Abstract
Small molecule activation is a topic of growing importance and the use of low-valent f-elements to perform these reactions is nowadays well established. The complex Cptt2Sm(thf) (1, Cptt = 1,3-(tBu)2Cp) is shown to activate the alkyne C–H bond of phenylacetylene to form the Cptt2Sm(C≡C–Ph)(thf) complex. The subsequent reaction of this Sm(III) complex with CO2 leads to the CO2 insertion, yielding a dimeric [Cptt2Sm(O2C–C≡C–Ph)]2 complex (2), in which the carbon dioxide has been inserted in the Sm–C bond. Along with the experimental chemical structure analysis, theoretical calculations have been performed in order to rationalize the formation of 1 and 2.
1. Introduction
Small-molecule activation is a topic of growing importance from both academic and societal aspects [1,2,3]. The transformation of pollutant gases such as nitrous oxide (N2O), methane (CH4), carbon monoxide (CO), and carbon dioxide (CO2) into fine chemicals by low-valent coordination complexes is of particular interest and has been well investigated with both electron-rich transition metal compounds and low-valent f-elements, but the topic is still very active [4,5,6,7,8,9]. In this matter, the use of soluble divalent samarium complexes is well established, since the early 1980s, with the reports of the useful Cp*2Sm(L) complexes by W. J. Evans [10].
For example, in 1985, Evans reported the reaction between the Cp*2Sm(thf)2 (Cp* = C5Me5) and N2O to form an oxo-bridged bimetallic organosamarium complex [11]. A decade later, the same group reported the reaction of the Cp*2Sm(thf)2 with CO2, which led to a bridged oxalate dimer [12]. This seminal report of CO2 activation with divalent lanthanide marked out the field very strongly since the latter reaction is a single electron transfer followed by a radical-radical coupling, a very appealing transformation that is rare with transition metal complexes, which would prefer two electron processes. However, in 2006, Gardiner reported the reaction between a macrocyclic divalent organosamarium complex and CO2, yielding the formation of a carbonate complex [13] in strong contrast with Evans initial report with Cp*2Sm(thf)2 and indicating the possibility of multiple mechanisms pathways. As such, initially, the different steric demand of the complexes has been incriminated.
The mechanisms have been studied [14] while the studies with different complexes having various steric demands have continued and recently, the formation of both oxalate and carbonate compounds from the reaction of a divalent ytterbium complex and CO2 has been demonstrated by Mazzanti, reinforcing the subtle differences between the different pathways and the crucial role of the sterics in these complexes [15]. In 2017, Cloke made a similar reaction with a low-valent uranium complex [16].
Based on these very important studies, we recently have reported the reductive disproportionation of CO2 with bulky divalent samarium incorporating Cptt and Cpttt ligand (Cptt = 1,3-(tBu)2Cp and Cpttt = 1,2,4-tBu)3Cp) [17]. In both cases, we observed the clean formation of dimeric carbonate complexes while the steric bulk between the Cptt and Cpttt is obviously different. The theoretical studies helped us to rationalize this observation and led us to conclude that the differentiation between the oxalate and carbonate formation mostly comes from the negative charge delocalization (on the carbon for the oxalate or on the oxygen for the carbonate) and therefore has both steric and electronic contributions.
In a different direction, this kind of divalent samarium complexes can also undergo C–H bond activation mediated by single-electron transfer reactivity. In 1990, Evans reported the dehydrocoupling of phenylacetylene by the Cp*2Sm complex yielding a dinuclear samarium (III) trienediyl complexes [18]. In 2015, Maron investigated the mechanism of this interesting reaction [19]. An important note in Evans work was the role of solvent in the product formation as well as the bulk of the true alkene in the outcome of the reaction. As we were interested in these intermediates, we wondered what role would play a different bulk on the samarocene complexes and were curious of the possibility in sequencing C–H activation and CO2 activation to yield organic carboxylate derivatives.
Therefore, the present work intents to report the C–H bond activation by an organometallic divalent samarium complex containing bulkier substituted cyclopentadienyl ligand, Cptt2Sm(thf), to form the monomeric Cptt2Sm(C≡C–Ph)(thf) complex, which then reacts with CO2, leading to the CO2 insertion in the samarium-alkynyl bond yielding a dimeric organic carboxylate complex (Scheme 1).
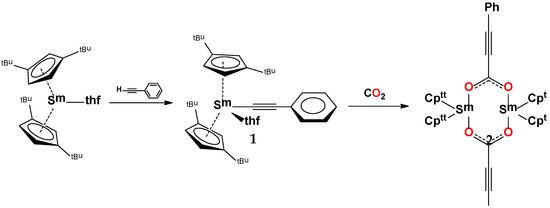
Scheme 1.
Synthetic pathway to the dimeric samarium complex.
2. Results and Discussion
2.1. Synthesis, Solid-State and Solution Structures
The synthesis of the Cptt2Sm(thf) complex was reported in 1990 by Bel’sky [20]. The reaction between Cptt2Sm(thf) and phenylacetylene was performed in toluene. A color change from deep brown to light yellow is accompanied by gas evolution. Cooling the solution at −35 °C overnight, led to the formation of yellow crystals, which were analyzed as the Cptt2Sm(C≡C–Ph)(thf) complex, 1. An ORTEP (Oak Ridge Thermal Ellipsoid Plot) of 1 is presented in Figure 1 while the metrics are reported in Supplementary Materials. A similar reaction has been observed by Evans starting from Cp*2Sm(thf)2 complex [21].
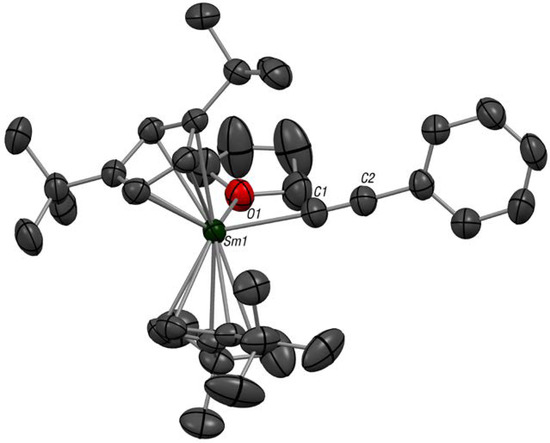
Figure 1.
ORTEP (Oak Ridge Thermal Ellipsoid Plot) of 1. Thermal ellipsoids are set to 50% probability level; H-atoms have been removed for clarity. Selected distances (in Å): Sm1–ctr (av.) 2.48(1); Sm1–C1 2.450(3); Sm1–O1 2.504(2); C1–C2 1.202(4). Ctr stands for centroid.
The Cptt–Sm average distance in 1 is 2.48(1) Å and is indicative of the trivalent oxidation state of the samarium metal center. Indeed, the Cptt–Sm distance is shorter than these found in divalent samarocene complexes (2.54 Å in Cptt2Sm(thf) complex). The Cptt–Sm–Cptt angle is of 130° at the lower side of the 130–138° range usually observed for similar complexes [21]. In the Cp*2Sm(C≡C–Ph)(thf) complex, the Cp*–Sm–Cp* angle is at the opposite end of the range with 138°. The Sm–O distance is 2.504(2) Å close to Evans observation for Cp*2Sm–O (2.47(2) Å) and similar to what has already been observed for this type of complexes [22,23]. The Sm–C≡C–Ph distance is a little shorter than that reported in Evans complex (2.450(3) Å in 1, vs. 2.49(2) Å in Cp*2Sm(C≡C–Ph)(thf) complex). On the contrary the C≡C bond is really different between complex 1 and Cp*2Sm(C≡C–Ph)(thf). In Cp*2Sm(C≡C–Ph)(thf), the C≡C distance is 1.11(2) Å that is shorter than the 1.202(4) Å observed in complex 1. This distance is similar to the 1.19–1.21 Å distances in free alkynes. This is one of the major difference between Cp*2Sm(C≡C–Ph)(thf) and Cptt2Sm(C≡C–Ph)(thf). Whereas most of the small differences between the metric of 1 and the one of Cp*2Sm(C≡C–Ph)(thf) can find their source in the variation of the hindrance caused by the Cptt ligand, the different C≡C distance would better be explained by different electronic properties of the two complexes. This difference of electronic density between 1 and Evans complex would indeed impact the C≡C distance and the difference in the sterics would impact less this distance, especially since the distance in 1 is shorter compared to that in Cp*2Sm(C≡C–Ph)(thf) but the bulk is slightly larger.
The complex has also been studied by 1H NMR and is consistent with the crystalline structure obtained. 1H NMR spectrum of 1 (Figure S1) was recorded in toluene-d8 on crystals and shows two signals at 13.11 ppm and 9.22 ppm attributed to the protons of the Cp ring and another one at 0.60 ppm attributed to tBu substituent. The phenyl ring exhibits two signals at 8.36 ppm and 7.40 ppm. The two signals attributed to the coordinated thf are at −0.27 ppm and −1.43 ppm.
Since the Sm–C bond is likely to be highly polarized, we reasoned that an insertion would be favored and investigated the opportunity with CO2. Thus, the reaction of 1 with 1 atmosphere of CO2 was performed in deuterated toluene. No color evolution was observed but after 12 h of reaction, the yellow solution was cooled at −35 °C and yellow X-ray suitable crystals were obtained. The structure revealed a dimeric structure, in which the CO2 is inserted in the Sm–alkene bond yielding an organic carboxylate Ph–C≡C–CO2− species that bridged two samarocene fragments (Figure 2). A planar eight-membered ring structure is obtained and is similar to other reported lanthanides–carboxylates complexes [Cp*2Sm(O2C6H4Me-m)]2 and [(Cp*2Sm(O2CCH2Ph)]2 [24]. Evans complexes were obtained from a different synthetic pathway than complex 2. The complexes were synthesized from Cp*2Sm(thf)2 and m-toluic acid or phenylacetic acid to form [Cp*2Sm(O2C6H4Me-m)]2 and [(Cp*2Sm(O2CCH2Ph)]2. Those two complexes have been prepared to identify the several products obtained from the reaction between [Cp*2Sm][(µ-Ph)2BPh2]/LiMe/toluene and CO2 [24].
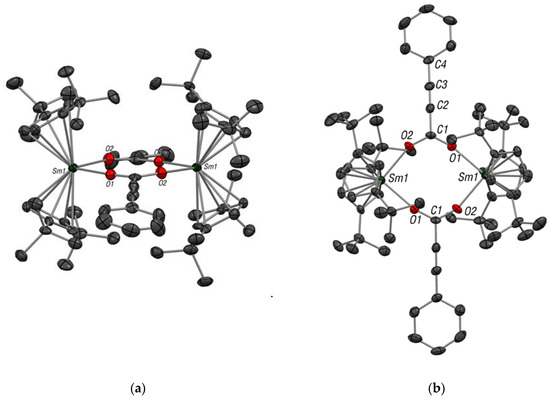
Figure 2.
ORTEP of 2: (a) side view; (b) top view. Thermal ellipsoids are set to 50% probability level; H-atoms have been removed for clarity. Selected distances (in Å): Sm1–ctr (av.) 2.45(1); Sm1–O1 2.312(4); Sm1–O2 2.315(5); C1–C2 1.447(10).
The Cptt–Sm average distance in 2 is similar to the average distance in the complex 1 (2.45(1) Å vs. 2.48(1) Å). The Sm–O average distance is 2.31(1) Å and is consistent with that have been already observed for [Cp*2Sm(O2C6H4Me-m)]2 and [(Cp*2Sm(O2CCH2Ph)]2. The C–O distance of the carboxylate moiety in the complex 2 is also in good agreement with the average C–O distance observed in Evans complexes.
The reaction was monitored by 1H NMR and 1H NMR of 2 showed shifted signals compared to the signals of 1 (Figure S2). The three signals attributed to the Cp ring and the tBu substituents are at 14.75 ppm, 12.44 ppm, and 0.10 ppm. The phenyl ring exhibits two signals at 5.95 ppm and 5.25 ppm.
Other alkynes such as 4-methyl-1-pentyne, trimethylsilylacetylene, and but-2-yne have been used to study the impact of the steric hindrance on the formation of the complex 1. Those reactions have been followed by 1H NMR and shown no reaction between Cptt2Sm(thf) and the other alkynes.
The importance of the samarium precursor has also been studied. Indeed, Evans published in 1990, the formation of a dimeric samarium complex by the reaction between the solvent-free precursor Cp*2Sm and the phenylacetylene (Figure 3) [18], i.e., a dehydrogenative coupling. The reaction of Cptt2Sm with the phenylacetylene has been followed by 1H NMR and the presence of several products has been observed. Unsuccessful attempts have been made to isolate the dimeric complex obtained from a similar dehydrocoupling as the one observed for Cp*2Sm were unsuccessful.
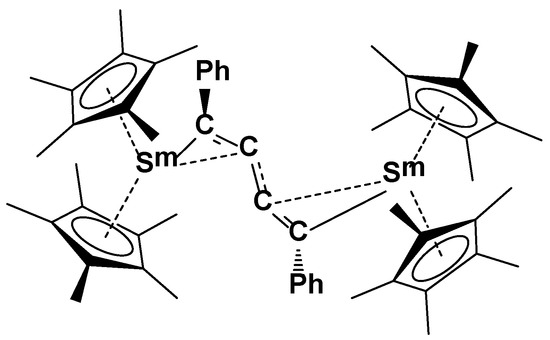
Figure 3.
Dimeric complex [(Cp*2Sm)2(µ-η2:η2–PhC4–Ph)] obtained from the reaction between solvent-free samarocene and phenylacetylene [18].
The difference of steric hindrance of the samarocene complexes incorporating Cp* and Cptt ligand plays a role in the formation of stable dimeric samarium complexes. Thus, theoretical calculations have been performed in order to study the mechanism leading to 2.
2.2. Theoretical Studies
Theoretical calculations have been performed on the formation of 1 and 2. The addition of phenylacetylene on the divalent samarium complex is favorable as shown in Figure 4. Two samarium centers are needed to achieve the formation of 1.
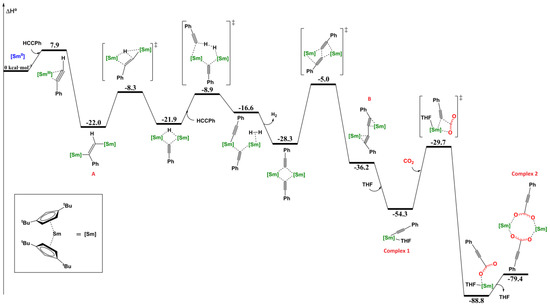
Figure 4.
Energy profile of CO2 insertion pathway for the complex Cptt2Sm(C≡C–Ph)(thf) (1).
The intermediate A at −22 kcal·mol−1 is the initiation step in the dehydrocoupling of phenylacetylene by Cp*2Sm as shown by Maron in 2015. This intermediate A is formed by a reduced phenylacetylene coordinated to two samarium through an exothermic process. The dimeric complex B is similar to the isolated complex of Evans [18] with Cp*Sm and is also proposed as an intermediate to the formation of 1. Three different types of reaction pathways have been investigated by Maron for the formation of this intermediate B with Cp*2Sm in a previous article [18]. All the mechanisms studied have the initiation step in common, which leads to the formation of the intermediate A (Figure 4). From this intermediate, three pathways have been studied depending on the first step: a C–C coupling, an H–H coupling, or a C–H coupling [18]. According to the calculations performed in this article, the overall mechanism proposed for the formation of 1 is very similar to the one observed for the reaction of Cp*2Sm and phenylacetylene. The first step is a C–H activation, then an isomerization and a C–C coupling in order to obtain the intermediate B. This intermediate cannot be isolated from the reaction of the solvent free Cptt2Sm complex and phenylacetylene. Indeed, the complex 1 lays −54.3 kcal·mol−1 below the starting material and −18.1 kcal·mol−1 below the intermediate B. This would explain the formation of the monomeric complex 1 instead of the dimeric complex B: the presence of coordinating solvent (thf) easily breaks this dimeric complex and leads to the formation of 1.
Then CO2 insertion on 1 shows two monomeric intermediates before the formation of 2. Theoretical calculations show the easy insertion of the CO2 into the Sm–C bond with a moderate energy barrier of 24.6 kcal·mol−1 before the elimination of the solvent to form 2.
In conclusion, we have successfully obtained Cptt2Sm(C≡C–Ph)(thf) by C–H bond activation mediated by single-electron transfer reactivity. The subsequent CO2 insertion in the Sm–C bond forms a dimeric carboxylate complex. The mechanism for those reactions has been investigated by theoretical computations and led to a similar mechanism as the one calculated for Cp*2Sm. The structural difference between Cp*2Sm(C≡C–Ph)(thf) studied by Evans [21] and Cptt2Sm(C≡C–Ph)(thf) have also been investigated. This work has also shown the importance of the steric hindrance of the samarocene complexes on the reactivity of those precursors with alkynes. The possibility to release the obtained carboxylic acid to recycle the active SmII complex is under study; it would then be an elegant pathway to the synthesis of carboxylic acid from CO2 activation [25].
3. Experimental Section
3.1. General Considerations
All reactions were performed using standard Schlenk-line techniques or in an argon-filled glovebox (MBraun, Garching, Germany). All glassware has been dried at 120 °C for at least 12 h prior to use. KCptt and Cptt2Sm(thf) were prepared according to published procedures [20]. Toluene and toluene-d8 were dried over sodium and transferred under reduced pressure in a cold flask and all solvents were degassed prior to use. Phenylacetylene was purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany) and purified prior to use. CO2 was purchased from Air liquid (Air liquid, Paris, France) as Alphagaz N48.
NMR spectra were recorded in 5 mm tubes adapted with a J. Young valve on Bruker 300 MHz Avance III spectrometers (Bruker, Billerica, MA, USA). Chemical shifts are expressed relative to TMS in ppm. Elemental analyses were performed at Mikroanalytisches Labor Pascher (Remagen, Germany).
3.2. Synthesis of Cptt2Sm(C≡C–Ph)(thf) (1)
To a brown solution of Cptt2Sm(thf) (75.7 mg, 0.13 mmol, 1 equiv.) in toluene was added phenylacetylene (14.3 µL, 0.13 mmol, 1 equiv.). A discoloration of the solution from brown to yellow was observed. After storage overnight at −35 °C, yellow crystals (69.1 mg, 71%) suitable for X-ray diffraction crashed out from the mother liquor.
1H NMR (300 MHz, toluene-d8, 293 K): 12.96 (s, 2H, Cp), 9.10 (s, 4H, Cp), 8.32 (d, J = 6 Hz, 2H, Ph), 7.39 (t, J = 8 Hz, 2H, Ph), 7.19 (t, J = 9 Hz, 1H, Ph), 0.60 (m, 36H, tBu), 0.18 (s, 4H, thf), −0.35 (s, 4H, thf).
Elemental analysis: calculated for C38H55OSm: C: 67.29; H: 8.17. Found: C: 64.28; H: 8.09. The elemental analysis is low for carbon even when measured on crystals.
3.3. Synthesis of [Cptt2Sm(O2C–C≡C–Ph)]2 (2)
Cptt2Sm(C≡C–Ph)(thf) (20 mg, 0.04 mmol) was dissolved in 1 mL of toluene-d8. The obtained solution was transferred into a J. Young tapered NMR tube and degassed by three freeze-pump-thaw cycles, before being reacted with an atmosphere of CO2. After cooling the solution to −35 °C, yellow crystals (12 mg, 28%) suitable for X-ray diffraction were obtained.
1H NMR (300 MHz, toluene-d8, 293 K): 14.75 (s, 4H, Cp), 12.44 (s, 8H, Cp), 6.16 (s, br, 2H), 5.93 (s, br, 4H, Ph), 5.25 (s, 4H, Ph), 0.10 (s, 72H, tBu).
Elemental analysis: calculated for C70H94O4Sm2: C: 64.80; H: 7.07. Found: C: 52.95; H: 6.18. The elemental analysis is low for carbon and hydrogen even when measured on crystals.
3.4. X-Ray Diffraction
Single crystals of the compounds 1 and 2 were mounted on a Kapton loop using a Paratone-N oil. An APEX II CCD BRUKER detector and a graphite Mo-Kα monochromator (Nonius, Delft, Netherlands) were used for the data acquisition. All measurements were done at 150 K and a refinement method was used for solving the structure. The structure resolution was accomplished using the SHELXT-2014 [26,27] program and the refinement was done with the SHELXL-2014/7 [28] program. The structure solution and the refinement were achieved with the PLATON software [29]. Finally, ORTEP of the compounds were obtained using the MERCURY software [30]. During the refinement steps, all atoms—except hydrogens—were refined anisotropically. The position of the hydrogens was determined using residual electronic densities which are calculated by a Fourier difference. Finally, in order to obtain a complete refinement, a weighting step followed by multiples loops of refinement was done. Details on crystal data and structure refinements are summarized in Table S1 in the Supplementary Material. CIF files are deposited at the Cambridge Data Base Centre under the reference CCDC numbers 1852482–1852483.
3.5. Theoretical Computations Diffraction
Calculations were performed with the Gaussian 09 program [31] at the DFT level of theory using the hybrid functional B3PW91 [32,33]. Samarium was treated with a large-core Stuttgart-Dresden relativistic effective core potential (RECP), adapted to the +3 oxidation state, used in combination with its optimized basis set augmented by set of f polarization functions (α = 1.000) [32]. Hydrogen, oxygen and carbon atoms were described with a 6-31G+(d,p) double-ζ-quality basis set. Electronic energies and enthalpies were computed at T = 298 K in the gas phase. Geometry optimizations were performed without any symmetry constraints, and analytical frequency calculations allowed verification of the nature of the extrema. Intrinsic reaction coordinate (IRC) calculations were carried out to verify the connections of the optimized transition states.
Supplementary Materials
The following are available online at http://www.mdpi.com/2304-6740/6/3/82/s1, Figures S1 and S2: 1H NMR of 1 and 2 in toluene-d8, X-ray Crystal Structure details, Table S1: Selected crystal data collection parameters for 1 and 2, Tables S2 and S3: Bond length and angles for 1 and 2, Figures S3 and S4: ORTEP of 1 and 2 with 50% probability ellipsoids, Cif and checkcif files of complexes 1 and 2. References [26,27,28,29] are cited in the supplementary materials.
Author Contributions
V.G., M.X., and S.K. performed the experimental work. M.C. carried out the crystal structure determination. C.A.L. and L.M. performed the theoretical computations. V.G. and G.N. wrote the paper.
Funding
This research was funded by the French National Agency with grant number ANR-15-CE29-0019.
Acknowledgments
The authors thank the CNRS and Ecole polytechnique for funding. M.X. is grateful to the DGA for funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Crutchley, R.J. Preface. Coord. Chem. Rev. 2017, 334, 1. [Google Scholar] [CrossRef]
- Milani, B.; Licini, G.; Clot, E.; Albrecht, M. Small Molecule Activation. Dalton Trans. 2016, 45, 14419–14420. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.-H.; Chuang, H.-J.; Lin, P.-H.; Ko, B.-T. Copolymerization of carbon dioxide with cyclohexene oxide catalyzed by bimetallic dysprosium complexes containing hydrazine-functionalized Schiff-base derivatives. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 321–328. [Google Scholar] [CrossRef]
- Tolman, W.B. Binding and Activation of N2O at Transition-Metal Centers: Recent Mechanistic Insights. Angew. Chem. Int. Ed. 2010, 49, 1018–1024. [Google Scholar] [CrossRef] [PubMed]
- Labinger, J.A.; Bercaw, J.E. Understanding and exploiting C–H bond activation. Nature 2002, 417, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Gunsalus, N.J.; Koppaka, A.; Park, S.H.; Bischof, S.M.; Hashiguchi, B.G.; Periana, R.A. Homogeneous Functionalization of Methane. Chem. Rev. 2017, 117, 8521–8573. [Google Scholar] [CrossRef] [PubMed]
- Summerscales, O.T.; Cloke, F.G.N.; Hitchcock, P.B.; Green, J.C.; Hazari, N. Reductive Cyclotrimerization of Carbon Monoxide to the Deltate Dianion by an Organometallic Uranium Complex. Science 2006, 311, 829–831. [Google Scholar] [CrossRef] [PubMed]
- Franke, R.; Selent, D.; Borner, A. Applied Hydroformylation. Chem. Rev. 2012, 112, 5675–5732. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wu, L.; Jackstell, R.; Beller, M. Using carbon dioxide as a building block in organic synthesis. Nat. Commun. 2015, 6, 5933. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.J.; Hugues, L.A.; Hanusa, T.P. Synthesis and crystallographic characterization of an unsolvated, monomeric samarium bis(pentamethylcyclopentadienyl) organolanthanide complex, (C5Me5)2Sm. J. Am. Chem. Soc. 1984, 106, 4270–4272. [Google Scholar] [CrossRef]
- Evans, W.J.; Grate, J.W.; Bloom, I.; Hunter, W.E.; Atwood, J.L. Synthesis and X-ray crystallographic characterization of an oxo-bridged bimetallic organosamarium complex, [(C5Me)2Sm]2(µ-O). J. Am. Chem. Soc. 1985, 107, 405–409. [Google Scholar] [CrossRef]
- Evans, W.J.; Seibel, C.A.; Ziller, J.W. Organosamarium-mediated transformations of CO2 and COS: Monoinsertion and disproportionation reactions and the reductive coupling of CO2 to [O2CCO2]2−. Inorg. Chem. 1998, 37, 770–776. [Google Scholar] [CrossRef]
- Davies, N.W.; Frey, A.S.P.; Gardiner, M.G.; Wang, J. Reductive disproportionation of carbon dioxide by a Sm(II) complex: Unprecedented f-block element reactivity giving a carbonate complex. Chem. Commun. 2006, 4853–4855. [Google Scholar] [CrossRef]
- Castro, L.; Labouille, S.; Kindra, D.R.; Ziller, J.W.; Nief, F.; Evans, W.J.; Maron, L. Insights into the mechanism of reaction of [(C5Me)2SmII(thf)2] with CO2 and COS by DFT studies. Chem. Eur. J. 2012, 18, 7886–7895. [Google Scholar] [CrossRef] [PubMed]
- Andrez, J.; Pécaut, J.; Bayle, P.; Mazzanti, M. Tuning Lanthanide Reactivity towards Small Molecules with Electron-Rich Siloxide Ligands. Angew. Chem. Int. Ed. 2014, 53, 10448–10452. [Google Scholar] [CrossRef] [PubMed]
- Tsoureas, N.; Castro, L.; Kilpatrick, A.F.R.; Cloke, F.G.N.; Maron, L. Controlling selectivity in the reductive activation of CO2 by mixed sandwich uranium(III) complexes. Chem. Sci. 2014, 5, 3777–3788. [Google Scholar] [CrossRef]
- Xemard, M.; Goudy, V.; Braun, A.; Tricoire, M.; Cordier, M.; Ricard, L.; Castro, L.; Louyriac, E.; Kefalidis, C.E.; Clavaguéra, C.; et al. Reductive disproportionation of CO2 with bulky divalent samarium complexes. Organometallics 2017, 36, 4660–4668. [Google Scholar] [CrossRef]
- Evans, W.J.; Keyer, R.A.; Ziller, J.W. Carbon–carbon bond formation by coupling of two phenylethynyl ligand in an organolanthanide system. Organometallics 1990, 9, 2628–2631. [Google Scholar] [CrossRef]
- Kefalidis, C.E.; Maron, L. On the dehydrocoupling of alkenylacetylenes mediated by various samarocene complexes: A charming story of metal cooperativity revealing a novel dual metal σ-bond metathesis type of mechanism (DM|σ-BM). Inorganics 2015, 3, 573–588. [Google Scholar] [CrossRef]
- Bel’sky, V.K.; Gunko, Y.K.; Bulychev, B.M.; Sizov, A.I.; Soloveichik, G.L. Dicyclopentadienylsamarium complexes with t-butyl substituents in the ring. Crystal and molecular structures of the solvate (η5-C5H3Bu2t)2Sm·OC4H8 and homoleptic ate complex [NaSm(η5:η2-C5H4But)3·OC4H8]n. J. Organomet. Chem. 1990, 390, 35–44. [Google Scholar] [CrossRef]
- Evans, W.J.; Keyer, R.A.; Ziller, J.W. Investigation of organolanthanide-based carbon-carbon bond formation: Synthesis, structure, and coupling reactivity of organolanthanide alkynide complexes, including the unusual structures of the trienediyl complex [(C5Me5)2Sm]2[µ-η2:η2-Ph(CH2)2C=C=C=C–(CH2)2Ph] and the unsolvated alkynide [(C5Me5)2Sm(C≡CCMe3)]2. Organometallics 1993, 12, 2618–2633. [Google Scholar] [CrossRef]
- Evans, W.J.; Ulibarri, T.A. Reactivity of (C5Me5)2Sm with cyclopentadiene and cyclopentadienide: Isolation of the mixed-valence complex (C5Me5)2Sm(III)(µ-C5H5)Sm(II)(C5Me5)2. J. Am. Chem. Soc. 1987, 109, 4292–4297. [Google Scholar] [CrossRef]
- Evans, W.J.; Chamberlain, L.R.; Ulibarri, T.A.; Ziller, J.W. Reactivity of trimethylaluminum with (C5Me5)2Sm(THF)2: Synthesis, structure, and reactivity of the samarium methyl complexes (C5Me5)2Sm[(µ-Me)AlMe2(µ-Me)]2Sm(C5Me5)2 and (C5Me5)2SmMe(THF). J. Am. Chem. Soc. 1988, 110, 6423–6432. [Google Scholar] [CrossRef]
- Evans, W.J.; Perotti, J.M.; Ziller, J.W. Synthetic Utility of [(C5Me5)2Ln][(μ-Ph)2BPh2] in Accessing [(C5Me5)2LnR]x Unsolvated Alkyl Lanthanide Metallocenes, Complexes with High C−H Activation Reactivity. J. Am. Chem. Soc. 2005, 127, 3894–3909. [Google Scholar] [CrossRef] [PubMed]
- Manjolinho, F.; Arndt, M.; Gooßen, K.; Gooßen, L.J. Catalytic C–H Carboxylation of Terminal Alkynes with Carbon Dioxide. ACS Catal. 2012, 2, 2014–2021. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. A Found Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Cryst. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; Van De Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision, D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Burke, K.; Perdew, J.P.; Wang, Y. Electronic Density Functional Theory: Recent Progress and New Directions; Dobson, J.F., Vignale, G., Das, M.P., Eds.; Plenum: New York, NY, USA, 1998. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Phys. Chem. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Dolg, M.; Stoll, H.; Savin, A.; Preuss, H. Energy-adjusted pseudopotentials for the rare earth elements. Theor. Chim. Acta 1989, 75, 173–194. [Google Scholar] [CrossRef]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).