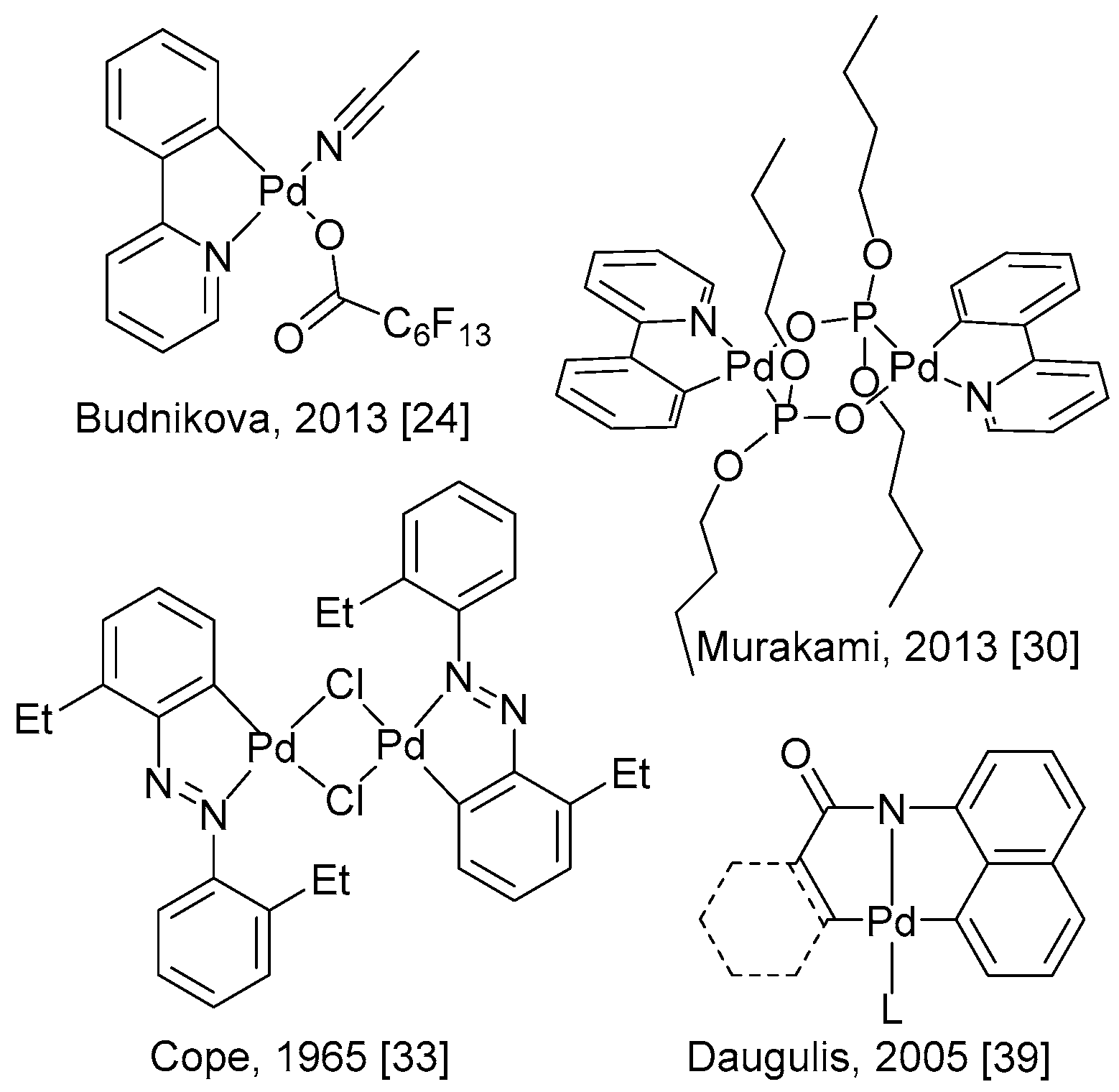
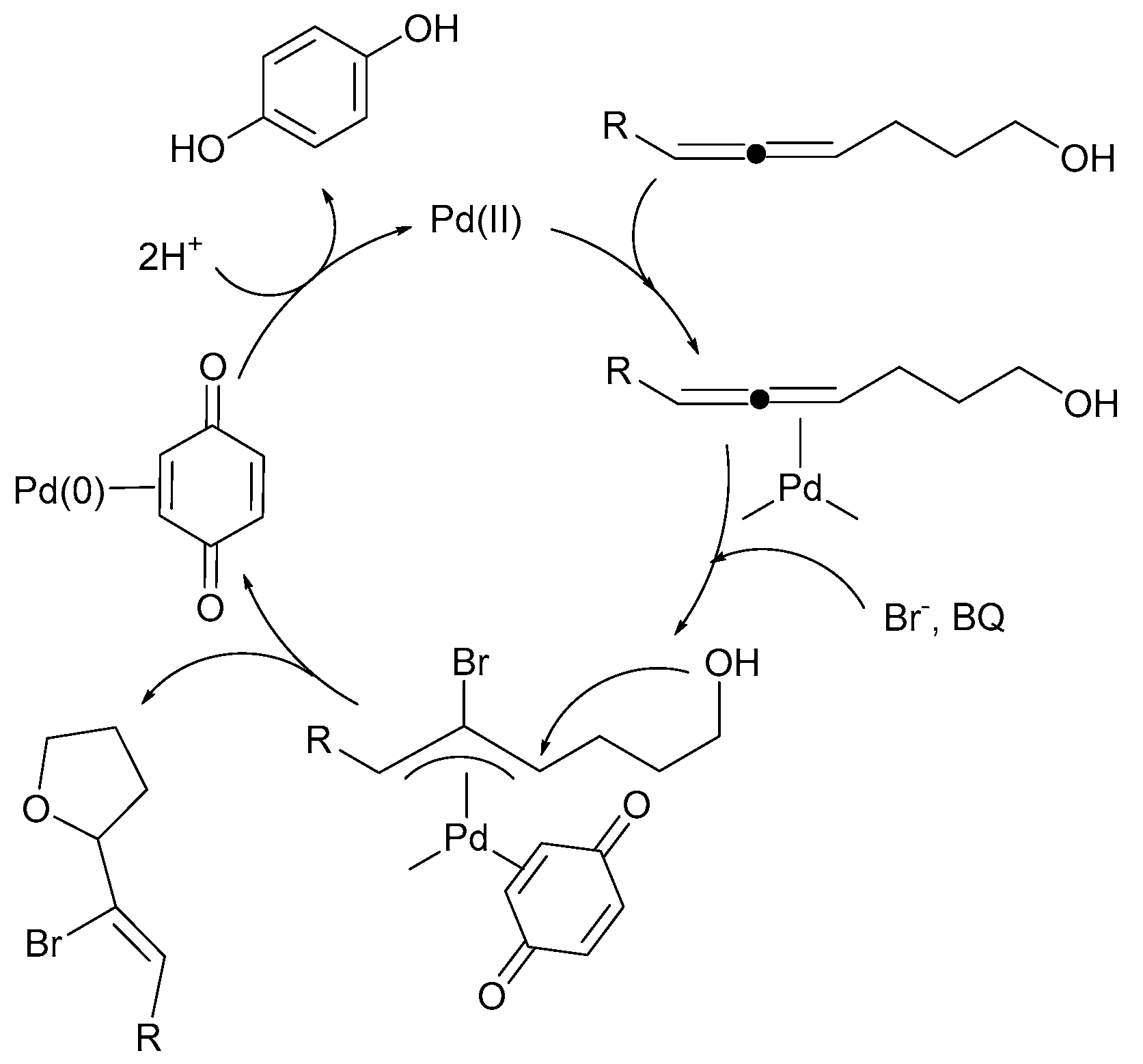
3. Mechanistic Considerations
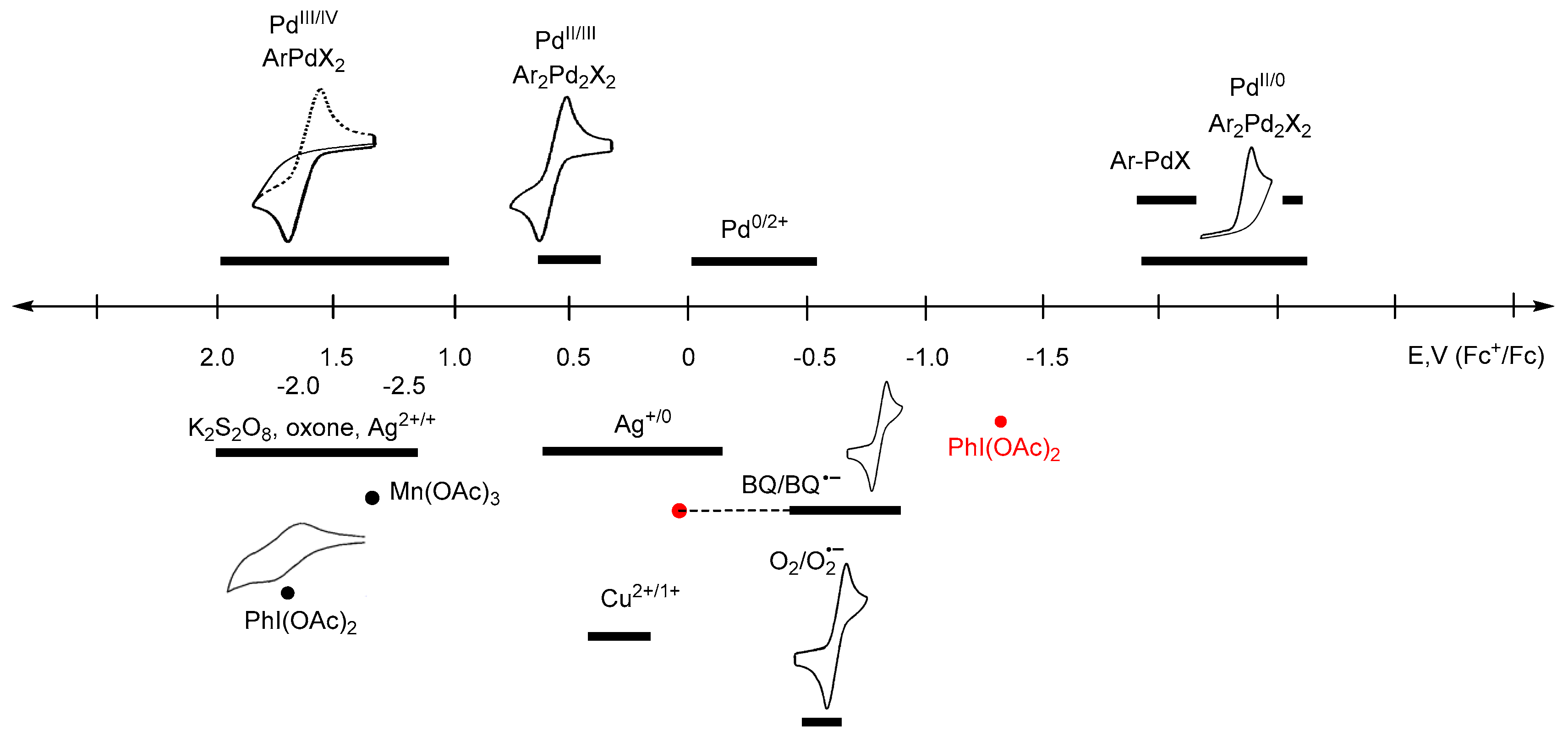
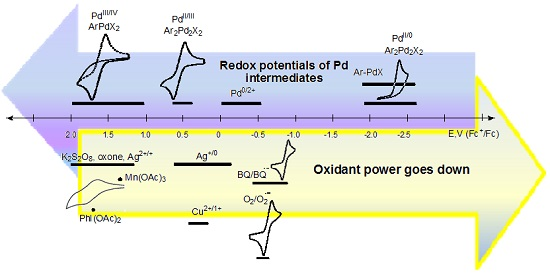
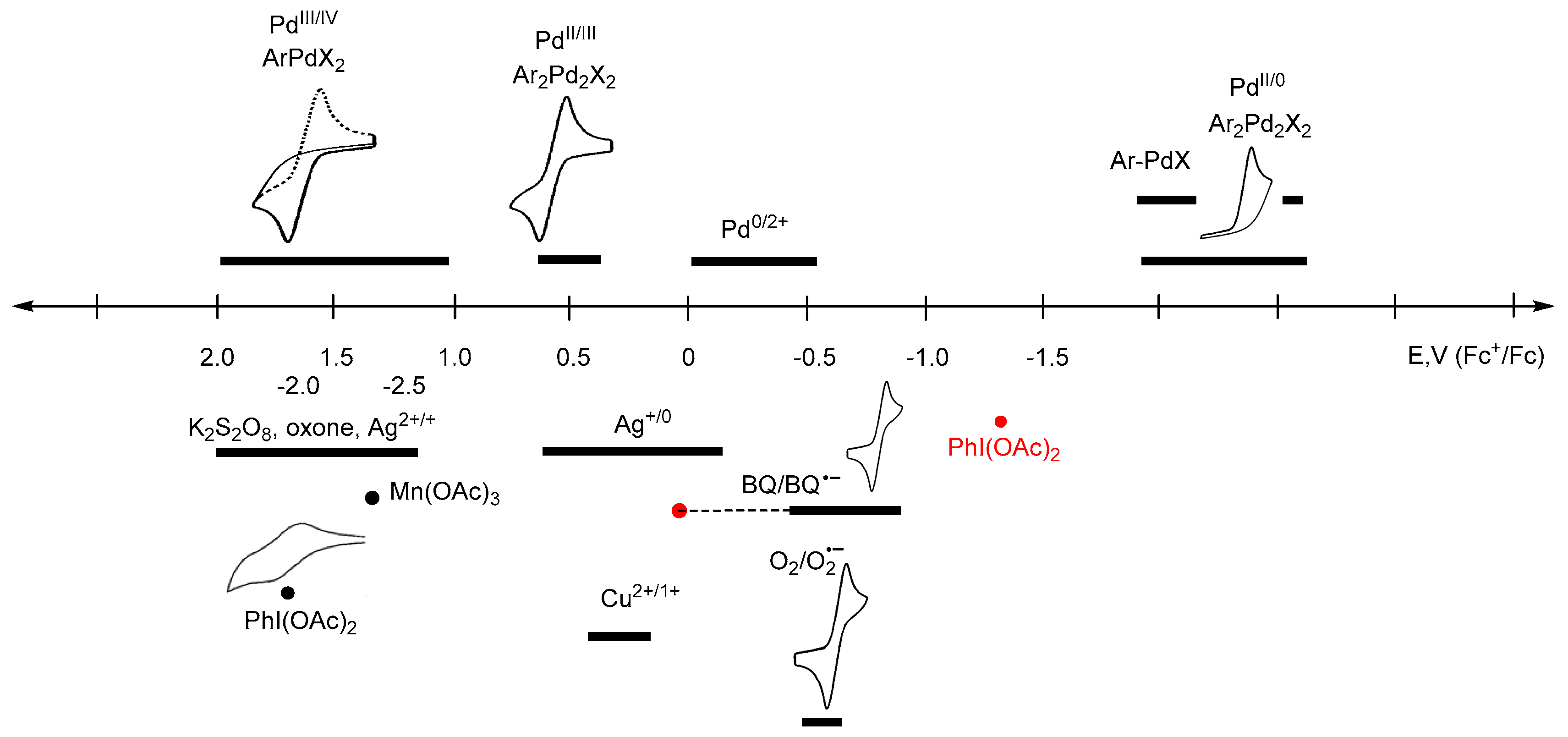
4. Electrode Potentials as the Measure of Oxidant/Reductant Strength
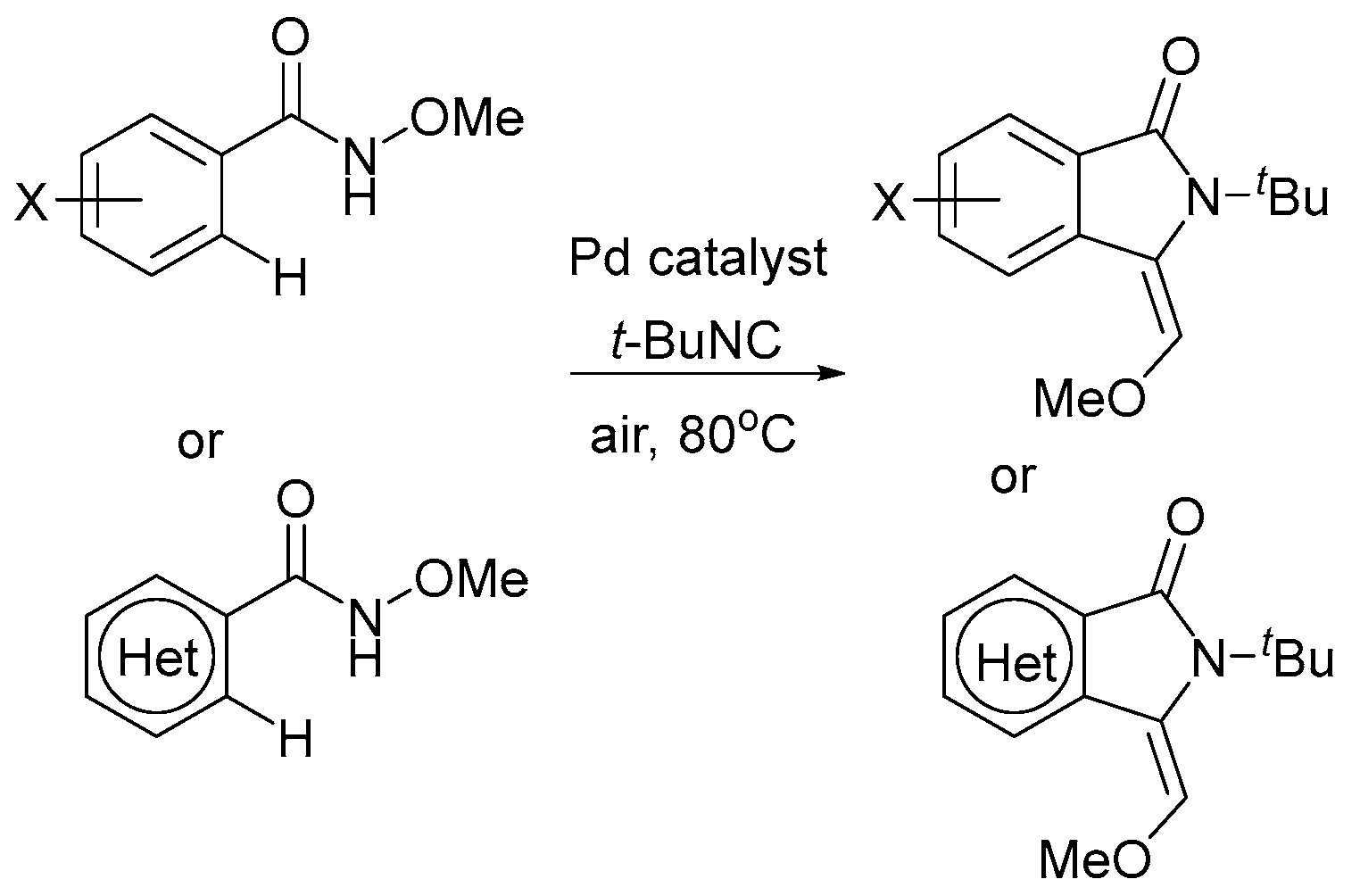
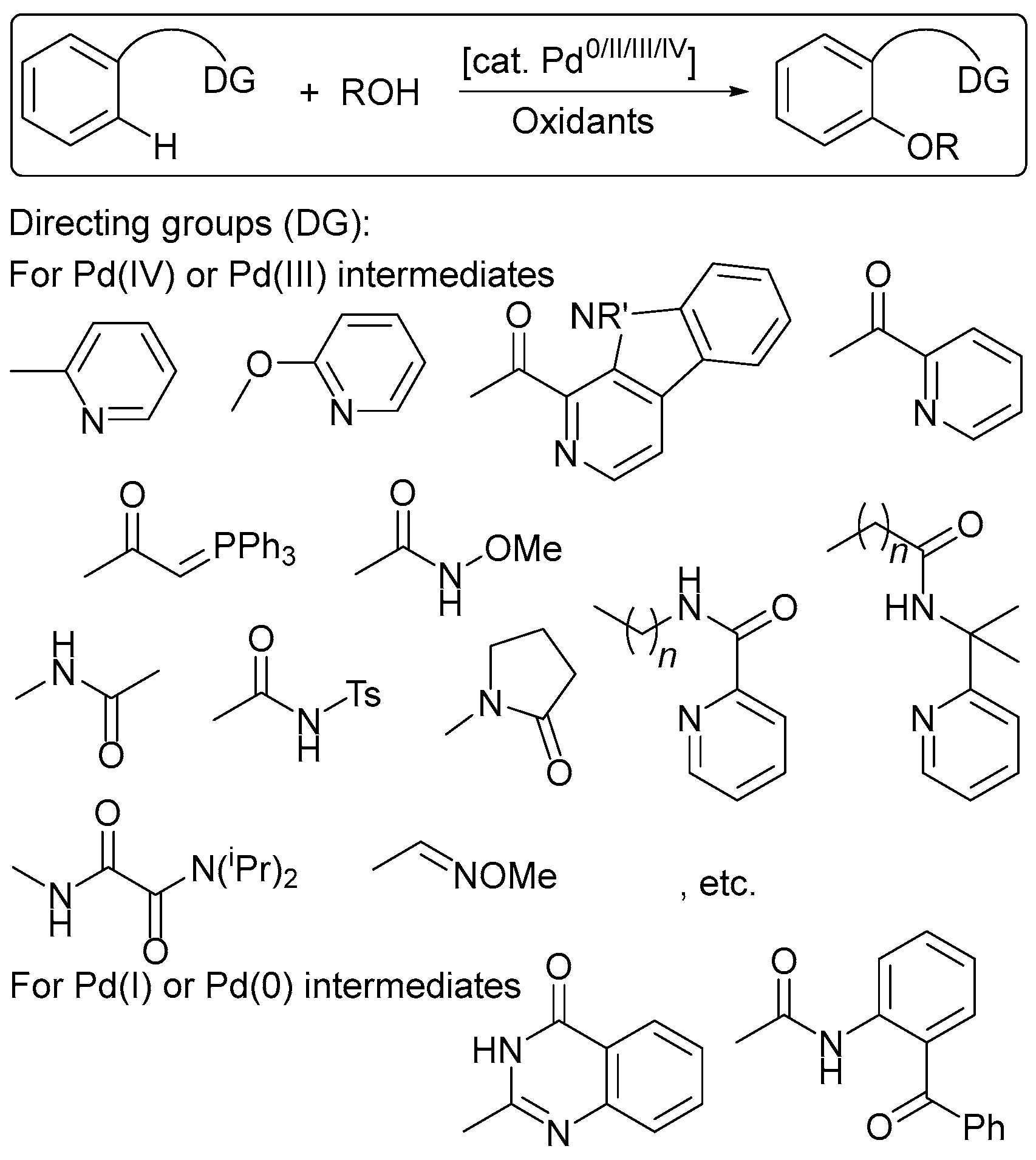
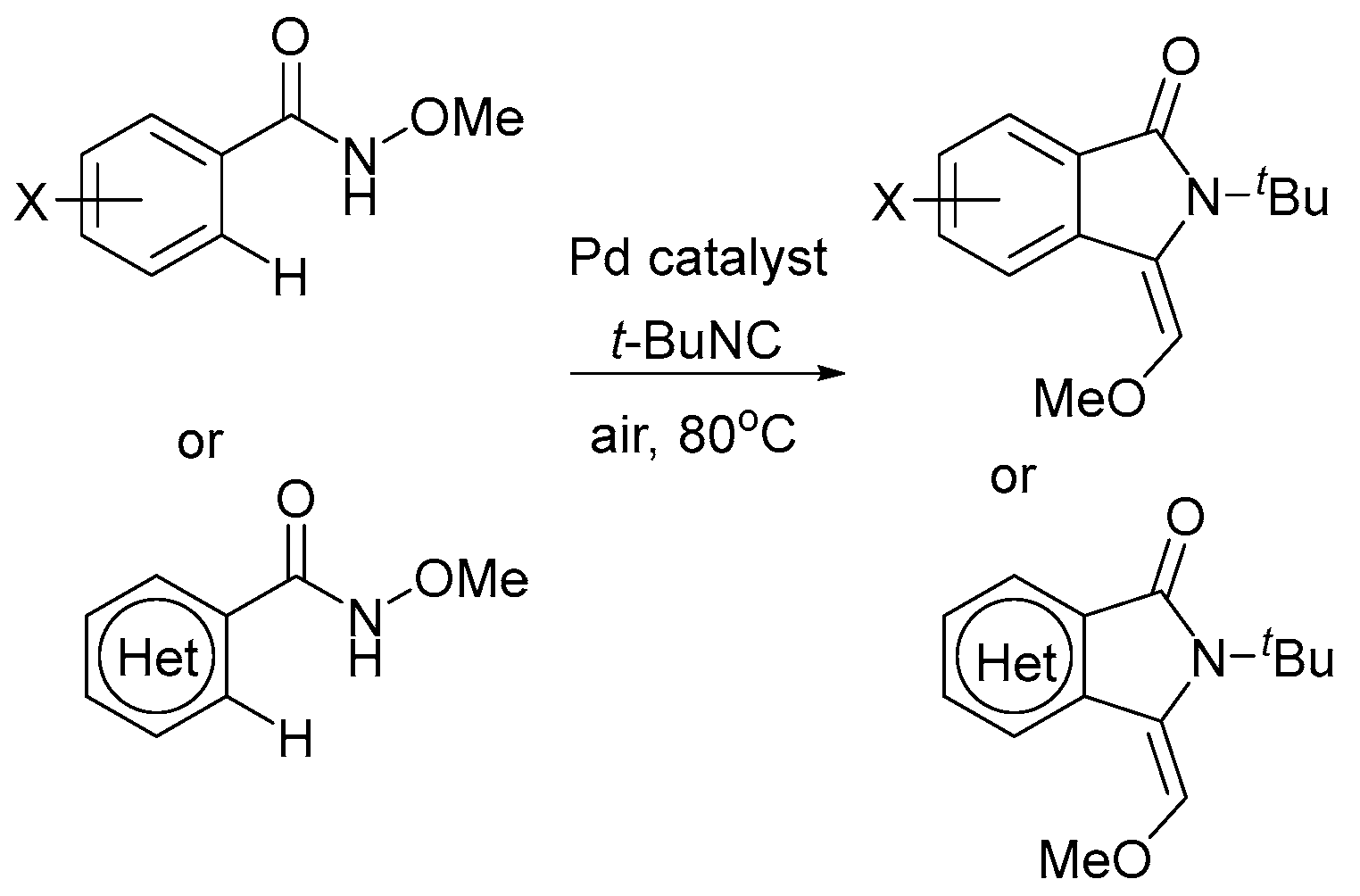
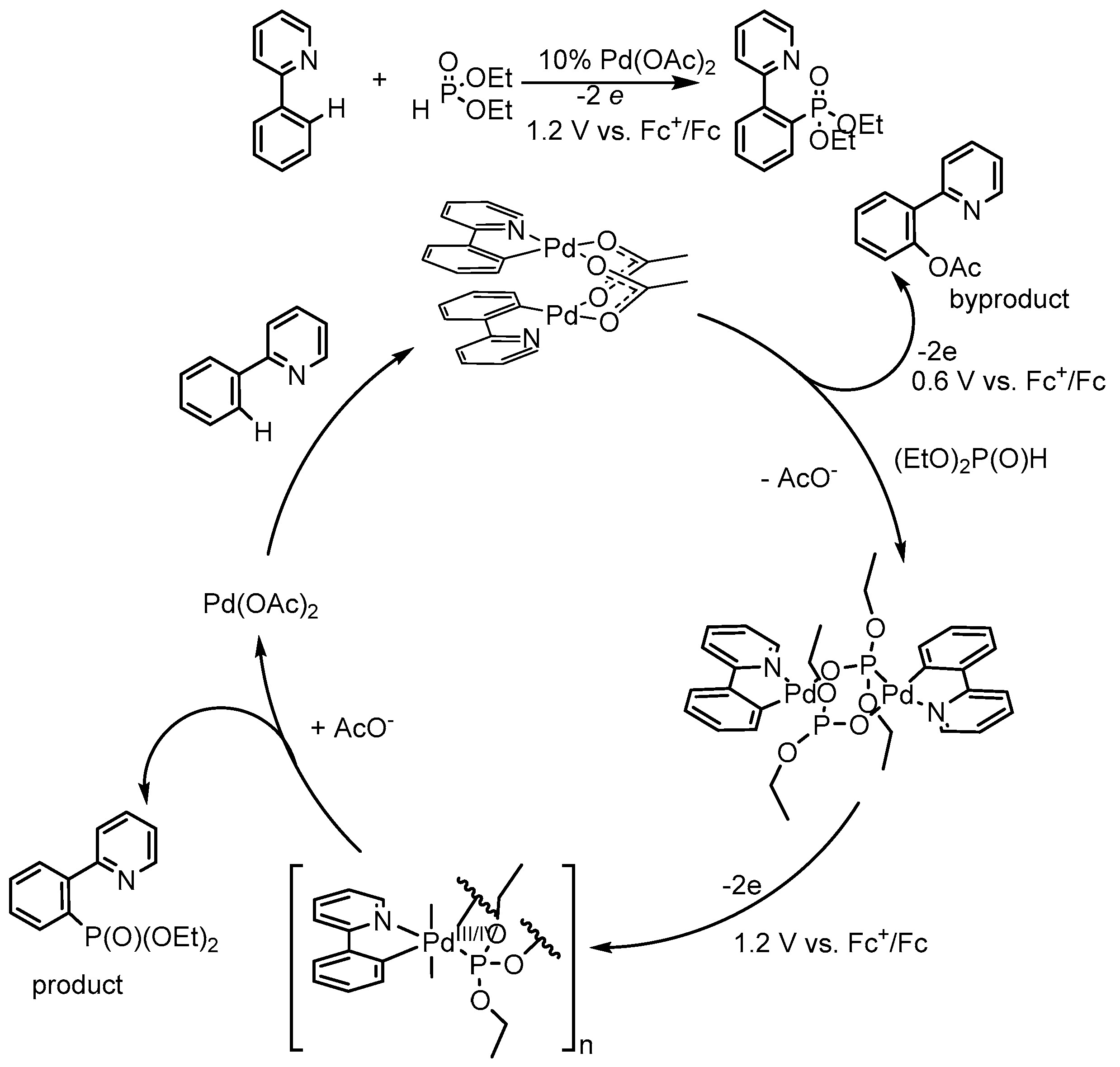
5. Electrocatalytic Ligand-Directed Substitution of C(sp2)–H Bonds
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gensch, T.; Hopkinson, M.N.; Glorius, F.; Wencel-Delord, J. Mild metal-catalyzed C–H activation: Examples and concepts. Chem. Soc. Rev. 2016, 45, 2900–2936. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Shi, Z.; Yuan, Y. Transition-metal-catalyzed Chelation-assisted C–H Functionalization of Aromatic Substrates. Chem. Rec. 2016, 16, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Lyons, T.W.; Sanford, M.S. Palladium-Catalyzed Ligand-Directed C−H Functionalization Reactions. Chem. Rev. 2010, 110, 1147–1169. [Google Scholar] [CrossRef] [PubMed]
- Wencel-Delord, J.; Kim, J.G.; Carr, K.J.T.; Bellina, F.; Soulé, J.-F.; Wang, G.-W.; Bruneau, C.; Dana, S.; Li, J.; Ackermann, L.; et al. C–H Bond Activation and Catalytic Functionalization I; Dixneuf, P.H., Doucet, H., Eds.; Springer: Cham, Switzerland, 2016. [Google Scholar]
- Ilies, L.; Nakamura, E.; Chatani, N.; Hirano, K.; Jones, W.; Sustac Roman, D.; Zhou, T.; Dailler, D.; Samantaray, M.K.; Bézier, D.; et al. C–H Bond Activation and Catalytic Functionalization II; Dixneuf, P.H., Doucet, H., Eds.; Springer: Cham, Switzerland, 2016. [Google Scholar]
- Liu, C.; Yuan, J.; Gao, M.; Tang, S.; Li, W.; Shi, R.; Lei, A. Oxidative Coupling between Two Hydrocarbons: An Update of Recent C–H Functionalizations. Chem. Rev. 2015, 115, 12138–12204. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.-M.; Li, B.-J.; Yang, Z.; Shi, Z.-J. Organopalladium(IV) chemistry. Chem. Soc. Rev. 2010, 39, 712–733. [Google Scholar] [CrossRef] [PubMed]
- Sehnal, P.; Taylor, R.J.K.; Fairlamb, I.J.S. Emergence of Palladium(IV) Chemistry in Synthesis and Catalysis. Chem. Rev. 2010, 110, 824–889. [Google Scholar] [CrossRef] [PubMed]
- Engle, K.M.; Mei, T.-S.; Wang, X.; Yu, J.-Q. Bystanding F+ Oxidants Enable Selective Reductive Elimination from High-Valent Metal Centers in Catalysis. Angew. Chem. Int. Ed. 2011, 50, 1478–1491. [Google Scholar] [CrossRef] [PubMed]
- Hickman, A.J.; Sanford, M.S. High-valent organometallic copper and palladium in catalysis. Nature 2012, 484, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Powers, D.C.; Geibel, M.A.L.; Klein, J.E.M.N.; Ritter, T. Bimetallic Palladium Catalysis: Direct Observation of Pd(III)−Pd(III) Intermediates. J. Am. Chem. Soc. 2009, 131, 17050–17051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deprez, N.R.; Sanford, M.S. Synthetic and Mechanistic Studies of Pd-Catalyzed C−H Arylation with Diaryliodonium Salts: Evidence for a Bimetallic High Oxidation State Pd Intermediate. J. Am. Chem. Soc. 2009, 131, 11234–11241. [Google Scholar] [CrossRef] [PubMed]
- Budnikova, Y.H.; Gryaznova, T.V.; Grinenko, V.V.; Dudkina, Y.B.; Khrizanforov, M.N. Eco-efficient electrocatalytic C–P bond formation. Pure Appl. Chem. 2017, 89, 311–330. [Google Scholar] [CrossRef]
- Grayaznova, T.V.; Dudkina, Y.B.; Islamov, D.R.; Kataeva, O.N.; Sinyashin, O.G.; Vicic, D.A.; Budnikova, Y.H. Pyridine-directed palladium-catalyzed electrochemical phosphonation of C(sp2)–H bond. J. Organomet. Chem. 2015, 785, 68–71. [Google Scholar] [CrossRef]
- Gryaznova, T.; Dudkina, Y.; Khrizanforov, M.; Sinyashin, O.; Kataeva, O.; Budnikova, Y. Electrochemical properties of diphosphonate-bridged palladacycles and their reactivity in arene phosphonation. J. Solid State Electrochem. 2015, 19, 2665–2672. [Google Scholar] [CrossRef]
- Khrizanforov, M.N.; Strekalova, S.O.; Kholin, K.V.; Khrizanforova, V.V.; Kadirov, M.K.; Gryaznova, T.V.; Budnikova, Y.H. Novel approach to metal-induced oxidative phosphorylation of aromatic compounds. Catal. Today 2017, 279, 133–141. [Google Scholar] [CrossRef]
- Grinenko, V.V.; Khrizanforov, M.N.; Strekalova, S.O.; Khrizanforova, V.V.; Kholin, K.V.; Gryaznova, T.V.; Budnikova, Y.H. Electrooxidative phosphorylation of coumarins by bimetallic catalytic systems Ni(II)/Mn(II) or Co(II)/Mn(II). Phosphorus Sulfur Silicon Relat. Elem. 2016, 191, 1660–1661. [Google Scholar] [CrossRef]
- Khrizanforov, M.N.; Strekalova, S.O.; Grinenko, V.V.; Khrizanforova, V.V.; Gryaznova, T.V.; Budnikova, Y.H. Various ways of C–P bonds formation via selective electrochemical phosphorylation of aromatic C–H bonds. Phosphorus Sulfur Silicon Relat. Elem. 2016, 191, 1491–1493. [Google Scholar] [CrossRef]
- Gryaznova, T.V.; Khrizanforov, M.N.; Strekalova, S.O.; Budnikova, Y.H.; Sinyashin, O.G. Electrochemical oxidative phosphonation of azoles. Phosphorus Sulfur Silicon Relat. Elem. 2016, 191, 1658–1659. [Google Scholar] [CrossRef]
- Strekalova, S.O.; Khrizanforov, M.N.; Shamsieva, A.V.; Grinenko, V.V.; Gryaznova, T.V.; Musina, E.I.; Karasik, A.A.; Budnikova, Y.H. Direct phosphorylation of pyridine in the presence of Ni(BF4)2bpy and CoCl2bpy metal complexes. Phosphorus Sulfur Silicon Relat. Elem. 2016, 191, 1545–1546. [Google Scholar] [CrossRef]
- Khrizanforova, V.V.; Khrizanforov, M.N.; Gryaznova, T.V.; Budnikova, Y.H. Electrochemical pathway to CH/PH functionalization of diphenylphosphine oxide. Phosphorus Sulfur Silicon Relat. Elem. 2016, 191, 1602–1603. [Google Scholar] [CrossRef]
- Khrizanforov, M.N.; Strekalova, S.O.; Kholin, K.V.; Khrizanforova, V.V.; Grinenko, V.V.; Gryaznova, T.V.; Budnikova, Y.H. One-stage Synthesis of FcP(O)(OC2H5)2 from Ferrocene and α-Hydroxyethylphosphonate. RSC Adv. 2016, 6, 42701–42707. [Google Scholar] [CrossRef]
- Dudkina, Y.B.; Mikhaylov, D.Y.; Gryaznova, T.V.; Sinyashin, O.G.; Vicic, D.A.; Budnikova, Y.H. MII/MIII-Catalyzed ortho-Fluoroalkylation of 2-Phenylpyridine. Eur. J. Org. Chem. 2012, 2114–2117. [Google Scholar] [CrossRef]
- Dudkina, Y.B.; Mikhaylov, D.Y.; Gryaznova, T.V.; Tufatullin, A.I.; Kataeva, O.N.; Vicic, D.A.; Budnikova, Y.H. Electrochemical Ortho Functionalization of 2-Phenylpyridine with Perfluorocarboxylic Acids Catalyzed by Palladium in Higher Oxidation States. Organometallics 2013, 32, 4785–4792. [Google Scholar] [CrossRef]
- Dudkina, Y.B.; Kholin, K.V.; Gryaznova, T.V.; Islamov, D.R.; Kataeva, O.N.; Rizvanov, I.K.; Levitskaya, A.I.; Fominykh, O.D.; Balakina, M.Y.; Sinyashin, O.G.; et al. Redox Trends in Cyclometalated Palladium(II) Complexes. Dalton Trans. 2017, 46, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Kalyani, D.; Dick, A.R.; Anani, W.Q.; Sanford, M.S. Scope and selectivity in palladium-catalyzed directed C–H bond halogenation reactions. Tetrahedron 2006, 62, 11483–11498. [Google Scholar] [CrossRef]
- Dick, A.R.; Hull, K.L.; Sanford, M.S. A Highly Selective Catalytic Method for the Oxidative Functionalization of C–H Bonds. J. Am. Chem. Soc. 2004, 126, 2300–2301. [Google Scholar] [CrossRef] [PubMed]
- Tsujihara, T.; Takenaka, K.; Onitsuka, K.; Hatanaka, M.; Sasai, H. PdII/PdIV Catalytic Enantioselective Synthesis of Bicyclo[3.1.0]hexanes via Oxidative Cyclization of Enynes. J. Am. Chem. Soc. 2009, 131, 3452–3453. [Google Scholar] [CrossRef] [PubMed]
- Lyons, T.W.; Sanford, M.S. Palladium(II/IV) catalyzed cyclopropanation reactions: Scope and mechanism. Tetrahedron 2009, 65, 3211–3221. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Yano, T.; Ishida, N.; Murakami, M. Pyridine-directed palladium-catalyzed phosphonation of C(sp2)–H bonds. Angew. Chem. Int. Ed. 2013, 52, 9801–9804. [Google Scholar] [CrossRef] [PubMed]
- Le Bras, J.; Muzart, J. Dehydrogenative (Hetero)arene Alkoxylations Triggered by PdII-Catalyzed C(sp2)–H Activation and Coordinating Substituent: PdII,III or PdIV Complex as Key Intermediate? Eur. J. Org. Chem. 2017, 3528–3548. [Google Scholar] [CrossRef]
- Hartwell, G.E.; Lawrence, R.V.; Smas, M.J. The formation of palladium(II)– and platinum(II)–carbon bonds by proton abstraction from benzo[h]quinoline and 8-Methylquinoline. J. Chem. Soc. Chem. Commun. 1970, 912. [Google Scholar] [CrossRef]
- Cope, A.C.; Siekman, R.W. Formation of Covalent Bonds from Platinum or Palladium to Carbon by Direct Substitution. J. Am. Chem. Soc. 1965, 87, 3272–3273. [Google Scholar] [CrossRef]
- Cope, A.C.; Friedrich, E.C. Electrophilic aromatttic substitution reactions by platinum(II) and palladium(II) chlorides on N,N-dimethylbenzylamines. J. Am. Chem. Soc. 1968, 90, 909–913. [Google Scholar] [CrossRef]
- Constable, A.G.; McDonald, W.G.; Sawkins, L.C.; Shaw, B.L. Palladation of dimethylhydrazones, oximes, and oxime O-allyl ethers: Crystal structure of [Pd3(ON=CPriPh)6]. J. Chem. Soc. Chem. Commun. 1978, 1061–1062. [Google Scholar] [CrossRef]
- Fuchita, Y.; Hiraki, K.; Uchiyama, T. Metallation of aliphatic carbon atoms. Part 1. Synthesis and characterization of the cyclopalladated complexes of 2-neopentylpyridine. J. Chem. Soc. Dalton Trans. 1983, 897–899. [Google Scholar] [CrossRef]
- Alsters, P.L.; Engel, P.F.; Hogerheide, M.P.; Copijn, M.; Spek, A.L.; van Koten, G. Rigid five- and six-membered C,N,N’-bound aryl-, benzyl-, and alkylorganopalladium complexes: sp2 vs. sp3 carbon-hydrogen activation during cyclopalladation and palladium(IV) intermediates in oxidative addition reactions with dihalogens and alkyl halides. Organometallics 1993, 12, 1831–1844. [Google Scholar] [CrossRef]
- Rouquet, G.; Chatani, N. Catalytic Functionalization of C(sp2)–H and C(sp3)–H Bonds by Using Bidentate Directing Groups. Angew. Chem. Int. Ed. 2013, 52, 11726–11743. [Google Scholar] [CrossRef] [PubMed]
- Zaitsev, V.G.; Shabashov, D.; Daugulis, O. Highly Regioselective Arylation of sp3 C−H Bonds Catalyzed by Palladium Acetate. J. Am. Chem. Soc. 2005, 127, 13154–13155. [Google Scholar] [CrossRef] [PubMed]
- Engle, K.M.; Mei, T.-S.; Wasa, M.; Yu, J.-Q. Weak Coordination as a Powerful Means for Developing Broadly Useful C–H Functionalization Reactions. Acc. Chem. Res. 2012, 45, 788–802. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.G.; Powers, D.C.; Raynaud, J.; Graham, M.J.; Xie, P.; Lee, E.; Ritter, T. Synthesis and structure of solution-stable one-dimensional palladium wires. Nat. Chem. 2011, 3, 949–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bercaw, J.E.; Durrell, A.C.; Gray, H.B.; Green, J.C.; Hazari, N.; Labinger, J.A.; Winkler, J.R. Electronic Structures of PdII Dimers. Inorg. Chem. 2010, 49, 1801–1810. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, B.N.; Adrio, L.A.; Albrecht, T.; White, A.J.P.; Newton, M.A.; Nachtegaal, M.; Figueroa, S.J.A.; Hii, K.K.M. Electronic structures of cyclometalated palladium complexes in the higher oxidation states. Dalton Trans. 2015, 44, 16586–16591. [Google Scholar] [CrossRef] [PubMed]
- Fabre, I.; von Wolff, N.; Le Duc, G.; Flegeau, E.F.; Bruneau, C.; Dixneuf, P.H.; Jutand, A. Autocatalytic Intermolecular versus Intramolecular Deprotonation in C–H Bond Activation of Functionalized Arenes by Ruthenium(II) or Palladium(II) Complexes. Chem. Eur. J. 2013, 19, 7595–7604. [Google Scholar] [CrossRef] [PubMed]
- Connelly, N.G.; Geiger, W.E. Chemical Redox Agents for Organometallic Chemistry. Chem. Rev. 1996, 96, 877–910. [Google Scholar] [CrossRef] [PubMed]
- Wacławek, S.; Grübel, K.; Cěrník, M. Simple spectrophotometric determination of monopersulfate. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 149, 928–933. [Google Scholar] [CrossRef] [PubMed]
- Block, P.A.; Brown, R.A.; Robinson, D. Novel Activation Technologies for Sodium Persulfate in Situ Chemical Oxidation. In Proceedings of the Fourth International Conference on Remediation of Chlorinated and Recalcitrant Compounds, Monterey, CA, USA, 24–27 May 2004. [Google Scholar]
- Haynes, W.M. CRC Handbook of Chemistry and Physics, 93rd ed.; CRC Press: Boca Raton, FL, USA, 2012. [Google Scholar]
- Wulfsberg, G. Inorganic Chemistry; University Science Books: Sausalito, CA, USA, 2000. [Google Scholar]
- Nayak, B.; Dash, U.N. The silver/silver acetate electrode Part I. Standard potential in formamide at 25 °C. J. Electroanal. Chem. 1973, 41, 323–328. [Google Scholar] [CrossRef]
- Felton, G.A.N.; Mebi, C.A.; Petro, B.J.; Vannucci, A.K.; Evans, D.H.; Glass, R.S.; Lichtenberger, D.L. Review of electrochemical studies of complexes containing the Fe2S2 core characteristic of [FeFe]-hydrogenases including catalysis by these complexes of the reduction of acids to form dihydrogen. J. Organomet. Chem. 2009, 694, 2681–2699. [Google Scholar] [CrossRef]
- Bour, J.R.; Camasso, N.M.; Meucci, E.A.; Kampf, J.W.; Canty, A.J.; Sanford, M.S. Carbon–carbon bond-forming reductive elimination from isolated Nickel(III) complexes. J. Am. Chem. Soc. 2016, 138, 16105–16111. [Google Scholar] [CrossRef] [PubMed]
- Wardman, P. Reduction Potentials of One-Electron Couples Involving Free Radicals in Aqueous Solution. J. Phys. Chem. Ref. Data 1989, 18, 1637–1755. [Google Scholar] [CrossRef]
- Prieto-Simón, B.; Fàbregas, E. Comparative study of electron mediators used in the electrochemical oxidation of NADH. Biosens. Bioelectron. 2004, 19, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Kitagawa, T. Solvent effects of 1,4-benzoquinone and its anion radicals probed by resonance Raman and absorption spectra and their correlation with redox potentials. J. Raman Spectrosc. 1998, 29, 773–780. [Google Scholar] [CrossRef]
- Jin, B.-K.; Li, L.; Huang, J.-L.; Zhang, S.-Y.; Tian, Y.-P.; Yang, J.-X. IR Spectroelectrochemical Cyclic Voltabsorptometry and Derivative Cyclic Voltabsorptometry. Anal. Chem. 2009, 81, 4476–4481. [Google Scholar] [CrossRef] [PubMed]
- Steckhan, E. Indirect Electroorganic Syntheses—A Modern Chapter of Organic Electrochemistry. Angew. Chem. Int. Ed. 1986, 28, 683–701. [Google Scholar] [CrossRef]
- Kokkinidis, G.; Papadopoulou, M.; Varvoglis, A. Electrochemical reduction of [bis(acyloxy)iodo]arenes. Electrochim. Acta 1989, 34, 133–139. [Google Scholar] [CrossRef]
- Song, C.; Zhang, J. Electrocatalytic Oxygen Reduction Reaction. In PEM Fuel Cell Electrocatalysts and Catalyst Layers. Fundamentals and Applications; Zhang, J., Ed.; Springer-Verlag: London, UK, 2008. [Google Scholar]
- Jeena, V.; Robinson, R.S. Convenient photooxidation of alcohols using dye sensitised zinc oxide in combination with silver nitrate and TEMPO. Chem. Commun. 2012, 48, 299–301. [Google Scholar] [CrossRef] [PubMed]
- Rychnosky, S.D.; Vaidyanathan, R.; Beauchamp, T.; Lin, R.; Farmer, P.J. AM1-SM2 Calculations Model the Redox Potential of Nitroxyl Radicals Such as TEMPO. J. Org. Chem. 1999, 64, 6745–6749. [Google Scholar] [CrossRef]
- Khrizanforov, M.N.; Arkhipova, D.M.; Shekurov, R.P.; Gerasimova, T.P.; Ermolaev, V.V.; Islamov, D.R.; Miluykov, V.A.; Kataeva, O.N.; Khrizanforova, V.V.; Sinyashin, O.G.; et al. Novel paste electrodes based on phosphonium salt room temperature ionic liquids for studying the redox properties of insoluble compounds. J. Sol. State Electrochem. 2015, 19, 2883–2890. [Google Scholar] [CrossRef]
- Favier, I.; Duñach, E. New protic salts of aprotic polar solvents. Tetrahedron Lett. 2004, 45, 3393–3395. [Google Scholar] [CrossRef]
- Amatore, C.; Jutand, A.; Lemaître, F.; Ricard, J.L.; Kozuch, S.; Shaik, S. Formation of anionic palladium(0) complexes ligated by the trifluoroacetate ion and their reactivity in oxidative addition. J. Organomet. Chem. 2004, 689, 3728–3734. [Google Scholar] [CrossRef]
- Amatore, C.; Jutand, A.; Khalil, F. Neutral palladium(0) complexes from Pd(OAc)2 and tri-2-furylphosphine and their reactivity in oxidative addition of iodobenzene. ARKIVOC 2006, 38–48. [Google Scholar] [CrossRef]
- Diculescu, V.C.; Chiorcea-Paquim, A.-M.; Corduneanu, O.; Oliveira-Brett, A.M. Palladium nanoparticles and nanowires deposited electrochemically: AFM and electrochemical characterization. J. Solid State Electrochem. 2007, 11, 887–898. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods. Fundamentals and Applications, 2nd ed.; John Wiley & Sons: New York, NY, USA, 2001; ISBN 978-0-471-04372-0. [Google Scholar]
- Stahl, S.S.; Alsters, P.L. Liquid Phase Aerobic Oxidation Catalysis: Industrial Applications and Academic Perspectives; Wiley-VCH: WeinHeim, Germany, 2016. [Google Scholar]
- Jutand, A. Contribution of Electrochemistry to Organometallic Catalysis. Chem. Rev. 2008, 108, 2300–2347. [Google Scholar] [CrossRef] [PubMed]
- Jutand, A.; Mosleh, A. Nickel- and Palladium-Catalyzed Homocoupling of Aryl Triflates. Scope, Limitation, and Mechanistic Aspects. J. Org. Chem. 1997, 62, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Lal, G.S.; Pez, G.P.; Syvret, R.G. Electrophilic NF Fluorinating Agents. Chem. Rev. 1996, 96, 1737–1755. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.D.; Kitamura, T. Alternative, Easy Preparation of (Diacetoxyiodo)arenes from Iodoarenes Using Potassium Peroxodisulfate as the Oxidant. Synthesis 2005, 12, 1932–1934. [Google Scholar] [CrossRef]
- Varvoglis, A. Aryliodine(III) dicarboxylates. Chem. Soc. Rev. 1981, 10, 377–407. [Google Scholar] [CrossRef]
- Reddy, B.V.S.; Revathi, G.; Reddy, A.S.; Yadav, J.S. Regioselective ortho-acetoxylation/methoxylation of N-(2-benzoylphenyl)benzamides via substrate directed C–H activation. Tetrahedron Lett. 2011, 52, 5926–5929. [Google Scholar] [CrossRef]
- Reddy, B.V.S.; Narasimhulu, G.; Umadevi, N.; Yadav, J.S. Quinazolinone-Directed C–H Activation: A Novel Strategy for the Acetoxylation–Methoxylation of the Arenes. Synlett 2012, 23, 1364–1370. [Google Scholar] [CrossRef]
- Reddy, B.V.S.; Umadevi, N.; Narasimhulu, G.; Yadav, J.S. Oxidative C–H functionalization: A novel strategy for the acetoxylation/alkoxylation of arenes tethered to 3,4-dihydroisoquinolines. Tetrahedron Lett. 2012, 53, 6091–6094. [Google Scholar] [CrossRef]
- Wang, G.-W.; Yuan, T.-T. Palladium-Catalyzed Alkoxylation of N-Methoxybenzamides via Direct sp2 C−H Bond Activation. J. Org. Chem. 2010, 75, 476–479. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Cui, X.; Yang, F.; Zhang, Q.; Zhu, Y.; Wu, Y. Palladium-catalyzed direct ortho C–O bond construction of azoxybenzenes with carboxylic acids and alcohols. Org. Chem. Front. 2015, 2, 951–955. [Google Scholar] [CrossRef]
- Piancatelli, G.; Leonelli, F. Oxidation of nerol to neral with iodobenzene diacetate and tempo. Org. Synth. 2006, 18–23. [Google Scholar] [CrossRef]
- Jonasson, C.; Horvath, A.; Backvall, J.E. Intramolecular Palladium(II)-Catalyzed 1,2-Addition to Allenes. J. Am. Chem. Soc. 2000, 122, 9600–9609. [Google Scholar] [CrossRef]
- Giri, R.; Lam, J.K.; Yu, J.-Q. Synthetic Applications of Pd(II)-Catalyzed C−H Carboxylation and Mechanistic Insights: Expedient Routes to Anthranilic Acids, Oxazolinones, and Quinazolinones. J. Am. Chem. Soc. 2010, 132, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Kuang, C. Palladium-Catalyzed Ortho-Alkoxylation of 2-Aryl-1,2,3-triazoles. J. Org. Chem. 2014, 79, 6105–6112. [Google Scholar] [CrossRef] [PubMed]
- Hull, K.L.; Lanni, E.L.; Sanford, M.S. Highly Regioselective Catalytic Oxidative Coupling Reactions: Synthetic and Mechanistic Investigations. J. Am. Chem. Soc. 2006, 128, 14047–14049. [Google Scholar] [CrossRef] [PubMed]
- Topczewski, J.J.; Sanford, M.S. Carbon–hydrogen (C–H) bond activation at PdIV: A Frontier in C–H functionalization catalysis. Chem. Sci. 2015, 6, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Han, J.; Chen, C.P.; Shi, D.Q.; Zhao, Y.S. Palladium-catalyzed oxygenation of C(sp2)–H and C(sp3)–H bonds under the assistance of oxalyl amide. RSC Adv. 2015, 5, 28430–28434. [Google Scholar] [CrossRef]
- Li, W.; Sun, P. Pd(OAc)2-Catalyzed Alkoxylation of Arylnitriles via sp2 C–H Bond Activation Using Cyano as the Directing Group. J. Org. Chem. 2012, 77, 8362–8366. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Sun, P. Palladium-Catalyzed Direct C(sp2)–H Alkoxylation of 2-Aryloxypyridines Using 2-Pyridyloxyl as the Directing Group. J. Org. Chem. 2014, 79, 8457–8461. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kim, M.; Park, J.; Kim, M.; Kwak, J.H.; Jung, Y.H.; Oh, J.S.; Lee, Y.; Kim, I.S. Palladium-Catalyzed Direct Acylation of Ketoximes and Aldoximes from the Alcohol Oxidation Level through C–H Bond Activation. Eur. J. Org. Chem. 2013, 6656–6665. [Google Scholar] [CrossRef]
- Gandeepan, P.; Cheng, C.-H. Allylic Carbon–Carbon Double Bond Directed Pd-Catalyzed Oxidative ortho-Olefination of Arenes. J. Am. Chem. Soc. 2012, 134, 5738–5741. [Google Scholar] [CrossRef] [PubMed]
- Engle, K.M.; Wang, D.-H.; Yu, J.-Q. Ligand-Accelerated C–H Activation Reactions: Evidence for a Switch of Mechanism. J. Am. Chem. Soc. 2010, 132, 14137–14151. [Google Scholar] [CrossRef] [PubMed]
- Baxter, R.D.; Sale, D.; Engle, K.M.; Yu, J.-Q.; Blackmond, D.G. Mechanistic Rationalization of Unusual Kinetics in Pd-Catalyzed C–H Olefination. J. Am. Chem. Soc. 2012, 134, 4600–4606. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-J.; Xu, H.; Kong, W.-J.; Shang, M.; Dai, H.-X.; Yu, J.-Q. Overcoming the limitations of directed C–H functionalizations of heterocycles. Nature 2014, 515, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Campbell, A.N.; Stahl, S.S. Overcoming the “Oxidant Problem”: Strategies to Use O2 as the Oxidant in Organometallic C–H Oxidation Reactions Catalyzed by Pd (and Cu). Acc. Chem. Res. 2012, 45, 851–863. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Jiang, H. Palladium-Catalyzed Oxidation of Unsaturated Hydrocarbons Using Molecular Oxygen. Acc. Chem. Res. 2012, 45, 1736–1748. [Google Scholar] [CrossRef] [PubMed]
- Gulzar, N.; Schweitzer-Chaput, B.; Klussmann, M. Oxidative coupling reactions for the functionalisation of C–H bonds using oxygen. Catal. Sci. Technol. 2014, 4, 2778–2796. [Google Scholar] [CrossRef]
- Baslé, O. Cross-Dehydrogenative-Coupling Reactions with Molecular Oxygen as the Terminal Oxidant. In From C–H to C–C Bonds: Cross-Dehydrogenative-Coupling; Li, C.-J., Ed.; Royal Society of Chemistry: Cambridge, UK, 2015; pp. 197–218. ISBN 978-1-84973-797-5. [Google Scholar]
- Wencel-Delord, J.; Colobert, F. A remarkable solvent effect of fluorinated alcohols on transition metal catalysed C–H functionalizations. Org. Chem. Front. 2016, 3, 394–400. [Google Scholar] [CrossRef]
- Dherbassy, Q.; Schwertz, G.; Chessé, M.; Hazra, C.K.; Wencel-Delord, J.; Colobert, F. 1,1,1,3,3,3-Hexafluoroisopropanol as a Remarkable Medium for Atroposelective Sulfoxide-Directed Fujiwara–Moritani Reaction with Acrylates and Styrenes. Chem. Eur. J. 2016, 22, 1735–1743. [Google Scholar] [CrossRef] [PubMed]
- Wesch, T.; Leroux, F.R.; Colobert, F. Atropodiastereoselective C–H Olefination of Biphenyl p-Tolyl Sulfoxides with Acrylates. Adv. Synth. Catal. 2013, 355, 2139–2144. [Google Scholar] [CrossRef]
- Villuendas, P.; Serrano, E.; Urriolabeitia, E.P. Pd-catalysed ortho-alkoxylation of benzamides N-protected with an iminophosphorane functionality. New J. Chem. 2015, 39, 3077–3083. [Google Scholar] [CrossRef]
- Yu, M.; Wang, Z.; Tian, M.; Lu, C.; Li, S.; Du, H. Purinyl N3-Directed Palladium-Catalyzed C–H Alkoxylation of N9-Arylpurines: A Late-Stage Strategy to Synthesize N9-(ortho-Alkoxyl)arylpurines. J. Org. Chem. 2016, 81, 3435–3442. [Google Scholar] [CrossRef] [PubMed]
- Dudkina, Y.B.; Khrizanforov, M.N.; Gryaznova, T.V.; Budnikova, Y.H. Prospects of Synthetic Electrochemistry in the Development of New Methods of Electrocatalytic Fluoroalkylation. J. Organomet. Chem. 2014, 751, 301–305. [Google Scholar] [CrossRef]
- Budnikova, Y.H. Metal complex catalysis in organic electrosynthesis. Russ. Chem. Rev. 2002, 71, 111–139. [Google Scholar] [CrossRef]
- Mikhaylov, D.Y.; Gryaznova, T.V.; Dudkina, Y.B.; Khrizanphorov, M.N.; Latypov, S.K.; Kataeva, O.N.; Vicic, D.A.; Sinyashin, O.G.; Budnikova, Y.G. Electrochemical nickel-induced fluoroalkylation: Synthetic, structural and mechanistic study. Dalton Trans. 2012, 41, 165–172. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strength | Oxidants | Reductants |
|---|---|---|
| Very strong | >0.8 | <−2.5 |
| Strong | 0.8–0.2 | −1.5 to −2.5 |
| Mild | 0.2 to −0.5 | −0.5 to −1.5 |
| Weak | <−0.5 | >−0.5 |
| Oxidant | Solvent | E° (V vs. Fc+/Fc) | Reference |
|---|---|---|---|
| Oxone | H2O | 1.98 | [46] |
| K2S2O8 | H2O | 1.48 | [47] |
| H2O | 1.39 | [48] | |
| H2O + [H+] | 1.50 | [48] | |
| CPE, solid | 1.58 | a | |
| [N(C6H2Br3-2,4,6)3]+ | CH3CN | 1.36 | [45] |
| Ag2+ | H2O | 1.36 | [49] |
| CPE, solid | 1.16 | a | |
| Mn(OAc)3 | CPE, solid | 1.32 | a |
| [N(C6H3Br2-2,4)3]+ | CH3CN | 1.14 | [45] |
| H2O2 | H2O | 1.18 | [47] |
| [NO]+ | CH2Cl2 | 1.00 | [45] |
| [Ru(phen)3]3+ | CH3CN | 0.87 | [45] |
| [NO]+ | CH3CN | 0.87 | [45] |
| [N(C6H4Br-4)3]+ | CH2Cl2 | 0.70 | [45] |
| CH3CN | 0.67 | [45] | |
| Ag+ | CH2Cl2 | 0.65 | [45] |
| THF | 0.41 | [45] | |
| acetone | 0.18 | [45] | |
| AgOAc | CPE, solid | 1.16 | a |
| AgOAc | formamide | −0.198 | [50] |
| AgNO3 | CH3CN | −0.08 | [51] |
| AgBF4 | CH3CN | −0.04 | [52] |
| Ag2O | CPE, solid | −1.26 | a |
| [Fe(η-C5H4COMe)2]+ | CH2Cl2 | 0.49 | [45] |
| [CuTf2] | CH3CN | 0.40 | [45] |
| Cu(OAc)2·H2O | CPE, solid | 0.33 | a |
| [Ni(tfd)2] | CH2Cl2 | 0.33 | [45] |
| [PtCl6]2− | H2O | 0.31 | [45] |
| Cl2 | CH3CN | 0.18 | [45] |
| DDQ | CH3CN | 0.13 | [45] |
| 1,4-BQ | H2O | 0.16 | [53,54] |
| H2O | −0.535 | [55] | |
| H2O | −0.526 | b | |
| CH3CN | −0.73 | [56] | |
| CH3CN | −0.86 | [55] | |
| CH3CN | −0.88 | c | |
| CH3CN + [H+] | d | ||
| CH2Cl2 | −0.805 | [55] | |
| Acetone | −0.875 | [55] | |
| Br2 | CH3CN | 0.07 | [45] |
| (FcBF4) [FeCp2]+ | 0 | [45] | |
| I2 | CH3CN | −0.14 | [45] |
| 0.0 | [57] | ||
| I+ | 0.33 | [56] | |
| TCNE | CH3CN | −0.27 | [45] |
| TCNQ | CH3CN | −0.30 | [45] |
| [FeCp*2]+ | CH3CN | −0.59 | [45] |
| CH2Cl2 | −0.48 | [45] | |
| PhI(OAc)2 | CH3CN | −1.293 | [43,58] |
| CH3CN | 1.70 | c | |
| CPE, solid | 1.70 | a | |
| O2 | H2O | −0.78 | [59] |
| H2O | −0.81 | b | |
| DMSO | −1.16 | [59] | |
| DMF | −1.24 | [59] | |
| Py | −1.24 | [59] | |
| MeCN | −1.25 | [59] | |
| Quinoline | −1.25 | [59] | |
| EMIBF4 | −1.23 | [59] | |
| PMIBF4 | −1.20 | [59] | |
| BMIBF4 | −1.24 | [59] | |
| [bmim]HFP | −1.26 | [59] | |
| TEMPO | CH2Cl2 | 0.014 | [60,61] |
| Palladium Complex | Solvent | E° (V vs. Fc+/Fc) | Reference |
|---|---|---|---|
| Pd2+/0 | DMF | −0.38 | [64] a |
| DMF | −0.02 | [65] b | |
| 0.1 M phosphate buffer | −0.64 | [66] | |
| −0.29 | [67] | ||
| H2O | −0.02 | [68] c | |
| PhPdIL | DMF | −0.88 reduction | [69] d |
| PdCl2(PPh3)2 | DMF | −1.29 reduction | [70] |
| Pd(OAc)2(TFP)2 | −1.34 reduction | [65] | |
| ArPdCl(PPh3) | DMF | >−2.03 reduction | [70] |
| [Pd(C^N)(OR)]2 | ACN | −2.03 to −2.37 reduction | [25] |
| 0.44–0.58 oxidation | [25] | ||
| DCM | −1.81 to −2.47 reduction | [25] | |
| 0.4 to 0.75 oxidation | [25] | ||
| [Pd(C^N)X]2 | ACN | −1.85 to −2.35 reduction | [25] |
| 0.63 to 1.00 oxidation | [25] | ||
| DCM | −2.29 to −2.43 reduction | [25] | |
| 0.73 to 0.74 oxidation | [25] | ||
| Pd(C^N)(CH3CN)ORF | ACN | −1.61 to −1.71 reduction | [25] |
| 1.19 to 1.32 oxidation | [25] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Budnikova, Y.H.; Dudkina, Y.B.; Khrizanforov, M.N. Redox-Induced Aromatic C–H Bond Functionalization in Metal Complex Catalysis from the Electrochemical Point of View. Inorganics 2017, 5, 70. https://doi.org/10.3390/inorganics5040070
Budnikova YH, Dudkina YB, Khrizanforov MN. Redox-Induced Aromatic C–H Bond Functionalization in Metal Complex Catalysis from the Electrochemical Point of View. Inorganics. 2017; 5(4):70. https://doi.org/10.3390/inorganics5040070
Chicago/Turabian StyleBudnikova, Yulia H., Yulia B. Dudkina, and Mikhail N. Khrizanforov. 2017. "Redox-Induced Aromatic C–H Bond Functionalization in Metal Complex Catalysis from the Electrochemical Point of View" Inorganics 5, no. 4: 70. https://doi.org/10.3390/inorganics5040070
APA StyleBudnikova, Y. H., Dudkina, Y. B., & Khrizanforov, M. N. (2017). Redox-Induced Aromatic C–H Bond Functionalization in Metal Complex Catalysis from the Electrochemical Point of View. Inorganics, 5(4), 70. https://doi.org/10.3390/inorganics5040070