Exploring Mechanisms in Ni Terpyridine Catalyzed C–C Cross-Coupling Reactions—A Review




Abstract
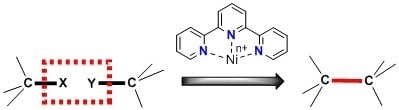
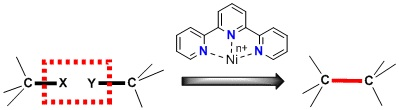
:
1. Introduction
2. Nickel–tpy Catalyzed C–C Cross-Coupling
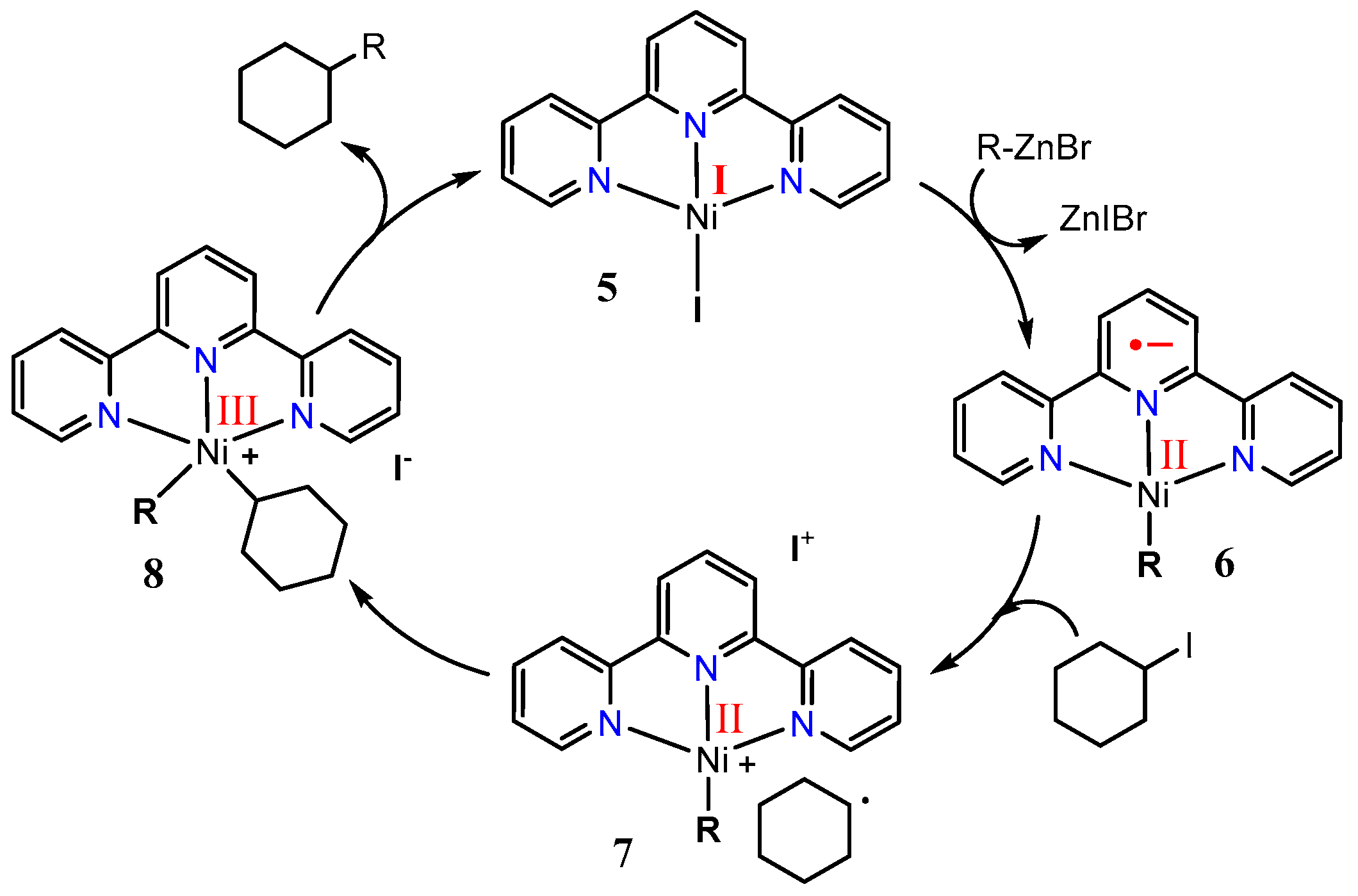
2.1. First Mechanistic Investigations on the [Ni(tpy)(Me)] Catalyst
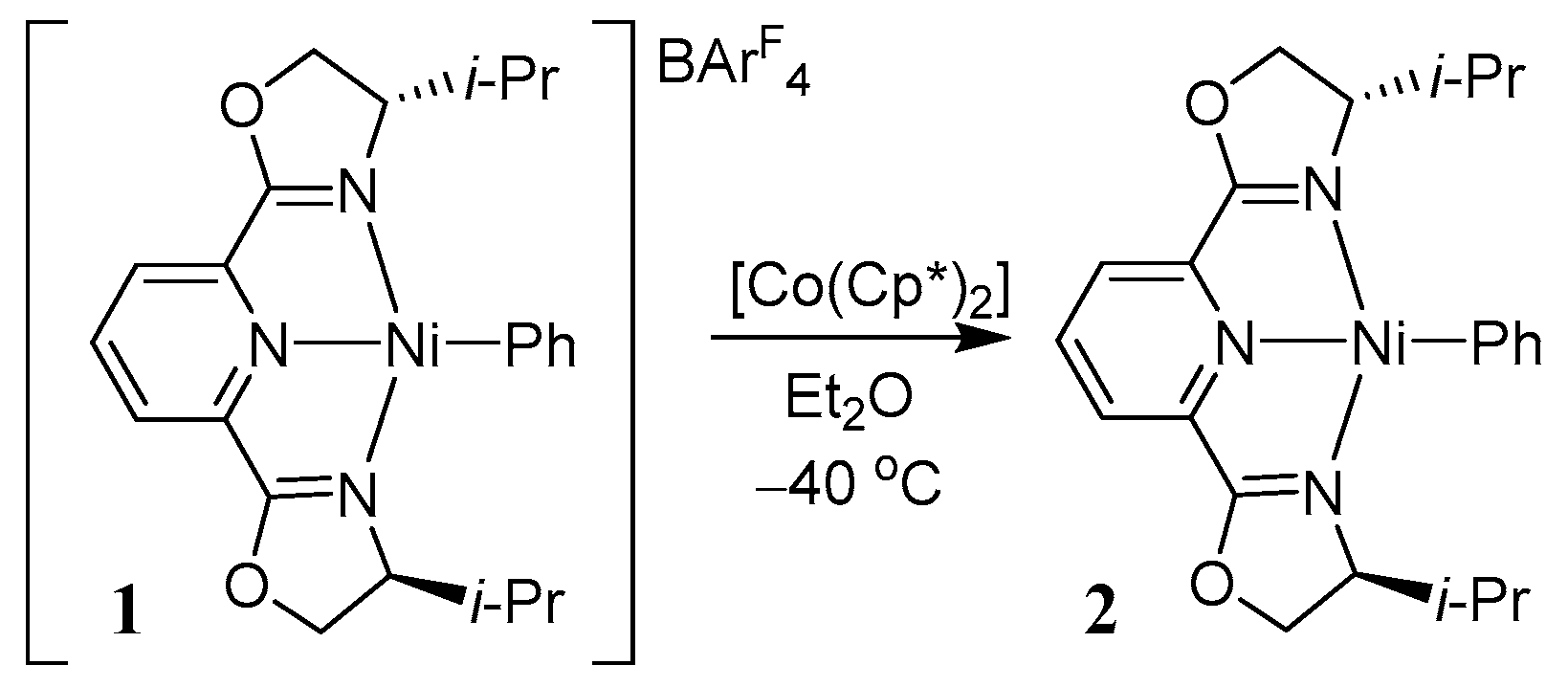
2.2. Investigations of the Low-Valent Systems [Ni(tpy)(aryl)], [Ni(tpy)I], [Ni(tpy)Br], and [Ni(tpy)Cl]
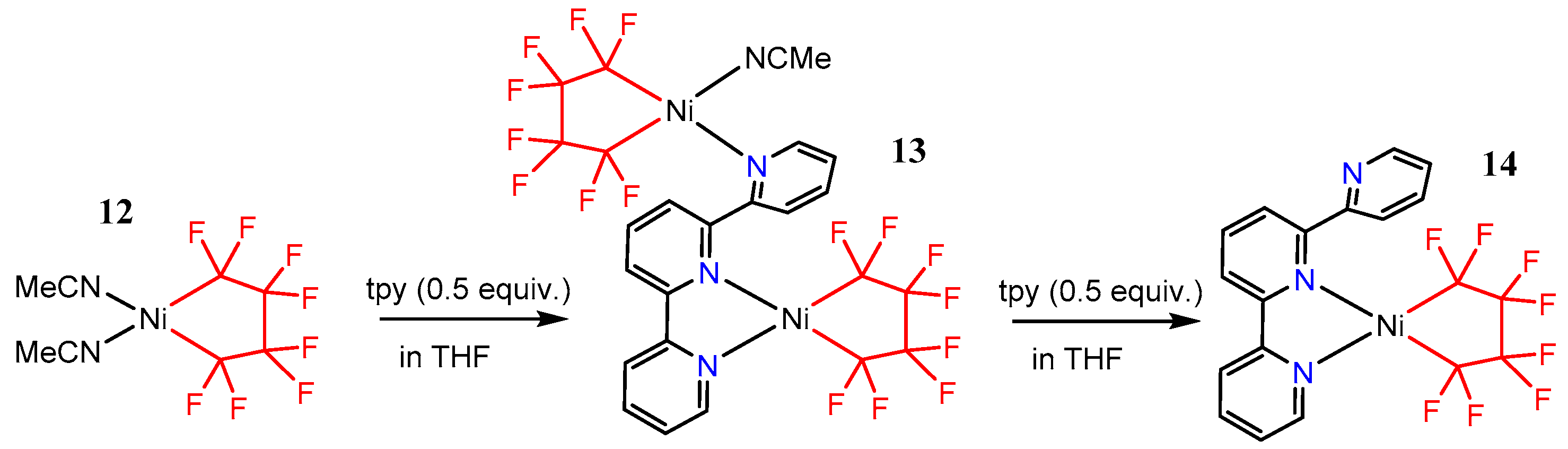
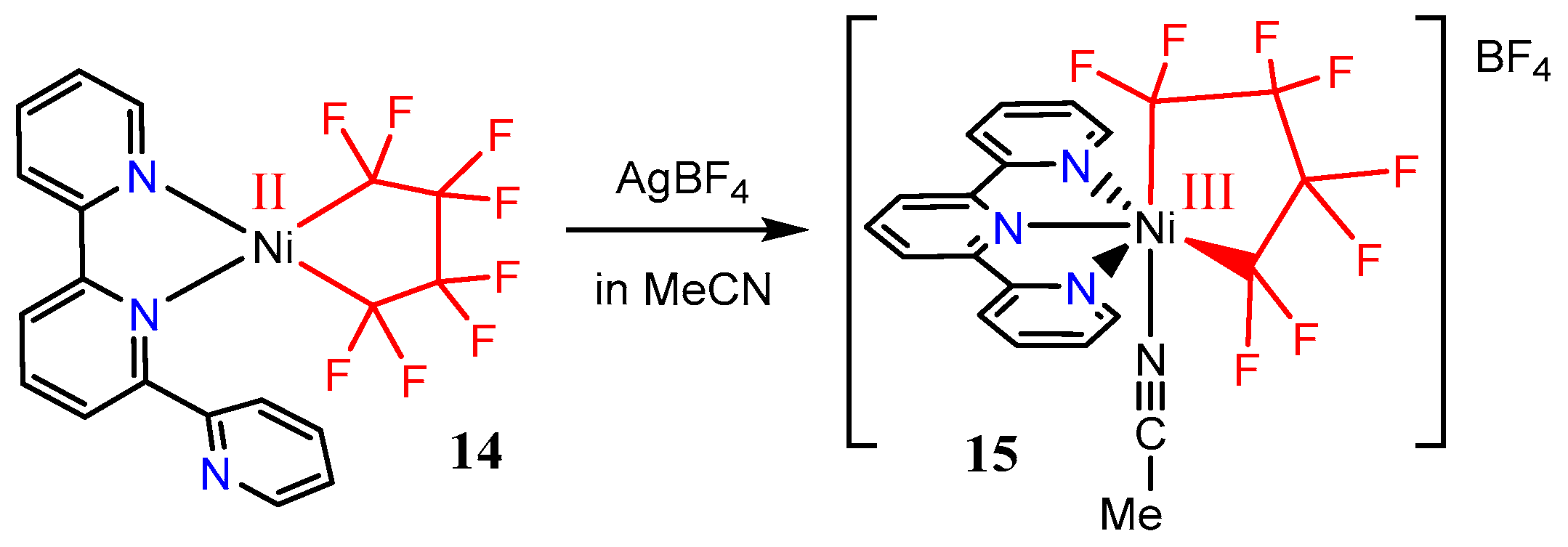
2.3. Studies on the Trivalent Species [Ni(tpy)(CnFm)2]+(CnFm = CF3 or C2F5)
2.4. Electrochemistry and Character of the Trivalent Species [Ni(tpy)(C4F8)]+
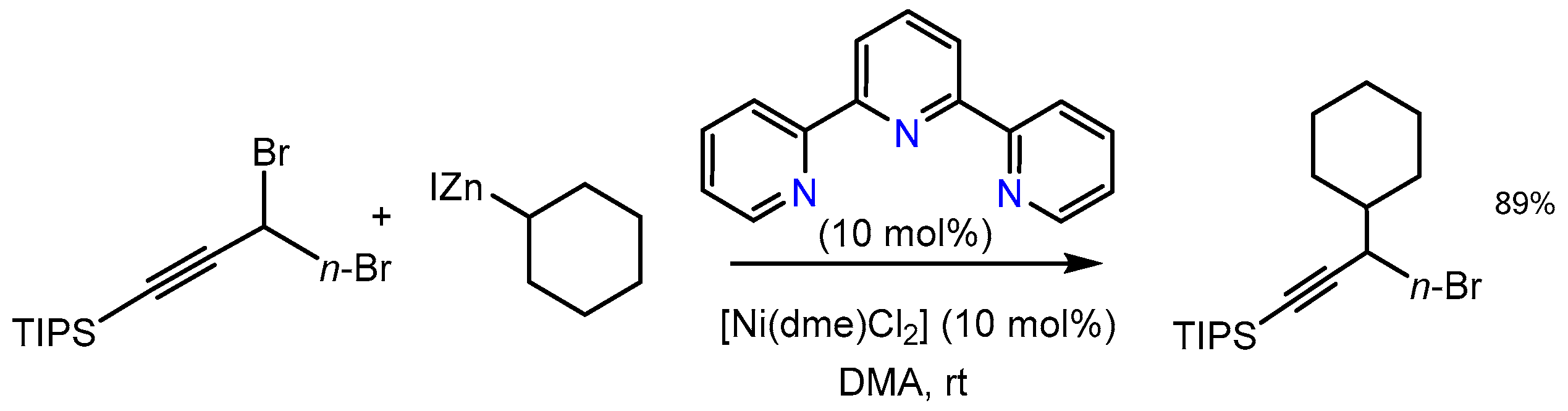
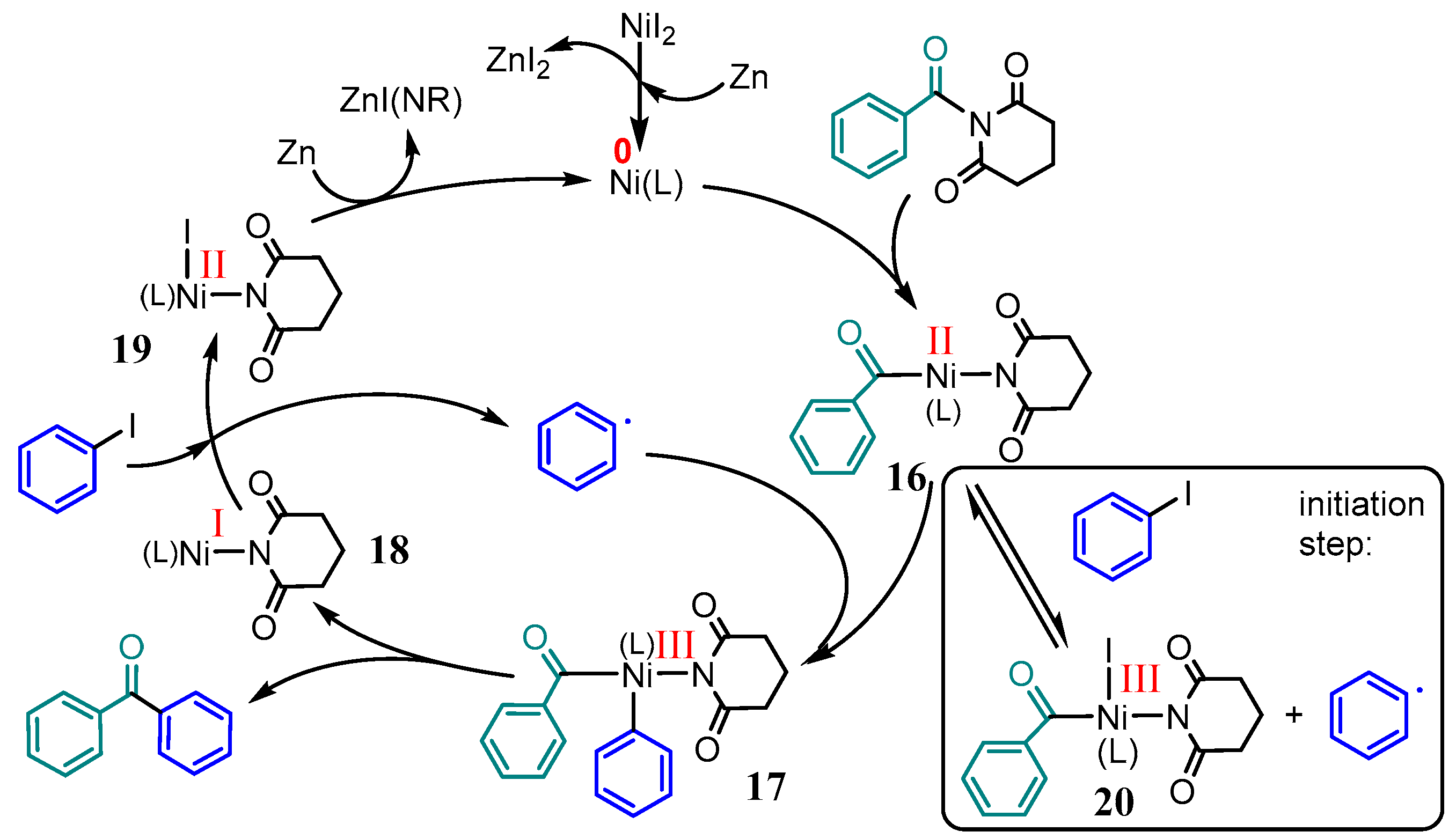
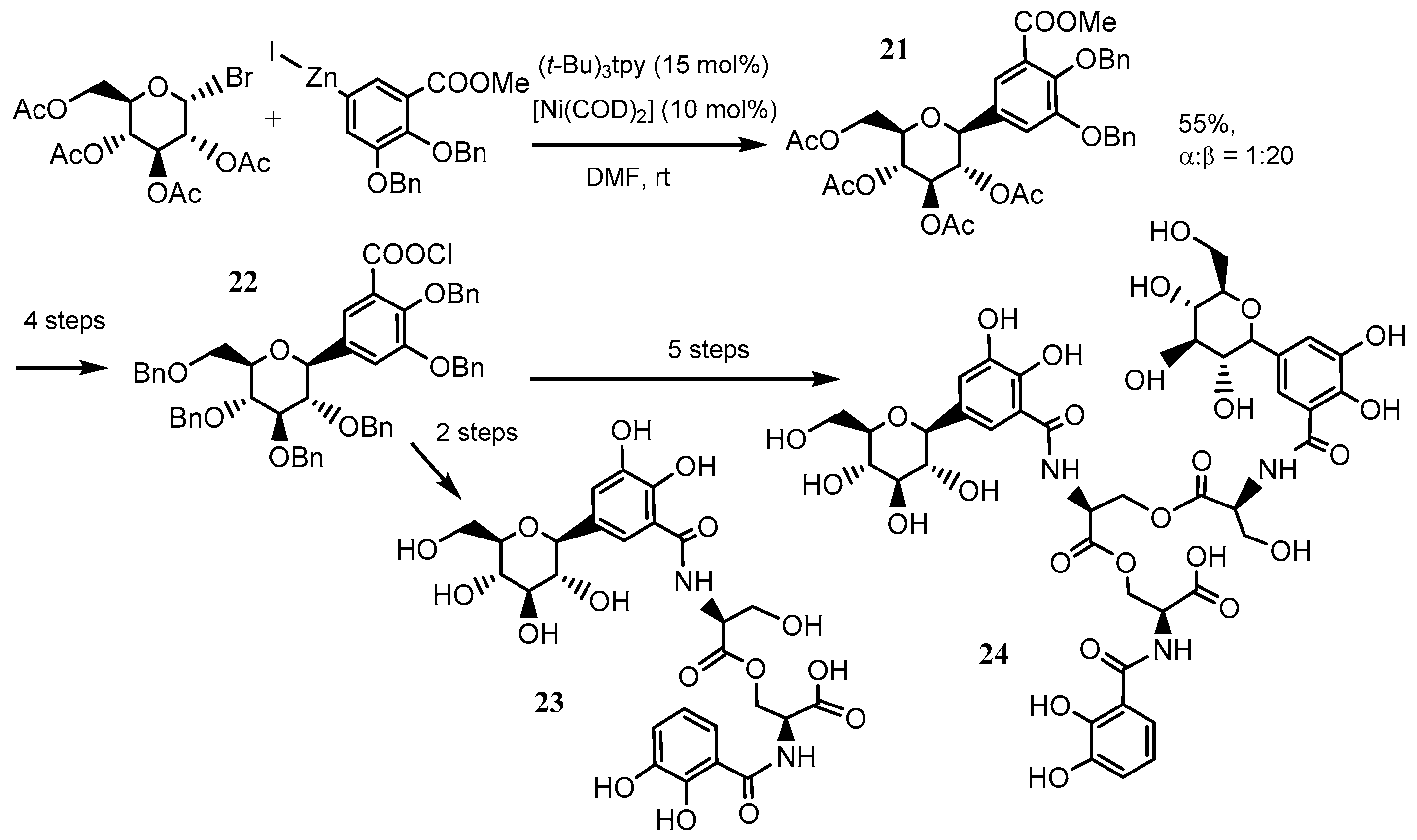
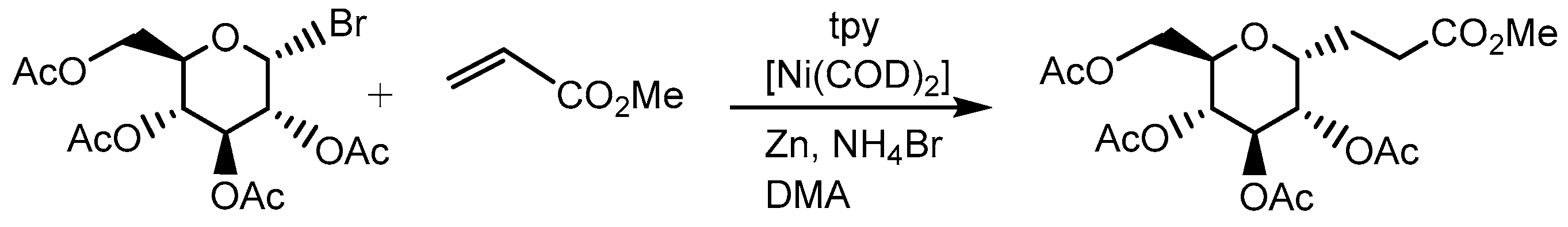
2.5. Further Ni–tpy Catalyzed C–C Cross-Coupling Reactions
3. Electrochemical Cross-Coupling Using Ni–tpy Systems
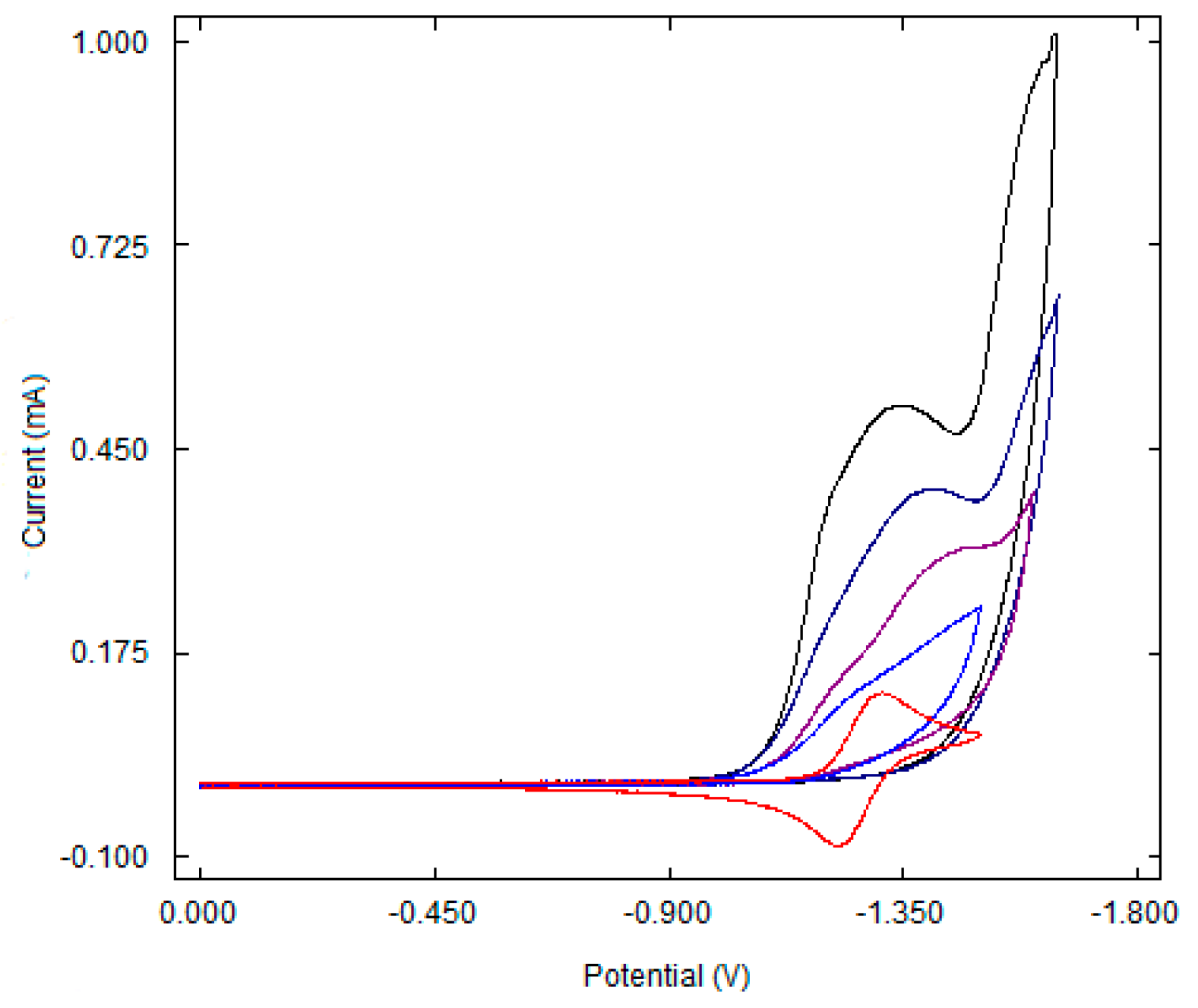
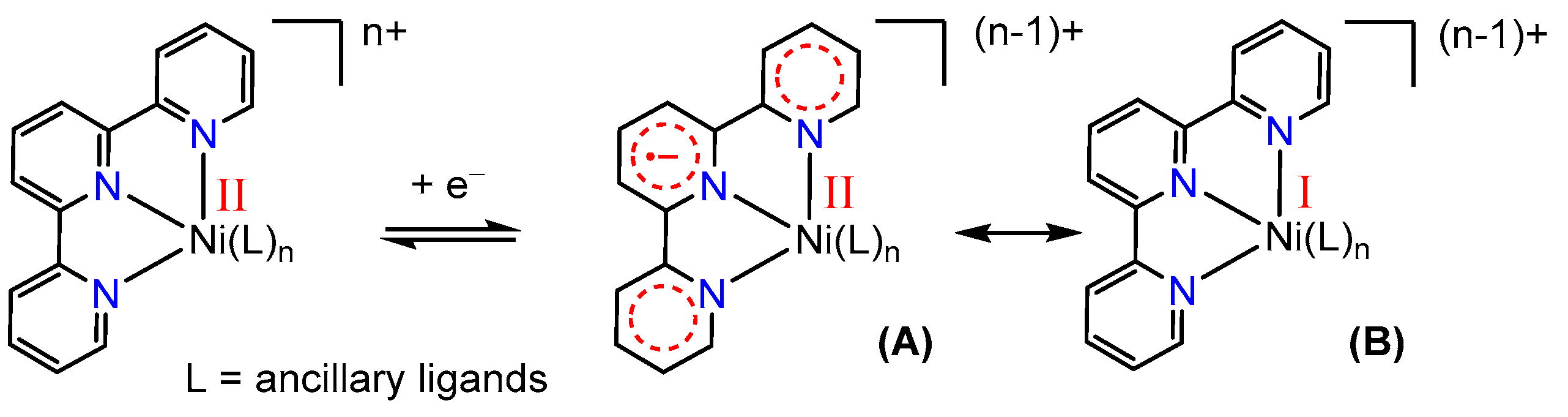
3.1. Electrochemical Properties of Ni–tpy Complexes
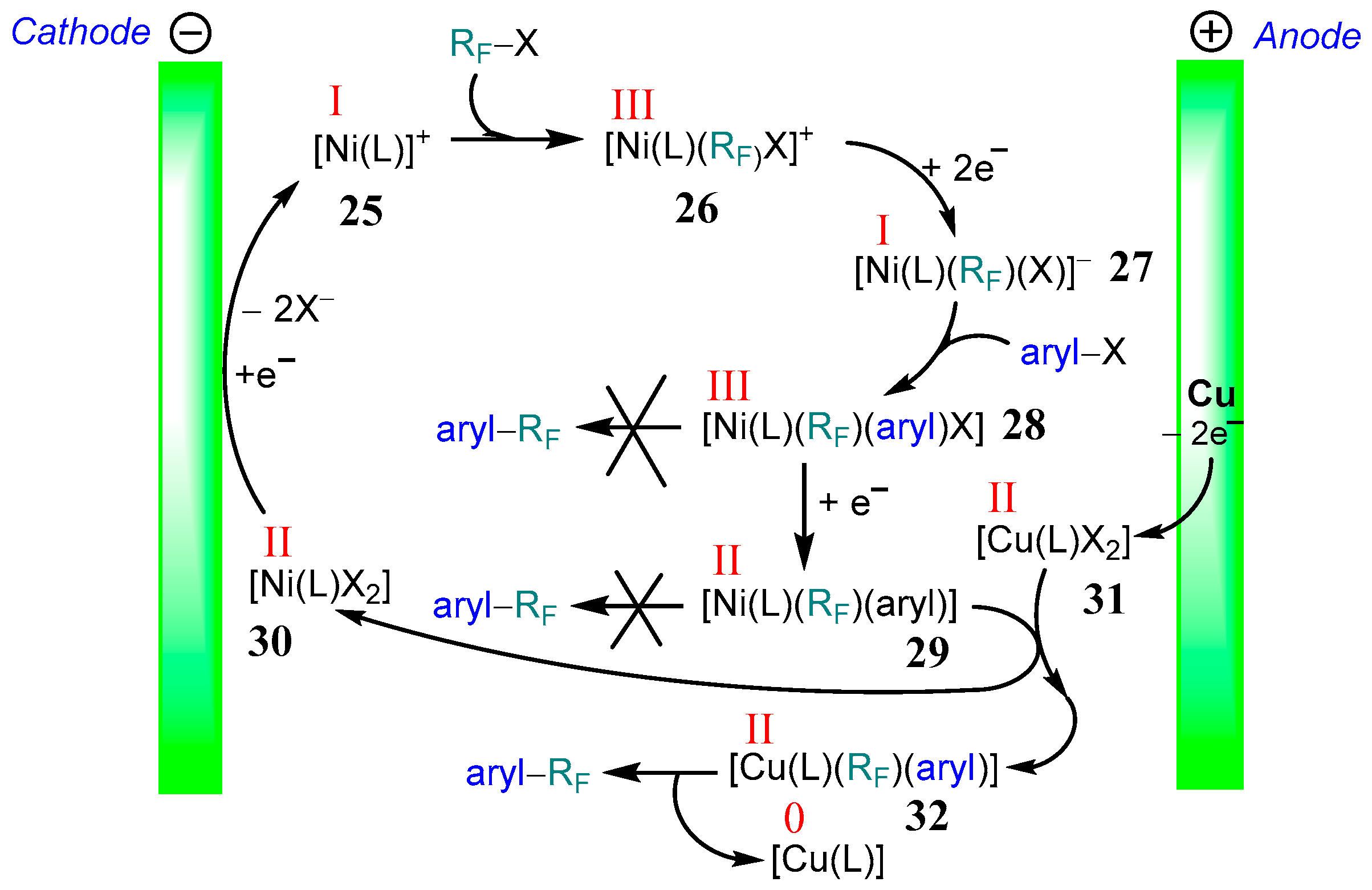
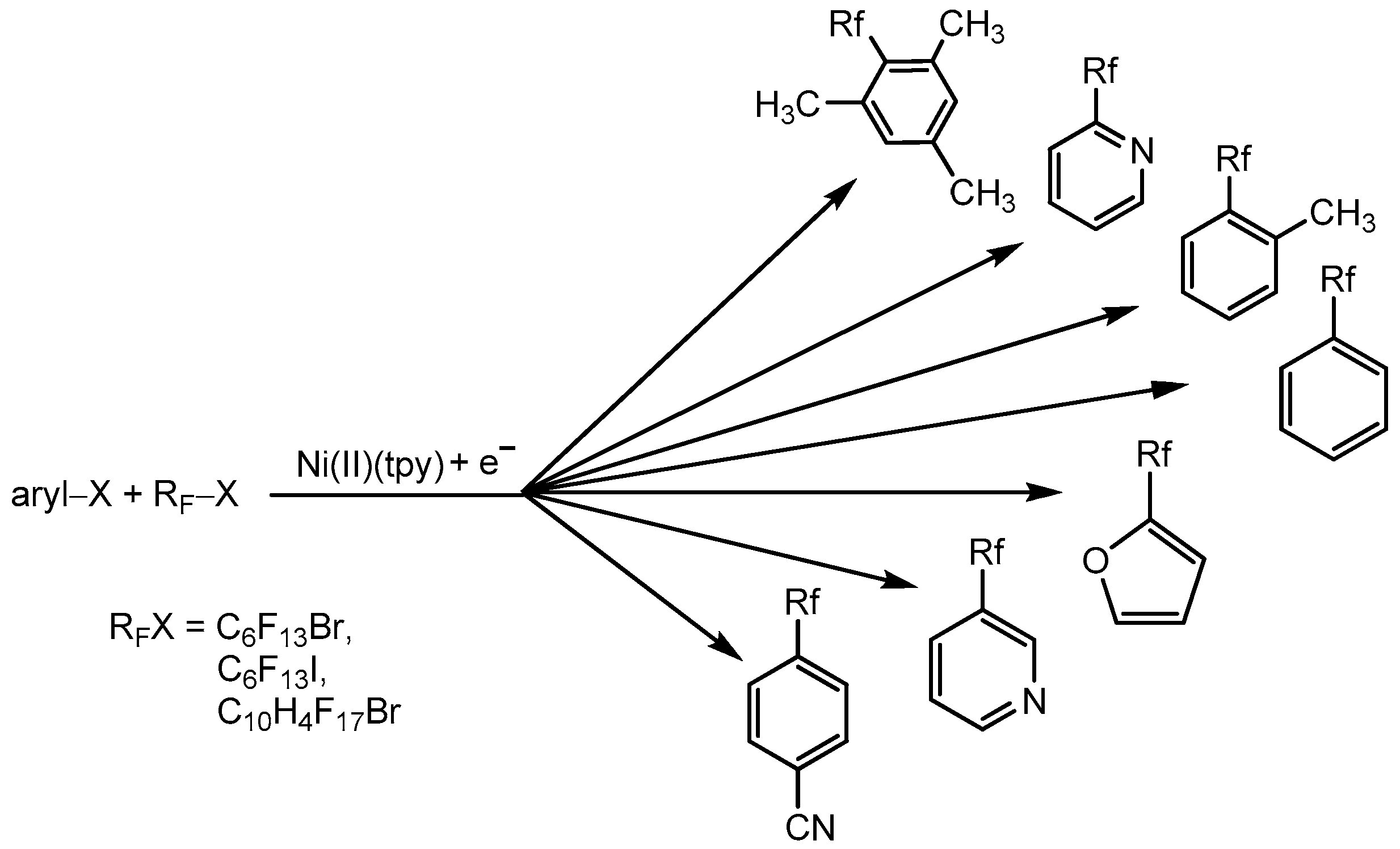
3.2. Aromatic Perfluoroalkylation Using Ni–tpy Under Electrocatalytic Conditions
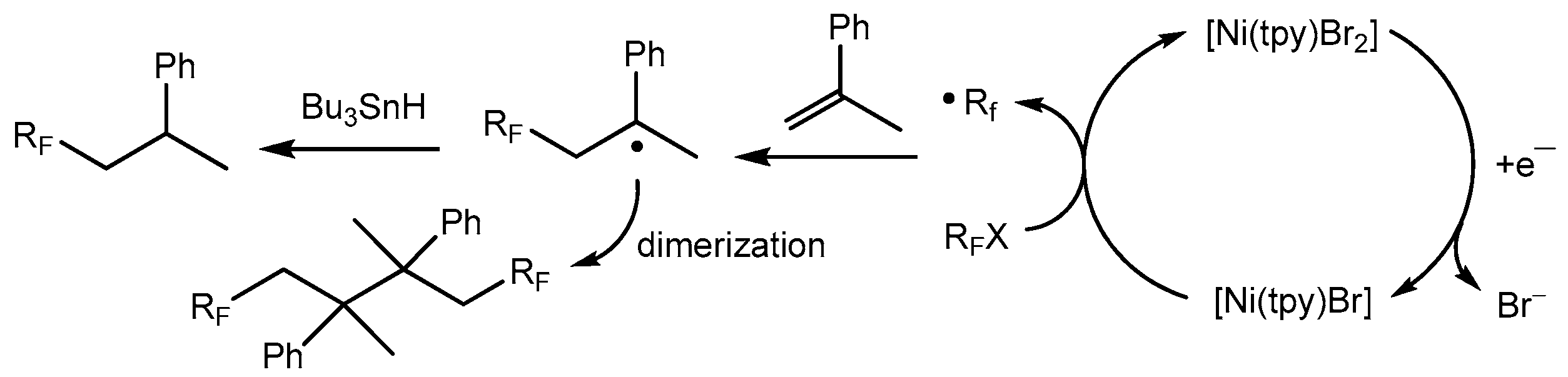
3.3. Electrochemical Nickel-Induced Fluoroalkylation of Olefins
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Winter, A.; Newkome, G.R.; Schubert, U.S. Catalytic Applications of Terpyridines and their Transition Metals. ChemCatChem 2011, 3, 1384–1406. [Google Scholar] [CrossRef]
- Kaes, C.; Katz, A.; Hosseini, M.W. Bipyridine: The Most Widely Used Ligand. A Review of Molecules Comprising at Least Two 2,2′-Bipyridine Units. Chem. Rev. 2000, 100, 3553–3590. [Google Scholar] [CrossRef] [PubMed]
- Constable, E.C. 2,2′:6′,2″-Terpyridines: From chemical obscurity to common supramolecular motifs. Chem. Soc. Rev. 2007, 36, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Morgan, G.; Burstall, F.H. Dehydrogenation of Pyridine by Anhydrous Ferric Chloride. J. Chem. Soc. 1932, 20–30. [Google Scholar] [CrossRef]
- Morgan, G.; Burstall, F.H. Researches on Residual Affinity and Co-ordination. Part XXX VII. Complex Metallic Salts containing 2:6-Di-2′-pyridylpyridine (2:2′:2″-Tripyridyl). J. Chem. Soc. 1937, 1649–1655. [Google Scholar] [CrossRef]
- Judge, J.S.; Reiff, W.M.; Intille, G.M.; Ballway, P.; Baker, W.A., Jr. Five-Coordinate Complexes of First Transition Series Ions with 2,2′,2″-Terpyridine. J. Inorg. Nucl. Chem. 1967, 29, 1711–1716. [Google Scholar] [CrossRef]
- Hogg, R.; Wilkins, R.G. Exchange Studies of Certain Chelate Compounds of the Transitional Metals. Part VIII. 2,2′,2″-Terpyridine Complexes. J. Chem. Soc. 1962, 341–350. [Google Scholar] [CrossRef]
- Gorczynski, A.; Walesa-Chorab, M.; Kubicki, M.; Korabik, M.; Patroniak, V. New complexes of 6,6″-dimethyl-2,2′:6′,2″-terpyridine with Ni(II) ions: Synthesis, structure and magnetic properties. Polyhedron 2014, 77, 17–23. [Google Scholar] [CrossRef]
- Constable, E.C.; Phillips, D.; Raithby, P.R. Nickel(II) chloride adducts of 4′-phenyl-2,2′:6′,2″-terpyridine. Inorg. Chem. Commun. 2002, 5, 519–521. [Google Scholar] [CrossRef]
- Baidya, N.; Olmstead, M.; Mascharak, P.K. Pentacoordinated Nickel(II) Complexes with Thiolato Ligation: Synthetic Strategy, Structures, and Properties. Inorg. Chem. 1991, 30, 929–937. [Google Scholar] [CrossRef]
- Arriortua, M.I.; Cortes, A.R.; Lezam, L.; Rojo, T.; Solans, X.; Font-Bardia, M. Crystal Structure and Magnetic Properties of [Ni(terpy)(N3)2]2·2H2O, a Nickel(II) Dinuclear Complex with Ferromagnetic Interaction. Inorg. Chim. Acta 1990, 174, 263–269. [Google Scholar] [CrossRef]
- Cortes, R.; Arriortua, M.I.; Rojo, T.; Solans, X.; Miravitelles, C.; Beltran, D. Structure of Diaquachloro(2,2′:6′,2″-terpyridyl)nickel(II) Chloride Monohydrate. Acta Crystallogr. C 1985, 41, 1733–1736. [Google Scholar] [CrossRef]
- Constable, E.C.; Lewis, J.; Liptrot, M.C.; Raithby, P.R. The coordination chemistry of 4′-phenyl-2,2′:6′,2″-terpyridine; the synthesis, crystal and molecular structures of 4′-phenyl-2,2′:6′,2″-terpyridine and bis(4′-phenyl-2,2′:6′,2″-terpyridine)nickel(II) chloride decahydrate. Inorg. Chim. Acta 1990, 178, 47–54. [Google Scholar] [CrossRef]
- Echevarren, A.M.; Homs, A. Mechanistic aspects of metal-catalyzed C,C- and C,X-bond forming reactions. In Metal-Catalyzed Cross-Coupling Reactions and More, 1st ed.; de Meijere, A., Bräse, S., Oestreich, M., Eds.; Wiley-VCH: Weinheim, Germany, 2014; pp. 1–64. ISBN 978-3-52-733154-3. [Google Scholar]
- Liu, C.; Zhang, H.; Shi, W.; Lei, A. Bond formations between two nucleophiles: Transition metal catalysed oxidative cross-coupling reactions. Chem. Rev. 2011, 111, 1780–1824. [Google Scholar] [CrossRef] [PubMed]
- Tasker, S.Z.; Standley, E.A.; Jamison, T.F. Recent advances in homogeneous nickel catalysis. Nature 2014, 509, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Weix, D.J. Mechanism and selectivity in nickel-catalyzed cross-electrophile coupling of aryl halides with alkyl halides. J. Am. Chem. Soc. 2013, 135, 16192–16197. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-X.; Liu, N. Nickel-catalyzed cross-coupling with pincer ligands. Eur. J. Inorg. Chem. 2012, 901–911. [Google Scholar] [CrossRef]
- Rosen, B.M.; Quasdorf, K.W.; Wilson, D.A.; Zhang, N.; Resmerita, A.-M.; Garg, N.K.; Percec, V. Nickel-catalyzed cross-couplings involving carbon–oxygen bonds. Chem. Rev. 2011, 111, 1346–1416. [Google Scholar] [CrossRef] [PubMed]
- Phapale, V.B.; Guisan-Ceinos, M.; Bunuel, E.; Cardenas, D.J. Nickel-catalyzed cross-coupling of alkyl zinc halides for the formation of C(sp2)–C(sp2) bonds: Scope and mechanism. Chem. Eur. J. 2009, 15, 12681–12688. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Z.; Ahneman, D.T.; Chu, L.; Terrett, J.A.; Doyle, A.G.; MacMillan, S.W.C. Merging photoredox with nickel catalysis: Coupling of α-carboxyl sp3-carbons with aryl halides. Science 2014, 345, 437–440. [Google Scholar] [CrossRef] [PubMed]
- Tellis, J.C.; Primer, D.N.; Molander, G.A. Single-electron transmetalation in organoboron cross-coupling by photoredox/nickel dual catalysis. Science 2014, 345, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Jones, G.C.; Ball, L.T. Self-control tames the coupling of reactive radicals. Science 2014, 345, 381–382. [Google Scholar] [CrossRef] [PubMed]
- Han, F.-S. Transition-metal-catalyzed Suzuki-Miyaura cross-coupling reactions: A remarkable advance from palladium to nickel catalysts. Chem. Soc. Rev. 2013, 42, 5270–5298. [Google Scholar] [CrossRef] [PubMed]
- Jana, R.; Pathak, T.P.; Sigman, M.S. Advances in transition metal (Pd,Ni,Fe)-catalyzed cross-coupling reactions using alkyl-organometallics as reaction partners. Chem. Rev. 2011, 111, 1417–1492. [Google Scholar] [CrossRef] [PubMed]
- Schley, N.D.; Fu, G.C. Nickel-catalyzed Negishi arylation of propargylic bromides: A mechanistic investigation. J. Am. Chem. Soc. 2014, 136, 16588–16593. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.S.; Haque, A.; Al-Suti, M.K.; Raithby, P.R. Recent advances in the application of group-transition metal based catalysts in C–H activation and functionalization. J. Organomet. Chem. 2015, 793, 114–133. [Google Scholar] [CrossRef] [Green Version]
- Johansson Seechurn, C.C.C.; Deangelis, A.; Colacot, T.J. Introduction to new trends in cross-coupling. In New Trends in Cross-Coupling: Theory and Applications; RSC Catalysis Series; Colacot, T.J., Ed.; Royal Society of Chemistry: Cambridge, UK, 2015; pp. 1–19. ISBN 978-1-84-973896-5. [Google Scholar]
- Colacot, T.J. The Nobel Prize in chemistry: Palladium-catalysed cross-coupling. The importance of carbon–carbon coupling for real world applications. Plat. Met. Rev. 2011, 55, 84–90. [Google Scholar] [CrossRef]
- Labinger, J.A. Tutorial on oxidative addition. Organometallics 2015, 34, 4784–4795. [Google Scholar] [CrossRef]
- Su, B.; Cao, Z.-C.; Shi, Z.-J. Exploration of earth-abundant transition metals (Fe, Co, and Ni) as catalysts in unreactive chemical bond activations. Acc. Chem. Res. 2015, 48, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Diccianni, J.B.; Katigbak, J.; Hu, C.; Zhang, Y.; Diao, T. Bimetallic C–C Bond-Forming Reductive Elimination from Nickel. J. Am. Chem. Soc. 2016, 138, 4779–4786. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, P.T.; Xu, L.; Chen, B.; McGarry, K.R.; Yu, S.; Wang, H.; Vicic, D.A. Mild, safe, and versatile reagents for (CF2)n transfer and the construction of fluoroalkyl-containing rings. Organometallics 2013, 32, 7552–7558. [Google Scholar] [CrossRef]
- Lin, S.; Day, M.W.; Agapie, T. Nickel hydrides supported by a non-innocent diphosphine arene pincer: Mechanistic studies of nickel–arene H-migration and partial arene hydrogenation. J. Am. Chem. Soc. 2011, 133, 3828–3831. [Google Scholar] [CrossRef] [PubMed]
- Uyeda, C.; Steiman, T.J.; Pal, S. Catalytically active nickel–nickel bonds using redox-active ligands. Synlett 2016, 27, 814–820. [Google Scholar] [CrossRef]
- Kaim, W. The Shrinking World of Innocent Ligands: Conventional and Non-Conventional Redox-Active Ligands. Eur. J. Inorg. Chem. 2012, 343–348. [Google Scholar] [CrossRef]
- Lyaskovsky, V.; de Bruin, B. Redox Non-Innocent Ligands: Versatile New Tools to Control Catalytic Reactions. ACS Catal. 2012, 2, 270–279. [Google Scholar] [CrossRef]
- Chaudhuri, P.; Verani, C.N.; Bill, E.; Bothe, E.; Weyhermüller, T.; Wieghardt, K. Electronic structure of bis(o-iminobenzosemiquinonato)metal complexes (Cu, Ni, Pd). The art of establishing physical oxidation states in transition-metal complexes containing radical ligands. J. Am. Chem. Soc. 2001, 123, 2213–2223. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, C.K. Differences between the four halide ligands, and discussion remarks on trigonal-bipyramidal complexes, on oxidation states, and on diagonal elements of one-electron energy. Coord. Chem. Rev. 1966, 1, 164–187. [Google Scholar] [CrossRef]
- Arana, C.; Yan, S.; Keshavarz, M.; Potts, K.T.; Abruna, H.D. Electrocatalytic Reduction of Carbon Dioxide with Iron, Cobalt, and Nickel Complexes of Terdentate Ligands. Inorg. Chem. 1992, 31, 3680–3682. [Google Scholar] [CrossRef]
- Wang, M.; England, J.; Weyhermüller, T.; Wieghardt, K. Electronic Structures of “Low-Valent” Neutral Complexes [NiL2](S = 0; L = bpy, phen, tpy)—An Experimental and DFT Computational Study. Eur. J. Inorg. Chem. 2015, 1511–1523. [Google Scholar] [CrossRef]
- Choi, J.; Fu, G.C. Transition metal-catalyzed alkyl–alkyl bond formation: Another dimension in cross-coupling chemistry. Science 2017, 356, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Fu, G.C. Nickel-Catalyzed Alkyl–Alkyl Cross-Couplings of Fluorinated Secondary Electropphiles: A General Approach to the Synthesis of Compounds having a Perfluoroalkyl Substituent. Angew. Chem. Int. Ed. 2015, 54, 9047–9051. [Google Scholar] [CrossRef] [PubMed]
- Anderson, T.J.; Jones, G.D.; Vicic, D.A. Evidence for a NiI Active Species in the Catalytic Cross-Coupling of Alkyl Electrophiles. J. Am. Chem. Soc. 2004, 126, 8100–8101. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.D.; McFarland, C.; Anderson, T.J.; Vicic, D.A. Analysis of key steps in the catalytic cross-coupling of alkyl electrophiles under Negishi-like conditions. Chem. Commun. 2005, 4211–4213. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.D.; Martin, J.L.; McFarland, C.; Allen, O.R.; Hall, R.E.; Haley, A.D.; Brandon, R.J.; Konovalova, T.; Desrochers, P.J.; Pulay, P.; et al. Ligand Redox Effects in the Synthesis, Electronic Structure, and Reactivity of an Alkyl–Alkyl Cross-Coupling Catalyst. J. Am. Chem. Soc. 2006, 128, 13175–13183. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Phillips, D.L. Density Functional Theory Studies of Negishi Alkyl–Alkyl Cross-Coupling Reactions Catalyzed by a Methylterpyridyl-Ni(I) Complex. J. Org. Chem. 2008, 73, 3680–3688. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.W.; Fu, G.C. Nickel-Catalyzed Negishi Cross-Couplings of Secondary Nucleophiles with Secondary Propargylic Electrophiles at Room Temperature. Angew. Chem. Int. Ed. 2008, 47, 9334–9336. [Google Scholar] [CrossRef] [PubMed]
- Breitenfeld, J.; Ruiz, J.; Wodrich, M.D.; Hu, X. Bimetallic Oxidative Addition Involving Radical Intermediates in Nickel-Catalyzed Alkyl—Alkyl Kumada Coupling Reactions. J. Am. Chem. Soc. 2013, 135, 12004–12012. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.; Budnikova, Y.H.; Sinyashin, O.G. Electron transfer in organonickel complexes of α-diimines: Versatile redox catalysts for C–C or C–P coupling reactions—A review. J. Organomet. Chem. 2007, 692, 3156–3166. [Google Scholar] [CrossRef]
- Hamacher, C.; Hurkes, N.; Kaiser, A.; Klein, A.; Schüren, A. Electrochemistry and Spectroscopy of Organometallic Terpyridine Nickel Complexes. Inorg. Chem. 2009, 48, 9947–9951. [Google Scholar] [CrossRef] [PubMed]
- Ciszewski, J.T.; Mikhaylov, D.Y.; Holin, K.V.; Kadirov, M.K.; Budnikova, Y.H.; Sinyashin, O.; Vicic, D.A. Redox Trends in Terpyridine Nickel Complexes. Inorg. Chem. 2011, 50, 8630–8635. [Google Scholar] [CrossRef] [PubMed]
- Mikhaylov, D.; Gryaznova, T.; Dudkina, Y.; Khrizanphorov, M.; Latypov, S.; Kataeva, O.; Vicic, D.A.; Sinyashin, O.G.; Budnikova, Y. Electrochemical nickel-induced fluoroalkylation: Synthetic, structural and mechanistic study. Dalton Trans. 2012, 41, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-P.; Wang, H.; Klein, A.; Biewer, C.; Stirnat, K.; Yamaguchi, Y.; Xu, L.; Gomez-Benitez, V.; Vicic, D.A. A Five-Coordinate Nickel(II) Fluoroalkyl Complex as a Precursor to a Spectroscopically Detectable Ni(III) Species. J. Am. Chem. Soc. 2013, 135, 8141–8144. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Dudkina, Y.; Wang, H.; Kholin, K.V.; Kadirov, M.K.; Budnikova, Y.H.; Vicic, D.A. Accessing perfluoroalkyl nickel(II), (III), and (IV) complexes bearing a readily attached [C4F8] ligand. Dalton Trans. 2015, 44, 19443–19446. [Google Scholar] [CrossRef] [PubMed]
- Coogan, M.P.; Fernandez-Moreira, V.; Kariuki, B.M.; Pope, S.J.A.; Thorp-Greenwood, F.L. A Rhenium Tricarbonyl 4′-Oxo-terpy Trimer as a Luminescent Molecular Vessel with a Removable Silver Stopper. Angew. Chem. Int. Ed. 2009, 48, 4965–4968. [Google Scholar] [CrossRef] [PubMed]
- Effendy; Marchetti, F.; Pettinari, C.; Pettinari, R.; Skelton, B.W.; White, A.H. Synthesis and structural characterization of adducts of silver(I) oxyanion salts, AgX (X = ClO4, NO3), with Ph2E(CH2)xEPh(‘dpex’; E = P, As; x = 1–3) and oligodentate aromatic N-bases derivative of 2,2′-bipyridyl, ‘L’, AgX:dpex:L (2:1:1) or (1:1:1). Inorg. Chim. Acta 2007, 360, 1414–1423. [Google Scholar] [CrossRef]
- Feng, H.; Zhou, X.-P.; Wu, T.; Li, D.; Yin, Y.-G.; Ng, S.W. Hydrothermal synthesis of copper complexes of 4′-pyridyl terpyridine: From discrete monomer to zigzag chain polymer. Inorg. Chim. Acta 2006, 359, 4027–4035. [Google Scholar] [CrossRef]
- Hannon, M.J.; Painting, C.L.; Plummer, E.A.; Childs, L.J.; Alcock, N.W. Competing Supramolecular Interactions Give a New Twist to Terpyridyl Chemistry: Anion- and Solvent-Induced Formation of Spiral Arrays in Silver(I) Complexes of a Simple Terpyridine. Chem. Eur. J. 2002, 8, 2225–2238. [Google Scholar] [CrossRef]
- Hou, L.; Li, D. A novel photoluminescent Ag–terpyridyl complex: One-dimensional linear metal string with double-helical structure. Inorg. Chem. Commun. 2005, 8, 128–130. [Google Scholar] [CrossRef]
- Ma, Z.; Xing, Y.; Yang, M.; Hu, M.; Liu, B.; da Silva, M.F.C.G.; Pombeiro, A.J.L. The double-helicate terpyridine silver(I) compound [Ag2L2](SO3CF3)(L = 4′-phenyl-terpyridine) as a building block for di- and mononuclear complexes. Inorg. Chim. Acta 2009, 362, 2921–2926. [Google Scholar] [CrossRef]
- Ni, S.; Zhang, W.; Mei, H.; Han, J.; Pan, Y. Ni-Catalyzed Reductive Cross-Coupling of Amides with Aryl Iodide Electrophiles via C–N Bond Activation. Org. Lett. 2017, 19, 2536–2539. [Google Scholar] [CrossRef] [PubMed]
- Joshi-Pangu, A.; Ganesh, M.; Biscoe, M.R. Nickel-Catalyzed Negishi Cross-Coupling Reactions of Secondary Alkylzinc Halides and Aryl Iodides. Org. Lett. 2011, 13, 1218–1221. [Google Scholar] [CrossRef] [PubMed]
- Prinsell, M.R.; Everson, D.A.; Weix, D.J. Nickel-catalyzed, sodium iodide-promoted reductive dimerization of alkyl halides, alkylpseudohalides, and allylic acetates. Chem. Commun. 2010, 46, 5743–5745. [Google Scholar] [CrossRef] [PubMed]
- Goldup, S.M.; Leigh, D.A.; McBurney, R.T.; McGonigal, P.R.; Plant, A. Ligand-assisted nickel-catalysed sp3–sphomocoupling of unactivated alkyl bromides and its application to the active template synthesis of rotaxanes. Chem. Sci. 2010, 1, 383–386. [Google Scholar] [CrossRef]
- Paul, A.; Smith, M.D.; Vannucci, A.K. Photoredox-Assisted Reductive Cross-Coupling: Mechanistic Insight into Catalytic Aryl–Alkyl Cross-Couplings. J. Org. Chem. 2017, 82, 1996–2003. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.; Hirano, K.; Satoh, T.; Miura, M. Palladium- and Nickel-Catalyzed Direct Alkylation of Azoles with Unactivated Alkyl Bromides and Chlorides. Chem. Eur. J. 2010, 16, 12307–12311. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.; Gagné, M.R. Diastereoselective Ni-Catalyzed Negishi Cross-Coupling Approach to Saturated, Fully Oxygenated C–Alkyl and C–Aryl Glycosides. J. Am. Chem. Soc. 2008, 130, 12177–12183. [Google Scholar] [CrossRef]
- Yu, X.; Dai, Y.; Yang, T.; Gagné, M.R.; Gong, H. Facile synthesis of salmochelin S1, S2, MGE, DGE, and TGE. Tetrahedron 2011, 67, 144–151. [Google Scholar] [CrossRef]
- Gong, H.; Andrews, R.S.; Zuccarello, J.L.; Lee, S.J.; Gagné, M.R. Sn-Free Ni-Catalyzed Reductive Coupling of Glycosyl Bromides with Activated Alkenes. Org. Lett. 2009, 11, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Khrizanforov, M.; Khrizanforova, V.; Mamedov, V.; Zhukova, N.; Strekalova, S.; Grinenko, V.; Gryaznova, T.; Sinyashin, O.; Budnikova, Y. Single-stage synthetic route to perfluoroalkylated arenes via electrocatalytic cross-coupling of organic halides using Co and Ni complexes. J. Organomet. Chem. 2016, 820, 82–88. [Google Scholar] [CrossRef]
- Khrizanforov, M.; Gryaznova, T.; Sinyashin, O.; Budnikova, Y. Aromatic perfluoroalkylation with metal complexes in electrocatalytic conditions. J. Organomet. Chem. 2012, 718, 101–104. [Google Scholar] [CrossRef]
- Sengmany, S.; Vasseur, S.; Lajnef, A.; Le Gall, E.; Léonel, E. Beneficial Effects of Electrochemistry in Cross-Coupling Reactions: Electroreductive Synthesis of 4-Aryl- or 4-Heteroaryl-6-pyrrolylpyrimidines. Eur. J. Org. Chem. 2016, 4865–4871. [Google Scholar] [CrossRef]
- Sengmany, S.; Vitu-Thiebaud, A.; Le Gall, E.; Condon, S.; Leonel, E.; Thobie-Gautier, C.; Pipelier, M.; Lebreton, J.; Dubreuil, D. An Electrochemical Nickel-Catalyzed Arylation of 3-Amino-6-Chloropyridazines. J. Org. Chem. 2013, 78, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Sengmany, S.; Le Gall, E.; Leonel, E. An Electrochemical Synthesis of Functionalized Arylpyrimidines from 4-Amino-6-Chloropyrimidines and Aryl Halides. Molecules 2011, 16, 5550–5560. [Google Scholar] [CrossRef] [PubMed]
- Sengmany, S.; Leonel, E.; Polissaint, F.; Nedelec, J.Y.; Pipelier, M.; Thobie-Gautier, C.; Dubreuil, D. Preparation of Functionalized Aryl- and Heteroarylpyridazines by Nickel-Catalyzed Electrochemical Cross-Coupling Reactions. J. Org. Chem. 2007, 72, 5631–5636. [Google Scholar] [CrossRef] [PubMed]
- Mikhaylov, D.Y.; Budnikova, Y.H. Fluoroalkylation of organic compounds. Russ. Chem. Rev. 2013, 835–864. [Google Scholar] [CrossRef]
- Budnikova, Y.H.; Gryaznova, T.V.; Grinenko, V.V.; Dudkina, Y.B.; Khrizanforov, M.N. Eco-efficient Electrocatalytic C–P bond Formation. Pure Appl. Chem. 2017, 89, 311–330. [Google Scholar] [CrossRef]
- Mikhaylov, D.Y.; Gryaznova, T.V.; Dudkina, Y.B.; Polyancev, F.M.; Latypov, S.K.; Sinyashin, O.G.; Budnikova, Y.H. Novel electrochemical pathway to fluoroalkyl phosphines and phosphine oxides. J. Fluor. Chem. 2013, 153, 178–182. [Google Scholar] [CrossRef]
- Budnikova, Y.G.; Kargin, Y.M.; Sinyashin, O.G. Electrosynthesis of mixed tertiary phosphines catalysed by nickel complexes. Mendeleev Commun. 1999, 9, 193–194. [Google Scholar] [CrossRef]
- Budnikova, Y.H.; Perichon, J.; Yakhvarov, D.G.; Kargin, Y.M.; Sinyashin, O.G. Highly reactive organonickel complexes in electrocatalytic processes. J. Organomet. Chem. 2001, 630, 185–192. [Google Scholar] [CrossRef]
- Milyukov, V.A.; Budnikova, Y.H.; Sinyashin, O.G. Organic chemistry of elemental phosphorus. Russ. Chem. Rev. 2005, 74, 781–805. [Google Scholar] [CrossRef]
- Budnikova, Y.H.; Sinyashin, O.G. Phosphorylation of aromatic C–H bonds involving metals and metal complexes. Russ. Chem. Rev. 2015, 84, 917–951. [Google Scholar] [CrossRef]
- Dudkina, Y.B.; Mikhaylov, D.Y.; Gryaznova, T.V.; Sinyashin, O.G.; Vicic, D.A.; Budnikova, Y.H. MII/MIII-Catalyzed ortho-Fluoroalkylation of 2-Phenylpyridine. Eur. J. Org. Chem. 2012, 2012, 2114–2117. [Google Scholar] [CrossRef]
- Jutand, A. Contribution of Electrochemistry to Organometallic Catalysis. Chem. Rev. 2008, 108, 2300–2347. [Google Scholar] [CrossRef] [PubMed]
- Francke, R.; Little, R.D. Redox catalysis in organic electrosynthesis: Basic principles and recent developments. Chem. Soc. Rev. 2014, 43, 2492–2521. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, J.-I.; Kataoka, K.; Horcajada, R.; Nagaki, A. Modern Strategies in Electroorganic Synthesis. Chem. Rev. 2008, 108, 2265–2299. [Google Scholar] [CrossRef] [PubMed]
- Sperry, J.B.; Wright, D.L. The application of cathodic reductions and anodic oxidations in the synthesis of complex molecules. Chem. Soc. Rev. 2006, 35, 605–621. [Google Scholar] [CrossRef] [PubMed]
- Budnikova, Y.H. Metal complex catalysis in organic electrosynthesis. Russ. Chem. Rev. 2002, 71, 111–139. [Google Scholar] [CrossRef]
- Kuehnel, M.F.; Orchard, K.L.; Dalle, K.E.; Reisner, E. Selective Photocatalytic CO Reduction in Water through Anchoring of a Molecular Ni Catalyst on CdS Nanocrystals. J. Am. Chem. Soc. 2017, 139, 7217–7223. [Google Scholar] [CrossRef] [PubMed]
- Elgrishi, N.; Chambers, M.B.; Artero, V.; Fontecave, M. Terpyridine complexes of first row transition metals and electrochemical reduction of CO2 to CO. Phys. Chem. Chem. Phys. 2014, 16, 13635–13644. [Google Scholar] [CrossRef] [PubMed]
- Arana, C.; Keshavarz, M.; Potts, K.T.; Abruna, H.D. Electrocatalytic reduction of CO and O with electropolymerized films of vinyl-terpyridine complexes of Fe, Ni and Co. Inorg. Chim. Acta 1994, 225, 285–295. [Google Scholar] [CrossRef]
- Prasad, R.; Scaife, D.B. Electro-oxidation and electro-reduction of some iron(II), cobalt(II) and nickel(II) polypyridyl complexes in acetonitrile. J. Electroanal. Chem. 1977, 84, 373–386. [Google Scholar] [CrossRef]
- Behrens, H.; Meyer, K. Über neue Darstellungsweisen von Nickel(0)-Komplexen aus Nickelocen. Z. Naturforsch. B 1966, 21, 489–490. [Google Scholar] [CrossRef]
- Churchill, M.R.; O’Brien, T.A. Crystal structure and molecular geometry of a trifluoromethyl complex of nickel: π-cyclopentadienyl-σ-trifluoromethyl(triphenylphosphine)nickel. J. Chem. Soc. A 1970, 161–167. [Google Scholar] [CrossRef]
- Dubinina, G.G.; Brennessel, W.W.; Miller, J.L.; Vicic, D.A. Exploring Trifluoromethylation Reactions at Nickel: A Structural and Reactivity Study. Organometallics 2008, 27, 3933–3938. [Google Scholar] [CrossRef]
- Savéant, J.-M. Molecular Catalysis of Electrochemical Reactions. Mechanistic Aspects. Chem. Rev. 2008, 108, 2348–2378. [Google Scholar] [CrossRef] [PubMed]
- Amatore, C.; Oturan, M.A.; Pinson, J.; Saveant, J.-M.; Thiebault, A. Electron-Transfer-Induced Reactions. A Novel Approach Based on Electrochemical Redox Catalysis. Application to Aromatic Nucleophilic Substitutions. J. Am. Chem. Soc. 1984, 106, 6318–6321. [Google Scholar] [CrossRef]
- Mikhaylov, D.Y.; Budnikova, Y.H.; Gryaznova, T.V.; Krivolapov, D.V.; Litvinov, I.A.; Vicic, D.A.; Sinyashin, O.G. Electrocatalytic fluoroalkylation of olefins. J. Organomet. Chem. 2009, 694, 3840–3843. [Google Scholar] [CrossRef]
- Mikhaylov, D.Y.; Budnikova, Y.H.; Gryaznova, T.V.; Sinyashin, O.G. Electrocatalytic fluoroalkylation of olefins. Russ. Chem. Bull. Int. Ed. 2010, 59, 1918–1920. [Google Scholar] [CrossRef]
- Dudkina, Y.B.; Gryaznova, T.V.; Osin, Y.N.; Salnikov, V.V.; Davydov, N.A.; Fedorenko, S.V.; Mustafina, A.R.; Vicic, D.A.; Sinyashin, O.G.; Budnikova, Y.H. Nanoheterogeneous catalysis in electrochemically induced olefin perfluoroalkylation. Dalton Trans. 2015, 44, 8833–8838. [Google Scholar] [CrossRef] [PubMed]






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
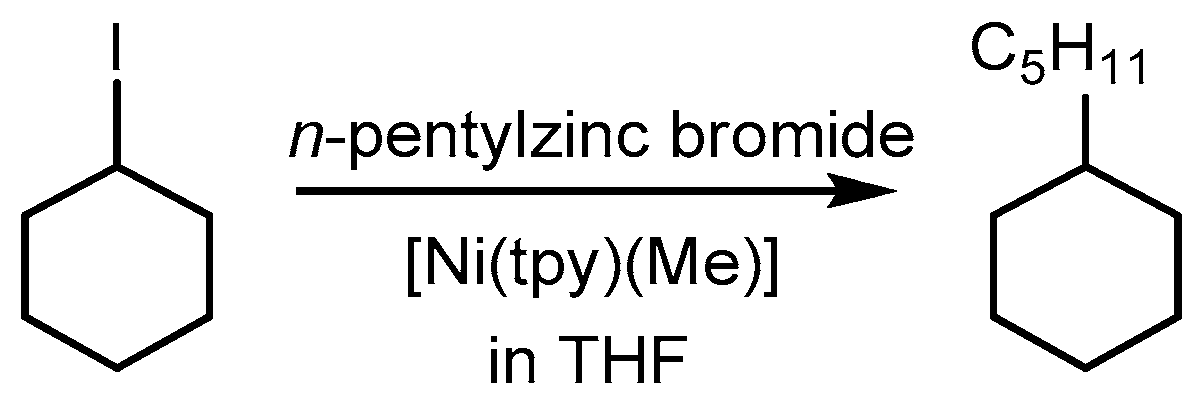{kind=link}
{kind=link}
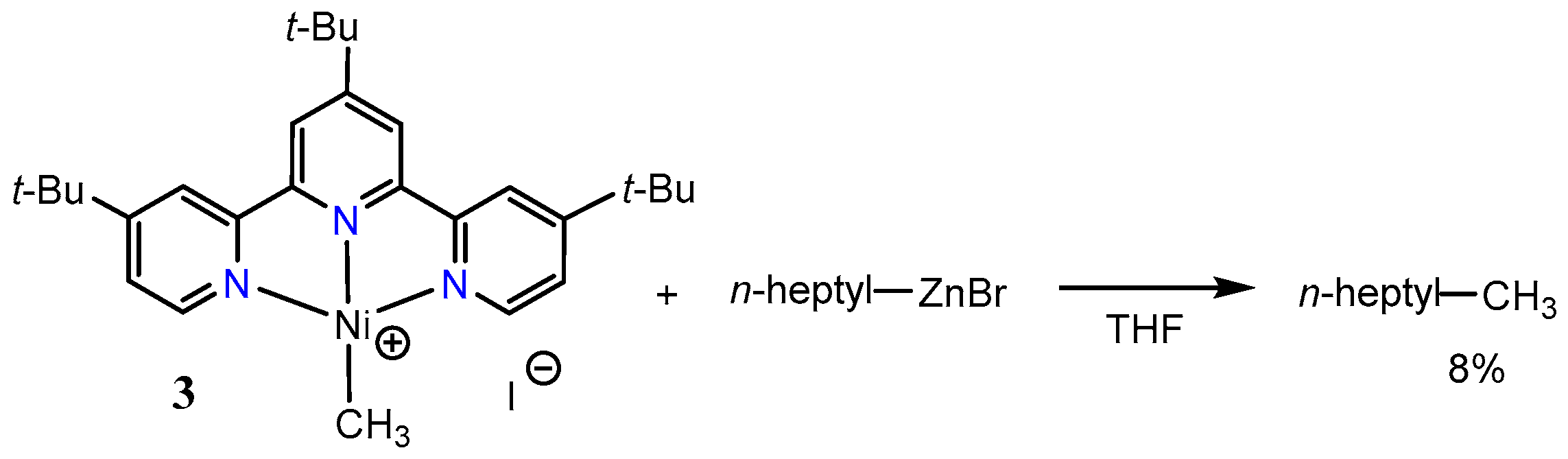{kind=link}
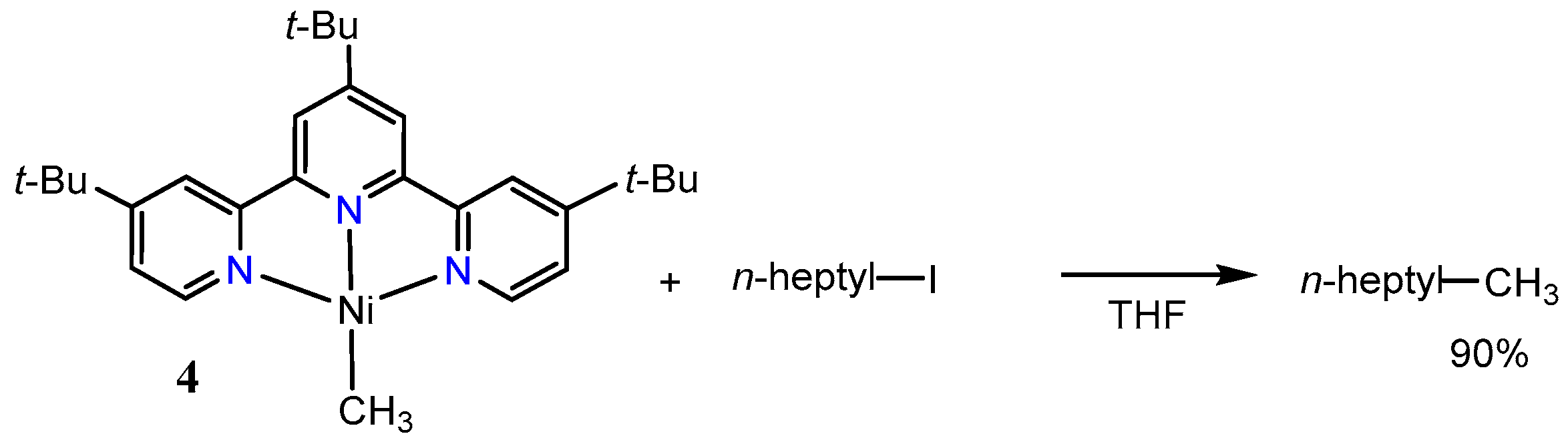{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
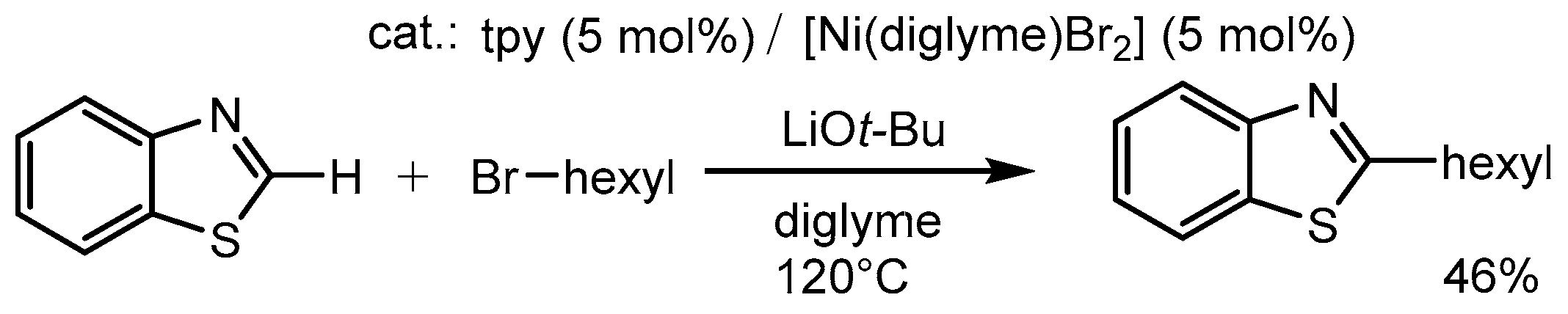{kind=link}
{kind=link}
{kind=link}
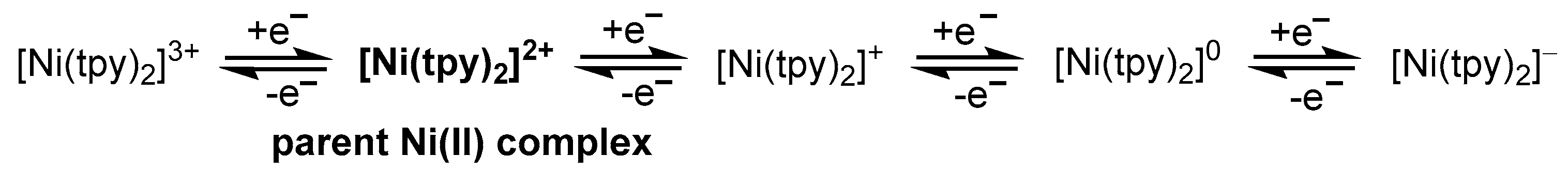{kind=link}
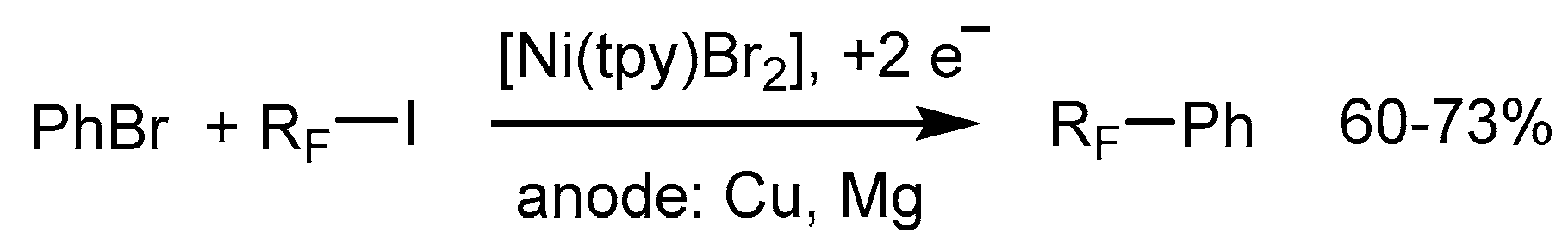{kind=link}
{kind=link}
{kind=link}
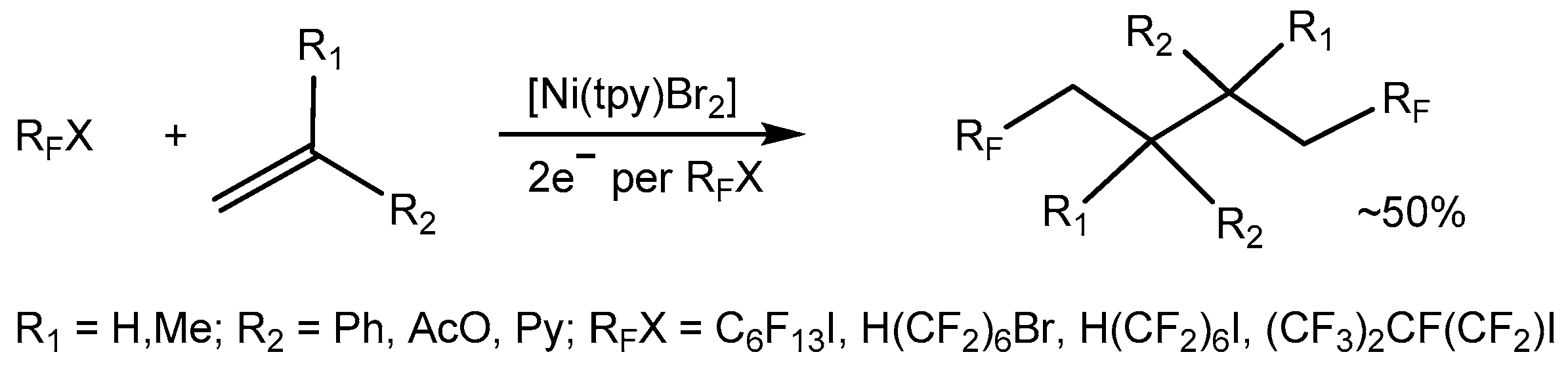{kind=link}
{kind=link}
| Complex | giso (298 K) | gav (LT) b | g1 (LT) | g2 (LT) | g3 (LT) | ∆g (LT) c |
|---|---|---|---|---|---|---|
| [Ni(tpy)Br]• | 2.139 | 2.146 | 2.256 | 2.091 | 2.091 | 0.165 |
| [Ni(tpy)(Me)]• | 2.021 | 2.025 | 2.056 | 2.021 | 1.999 | 0.026 |
| [Ni(tpy)(Mes)]• | 2.0006 | 2.0006 | 2.009 | 2.002 | 1.991 | 0.018 |
| [((t-Bu)3tpy)Ni(Xyl)]• d | 2.0006 | 2.0007 | 2.008 | 2.003 | 1.991 | 0.017 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Budnikova, Y.H.; Vicic, D.A.; Klein, A. Exploring Mechanisms in Ni Terpyridine Catalyzed C–C Cross-Coupling Reactions—A Review. Inorganics 2018, 6, 18. https://doi.org/10.3390/inorganics6010018
Budnikova YH, Vicic DA, Klein A. Exploring Mechanisms in Ni Terpyridine Catalyzed C–C Cross-Coupling Reactions—A Review. Inorganics. 2018; 6(1):18. https://doi.org/10.3390/inorganics6010018
Chicago/Turabian StyleBudnikova, Yulia H., David A. Vicic, and Axel Klein. 2018. "Exploring Mechanisms in Ni Terpyridine Catalyzed C–C Cross-Coupling Reactions—A Review" Inorganics 6, no. 1: 18. https://doi.org/10.3390/inorganics6010018
APA StyleBudnikova, Y. H., Vicic, D. A., & Klein, A. (2018). Exploring Mechanisms in Ni Terpyridine Catalyzed C–C Cross-Coupling Reactions—A Review. Inorganics, 6(1), 18. https://doi.org/10.3390/inorganics6010018