Room Temperature Ni(II) Catalyzed Hydrophosphination and Cyclotrimerization of Alkynes


Abstract
:
1. Introduction
2. Results and Discussion
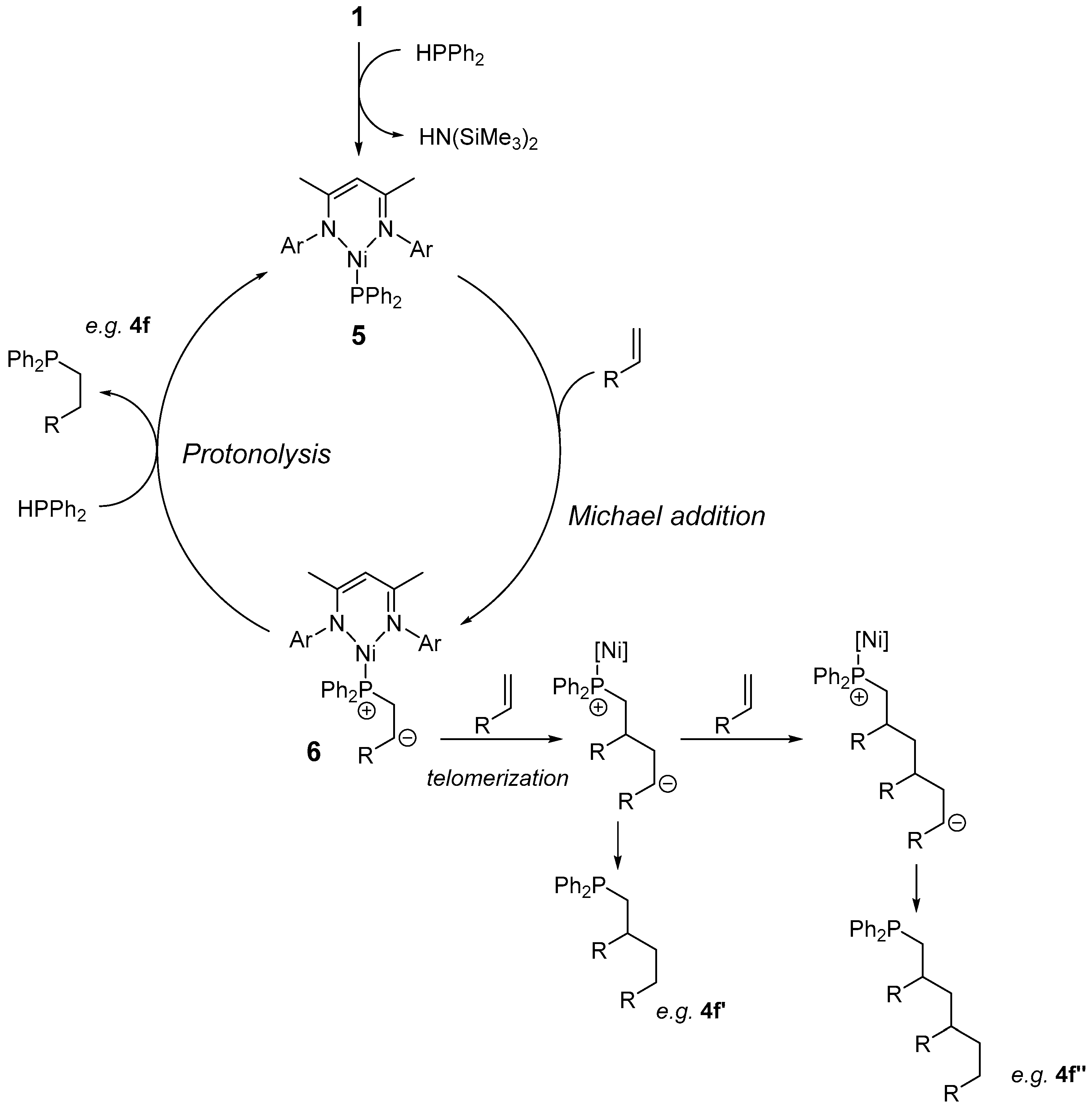
2.1. Reaction Optimization
2.2. Substrate Scope
3. Materials and Methods
3.1. General Method for Hydrophosphination
3.2. Analysis Data for Isolated Products
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Koshti, V.; Gaikwad, S.; Chikkali, S.H. Contemporary avenues in catalytic PH bond addition reaction: A case study of hydrophosphination. Coord. Chem. Rev. 2014, 265, 52–73. [Google Scholar] [CrossRef]
- Wauters, I.; Debrouwer, W.; Stevens, C.V. Preparation of phosphines through C–P bond formation. Beilstein J. Org. Chem. 2014, 10, 1064–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bange, C.A.; Waterman, R. Challenges in Catalytic Hydrophosphination. Chem. Eur. J. 2016, 22, 12598–12605. [Google Scholar] [CrossRef] [PubMed]
- Espinal-Viguri, M.; King, A.K.; Lowe, J.P.; Mahon, M.F.; Webster, R.L. Hydrophosphination of Unactivated Alkenes and Alkynes Using Iron(II): Catalysis and Mechanistic Insight. ACS Catal. 2016, 6, 7892–7897. [Google Scholar] [CrossRef] [Green Version]
- King, A.K.; Gallagher, K.J.; Mahon, M.F.; Webster, R.L. Markovnikov versus anti-Markovnikov Hydrophosphination: Divergent Reactivity Using an Iron(II) β-Diketiminate Pre-Catalyst. Chem. Eur. J. 2017, 23, 9039–9043. [Google Scholar] [CrossRef] [PubMed]
- Eckert, N.A.; Bones, E.M.; Lachicotte, R.J.; Holland, P.L. Nickel Complexes of a Bulky β-Diketiminate Ligand. Inorg. Chem. 2003, 42, 1720–1725. [Google Scholar] [CrossRef] [PubMed]
- Bezzenine-Lafollée, S.; Gil, R.; Prim, D.; Hannedouche, J. First-Row Late Transition Metals for Catalytic Alkene Hydrofunctionalisation: Recent Advances in C–N, C–O and C–P Bond Formation. Molecules 2017, 22, 1901. [Google Scholar] [CrossRef] [PubMed]
- Sadow, A.D.; Haller, I.; Fadini, L.; Togni, A. Nickel(II)-Catalyzed Highly Enantioselective Hydrophosphination of Methacrylonitrile. J. Am. Chem. Soc. 2004, 126, 14704–14705. [Google Scholar] [CrossRef] [PubMed]
- Sadow, A.D.; Togni, A. Enantioselective Addition of Secondary Phosphines to Methacrylonitrile: Catalysis and Mechanism. J. Am. Chem. Soc. 2005, 127, 17012–17024. [Google Scholar] [CrossRef] [PubMed]
- Dorfman, Y.A.; Levina, L.V.; Grekov, L.I.; Korolev, A.V. Hydroxymethylation of phosphine in the presence of Ni(II) amines. Kinet. Catal. 1989, 30, 578–583. [Google Scholar]
- Hoye, P.A.T.; Pringle, P.G.; Smith, M.B.; Worboys, K. Hydrophosphination of formaldehyde catalysed by tris-(hydroxymethyl)phosphine complexes of platinum, palladium or nickel. J. Chem. Soc. Dalton Trans. 1993, 269–274. [Google Scholar] [CrossRef]
- Kazankova, M.A.; Shulyupin, M.O.; Beletskaya, I.P. Catalytic Hydrophosphination of Alkenylalkyl Ethers. Synlett 2003, 2003, 2155–2158. [Google Scholar] [CrossRef]
- Kazankova, M.A.; Shulyupin, M.O.; Borisenko, A.A.; Beletskaya, I.P. Synthesis of Alkyl(diphenyl)phosphines by Hydrophosphination of Vinylarenes Catalyzed by Transition Metal Complexes. Russ. J. Org. Chem. 2002, 38, 1479–1484. [Google Scholar] [CrossRef]
- Kazankova, M.A.; Efimova, I.V.; Kochetkov, A.N.; Afanasev, V.V.; Beletskaya, I.P.; Dixneuf, P.H. New Approach to Vinylphosphines Based on Pd- and Ni-Catalyzed Diphenylphosphine Addition to Alkynes. Synlett 2001, 2001, 497–500. [Google Scholar] [CrossRef]
- Kazankova, M.A.; Efimova, I.V.; Kochetkov, A.N.; Afanas’ev, V.V.; Beletskaya, I.P. Synthesis of Vinylphosphines by Hydrophosphination of Alkynes in the Presence of Transition Metal Complexes. Russ. J. Org. Chem. 2002, 38, 1465–1474. [Google Scholar] [CrossRef]
- Yang, M.-J.; Liu, Y.-J.; Gong, J.-F.; Song, M.-P. Unsymmetrical Chiral PCN Pincer Palladium(II) and Nickel(II) Complexes with Aryl-Based Aminophosphine-Imidazoline Ligands: Synthesis via Aryl C–H Activation and Asymmetric Addition of Diarylphosphines to Enones. Organometallics 2011, 30, 3793–3803. [Google Scholar] [CrossRef]
- Ribière, P.; Bravo-Altamirano, K.; Antczak, M.I.; Hawkins, J.D.; Montchamp, J.-L. NiCl2-Catalyzed Hydrophosphinylation. J. Org. Chem. 2005, 70, 4064–4072. [Google Scholar] [CrossRef] [PubMed]
- Ananikov, V.P.; Khemchyan, L.L.; Beletskaya, I.P.; Starikova, Z.A. Acid-Free Nickel Catalyst for Stereo- and Regioselective Hydrophosphorylation of Alkynes: Synthetic Procedure and Combined Experimental and Theoretical Mechanistic Study. Adv. Synth. Catal. 2010, 352, 2979–2992. [Google Scholar] [CrossRef]
- Alonso, F.; Moglie, Y.; Radivoy, G.; Yus, M. Solvent- and catalyst-free regioselective hydrophosphanation of alkenes. Green Chem. 2012, 14, 2699–2702. [Google Scholar] [CrossRef]
- Moglie, Y.; Gonzalez-Soria, M.J.; Martin-Garcia, I.; Radivoy, G.; Alonso, F. Catalyst- and solvent-free hydrophosphination and multicomponent hydrothiophosphination of alkenes and alkynes. Green Chem. 2016, 18, 4896–4907. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, L. Mechanisms of Metal-Catalyzed Hydrophosphination of Alkenes and Alkynes. ACS Catal. 2013, 2845–2855. [Google Scholar] [CrossRef]
- Scriban, C.; Kovacik, I.; Glueck, D.S. A protic additive suppresses formation of byproducts in platinum-catalyzed hydrophosphination of activated olefins. Evidence for P–C and C–C bond formation by Michael addition. Organometallics 2005, 24, 4871–4874. [Google Scholar] [CrossRef]
- Scriban, C.; Glueck, D.S.; Zakharov, L.N.; Kassel, W.S.; DiPasquale, A.G.; Golen, J.A.; Rheingold, A.L. P–C and C–C bond formation by Michael addition in platinum-catalyzed hydrophosphination and in the stoichiometric reactions of platinum phosphido complexes with activated alkenes. Organometallics 2006, 25, 5757–5767. [Google Scholar] [CrossRef]
- Bai, G.; Wei, P.; Das, A.K.; Stephan, D.W. P–H and P–P bond activation by Ni(I) and Fe(I) β-diketiminato-complexes. Dalton Trans. 2006, 1141–1146. [Google Scholar] [CrossRef] [PubMed]
- Reppe, W.; Schlichting, O.; Klager, K.; Toepel, T. Cyclisierende Polymerisation von Acetylen 1. Uber Cyclooctatetraen. Ann. Chem. Justus Liebig 1948, 560, 1–92. [Google Scholar] [CrossRef]
- Reppe, W.; Schlicting, O.; Meister, H. Cyclisierende Polymerisation von Acetylen 2. Uber Die Kohlenwasserstoffe C10H10, C12H12 und Azulen. Ann. Chem. Justus Liebig 1948, 560, 93–104. [Google Scholar] [CrossRef]
- Reppe, W.; Schweckendiek, W.J. Cyclisierende Polymerisation von Acetylen 3. Benzol, Benzolderivate und Hydroaromatische Verbindungen. Ann. Chem. Justus Liebig 1948, 560, 104–116. [Google Scholar]
- Del Rosario, R.; Stuhl, L.S. Organonickel cyanide chemistry. Reactions of [(PhC.tplbond.CPh)Ni(CN)2]2−. An improved synthesis of [Ni(CN)2(CO)2]2−. Organometallics 1986, 5, 1260–1262. [Google Scholar] [CrossRef]
- Eisch, J.J.; Galle, J.E.; Aradi, A.A.; Bolesa̵wski, M.P. Organic chemistry of subvalent transition metal complexes: XI. Oxidative additions of nickel(0) complexes to carbon–carbon bonds in alkynes: Nickelirenes and nickeloles as catalytic carriers in the oligomerization of alkynes. J. Organomet. Chem. 1986, 312, 399–416. [Google Scholar] [CrossRef]
- Eisch, J.J.; Ma, X.; Han, K.I.; Gitua, J.N.; Krüger, C. Mechanistic Comparison of the Nickel(0)-Catalyzed Homo-Oligomerization and Co-Oligomerization of Alkynes and Nitriles. Eur. J. Inorg. Chem. 2001, 2001, 77–88. [Google Scholar] [CrossRef]
- Bennett, M.A.; Hockless, D.C.R.; Wenger, E. Generation of (2,3-η)-Naphthalyne–Nickel(0) Complexes and Their Reactions with Unsaturated Molecules. Organometallics 1995, 14, 2091–2101. [Google Scholar] [CrossRef]
- Bennett, M.A.; Wenger, E. Insertion Reactions of Benzyne–Nickel(0) Complexes with Acetylenes. Organometallics 1995, 14, 1267–1277. [Google Scholar] [CrossRef]
- Hollingsworth, R.L.; Bheemaraju, A.; Lenca, N.; Lord, R.L.; Groysman, S. Divergent reactivity of a new dinuclear xanthene-bridged bis(iminopyridine) di-nickel complex with alkynes. Dalton Trans. 2017, 46, 5605–5616. [Google Scholar] [CrossRef] [PubMed]
- Reinhard, S.; Šoba, P.; Rominger, F.; Blümel, J. New Silica-Immobilized Nickel Catalysts for Cyclotrimerizations of Acetylenes. Adv. Synth. Catal. 2003, 345, 589–602. [Google Scholar] [CrossRef]
- Haines, R.I.; McAuley, A. Synthesis and reactions of nickel(III) complexes. Coord. Chem. Rev. 1981, 39, 77–119. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Saha, B.; Dutta, A.; Banerjee, P. Electron transfer reactions of nickel(III) and nickel(IV) complexes. Coord. Chem. Rev. 1998, 170, 47–74. [Google Scholar] [CrossRef]
- Camp, C.; Arnold, J. On the non-innocence of “Nacnacs”: Ligand-based reactivity in β-diketiminate supported coordination compounds. Dalton Trans. 2016, 45, 14462–14498. [Google Scholar] [CrossRef] [PubMed]
- Sabater, S.; Page, M.J.; Mahon, M.F.; Whittlesey, M.K. Stoichiometric and Catalytic Reactivity of Ni(6-Mes)(PPh3)2. Organometallics 2017, 36, 1776–1783. [Google Scholar] [CrossRef]
- Duncan, M.; Gallagher, M.J. The 1H, 13C and 31P NMR spectra of EZ pairs of some phosphorus substituted alkenes. Org. Magn. Reson. 1981, 15, 37–42. [Google Scholar] [CrossRef]
- Hayashi, M.; Matsuura, Y.; Watanabe, Y. Regio- and Stereoselective Synthesis of Alkenylphosphines: A Rhodium-Catalyzed Hydrophosphination of Alkynes Using a Silylphosphine. J. Org. Chem. 2006, 71, 9248–9251. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, K.J.; Webster, R.L. Room temperature hydrophosphination using a simple iron salen pre-catalyst. Chem. Commun. 2014, 50, 12109–12111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilt, G.; Vogler, T.; Hess, W.; Galbiati, F. A simple cobalt catalyst system for the efficient and regioselective cyclotrimerisation of alkynes. Chem. Commun. 2005, 1474–1475. [Google Scholar] [CrossRef] [PubMed]
- Yoshikai, N.; Mashima, H.; Nakamura, E. Nickel-Catalyzed Cross-Coupling Reaction of Aryl Fluorides and Chlorides with Grignard Reagents under Nickel/Magnesium Bimetallic Cooperation. J. Am. Chem. Soc. 2005, 127, 17978–17979. [Google Scholar] [CrossRef] [PubMed]

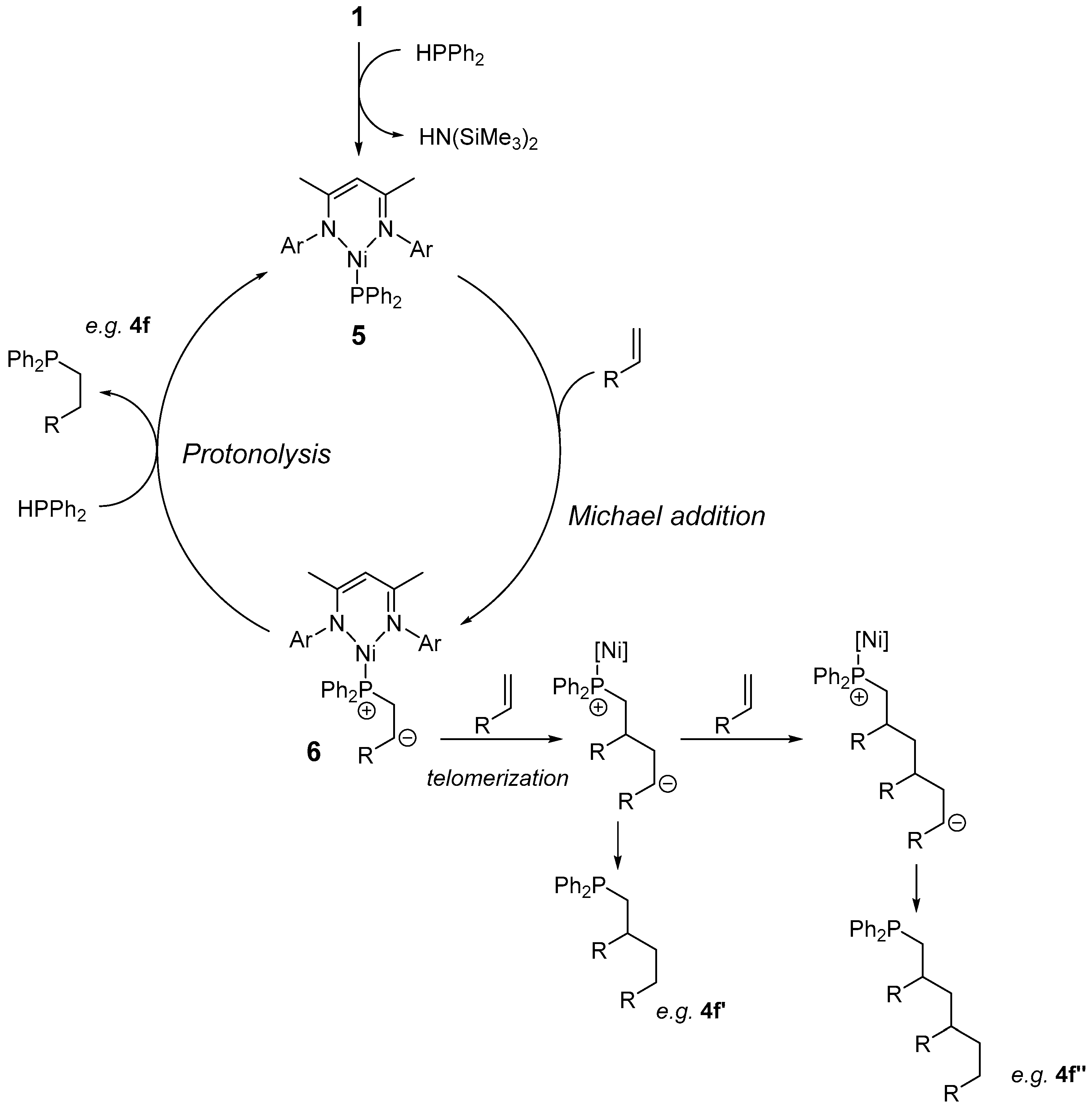

{kind=link}
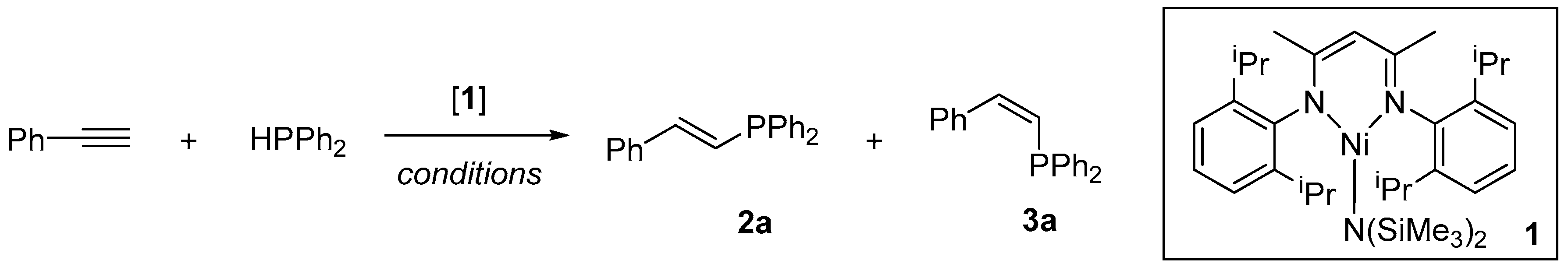{kind=link}
{kind=link}
| Entry | 1 (mol %) | Solvent | Conditions | Spectroscopic Yield 2a + 3a (%) a |
|---|---|---|---|---|
| 1 | 5 | C6H6 | 80 °C | 53 |
| 2 | 5 | C6H6 | RT | 5 |
| 3 | 5 | CH2Cl2 | 80 °C | No reaction |
| 4 | 5 | CH3CN | 80 °C | 90 |
| 5 | 5 | CH3CN | RT, 18 h | 77 |
| 6 | 2 | CH3CN | RT, 18 h | 33 |
| 7 b | 5 | CH3CN | RT, 4 h | 82 |
| 8 c | 5 | CH3CN | RT, 18 h | 74 |
| 9 b,d | 5 | CH3CN | RT, 18 h | 94 (81) {1:12 E:Z} |
| Entry | Olefin | Product | Ratio E:Z (2:3) | Spectroscopic Yield, % a (Isolated Yield, %) | |
|---|---|---|---|---|---|
| 1 |  |  | 2a 3a | 1:12 | 94 (81) |
| 2 |  |  | 2b 3b | 1:1 | 45 |
| 3 |  |  | 2c 3c | 2:1 | 52 |
| 4 |  |  | 2d 3d | 6:5 | 67 |
| 5 |  |  | 2e 3e | - | Complex mixture |
| 6 b,c |  |  | 2f 3f | - | No reaction |
| 7 b |  |  | 2g 3g | - | No reaction |
| 8 |  |  | 4a | - | 100 (92) |
| 9 |  |  | 4b | - | 100 (68) |
| 10 |  |  | 4c | - | 100 (49) |
| 11 |  |  | 4d | - | 11 |
| 12 |  |  | 4e | - | 85 (52/20) |
 | 4e’ | ||||
| 13 |  |  | 4f | - | 100 (43/17/4) |
 | 4f’ | ||||
 | 4f” | ||||
| 14 |  |  | 4g | - | Complex mixture |
| Entry | Olefin | Product | Ratio 1,2,4:1,3,5 | Spectroscopic Yield, % a (Isolated Yield, %) |
|---|---|---|---|---|
| 1 |  |  7a 7a | 93:7 | 100 (66) |
| 2 |  |  7b 7b | 85:15 | 100 |
| 3 b |  |  7c 7c | Not determined | 66 (41) |
| 4 b |  |  8 8 | - | 56 |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Webster, R.L. Room Temperature Ni(II) Catalyzed Hydrophosphination and Cyclotrimerization of Alkynes. Inorganics 2018, 6, 120. https://doi.org/10.3390/inorganics6040120
Webster RL. Room Temperature Ni(II) Catalyzed Hydrophosphination and Cyclotrimerization of Alkynes. Inorganics. 2018; 6(4):120. https://doi.org/10.3390/inorganics6040120
Chicago/Turabian StyleWebster, Ruth L. 2018. "Room Temperature Ni(II) Catalyzed Hydrophosphination and Cyclotrimerization of Alkynes" Inorganics 6, no. 4: 120. https://doi.org/10.3390/inorganics6040120
APA StyleWebster, R. L. (2018). Room Temperature Ni(II) Catalyzed Hydrophosphination and Cyclotrimerization of Alkynes. Inorganics, 6(4), 120. https://doi.org/10.3390/inorganics6040120