In Vitro Cytotoxicity and In Vivo Antitumor Efficacy of Tetrazolato-Bridged Dinuclear Platinum(II) Complexes with a Bulky Substituent at Tetrazole C5


Abstract
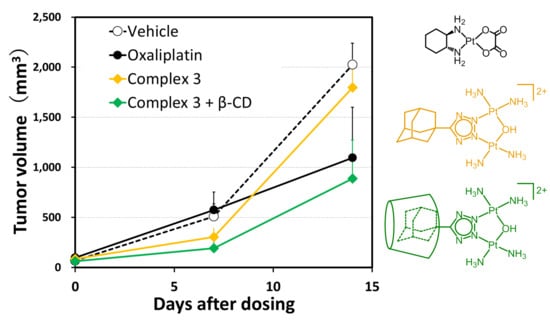
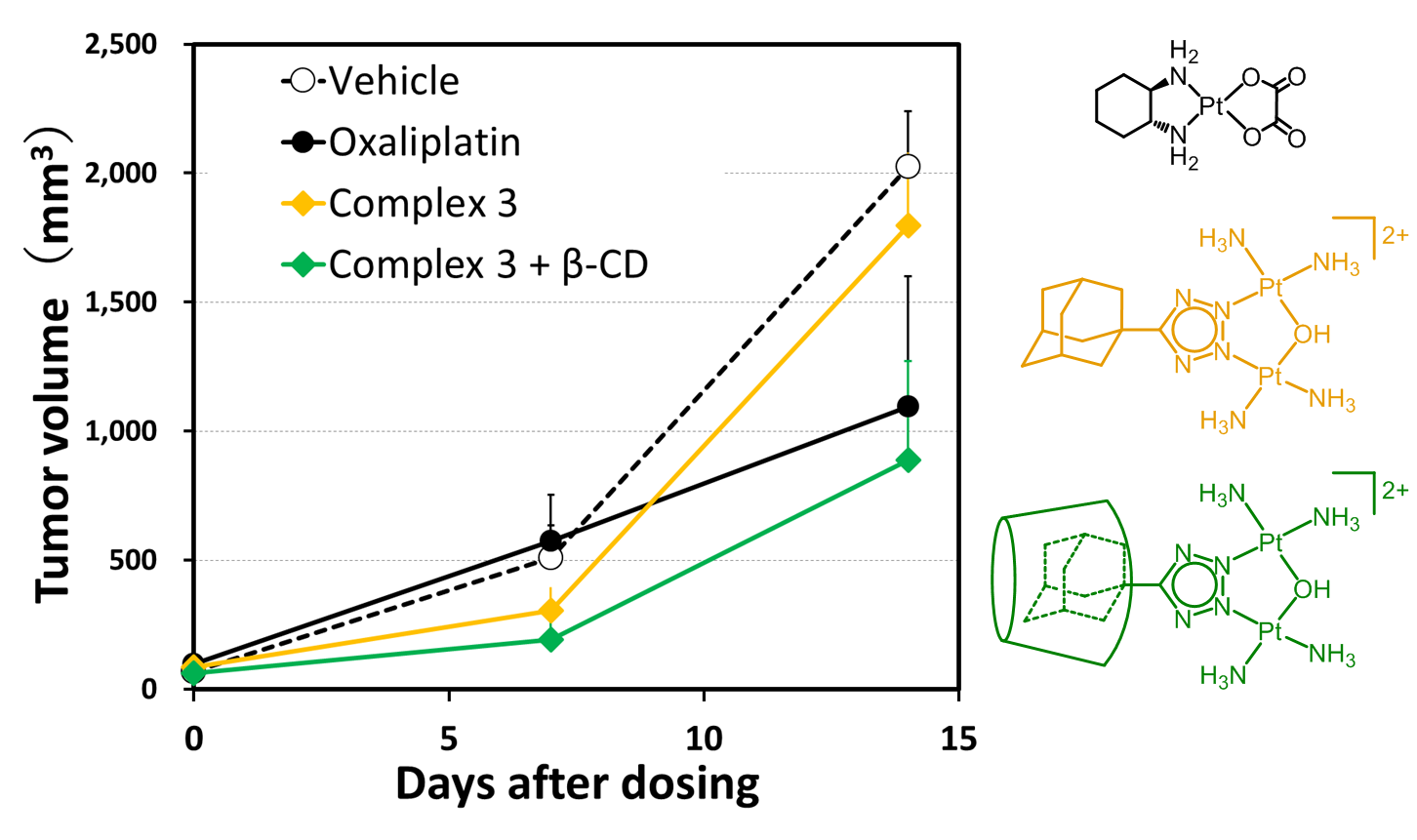
:
1. Introduction
2. Results
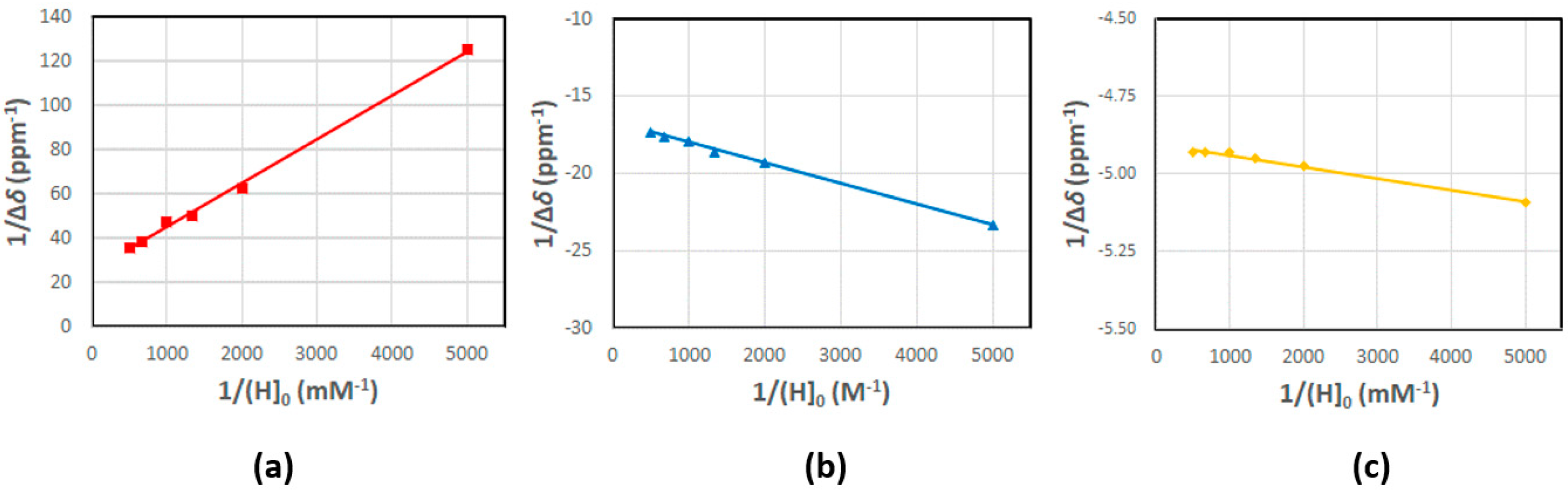
2.1. Determination of the Stability Constant of Inclusion Complexes with β-CD
2.2. In Vitro Cytotoxicity
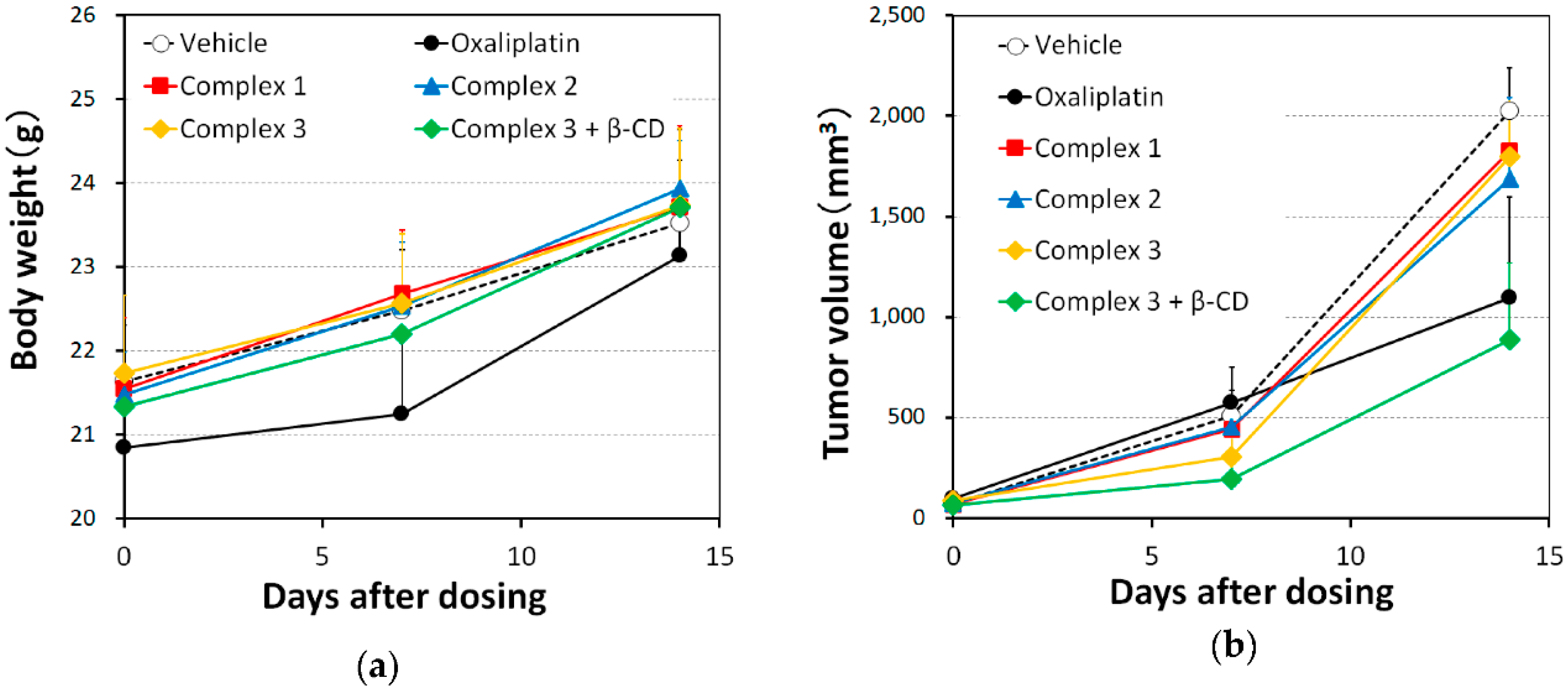
2.3. In Vivo Antitumor Efficacy
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Determination of Stability Constant
4.3. In Vitro Cytotoxicity Study
4.4. In Vivo Mouse Homografts
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rosenberg, B.; VanCamp, L.; Trosko, J.E.; Mansour, V.H. Platinum compounds: A new class of potent antitumour agents. Nature 1969, 222, 385–386. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, B.; VanCamp, L. The successful regression of large solid sarcoma 180 tumors by platinum compounds. Cancer Res. 1970, 30, 1799–1802. [Google Scholar]
- Gottlieb, J.A.; Drewinko, B. Review of the current clinical status of platinum coordination complexes in cancer chemotherapy. Cancer Chemother. Rep. 1975, 59, 621–628. [Google Scholar] [PubMed]
- Calvert, A.H.; Harland, S.J.; Newell, D.R.; Siddik, Z.H.; Jones, A.C.; McElwain, T.J.; Raju, S.; Wiltshaw, E.; Smith, I.E.; Baker, J.M.; et al. Early clinical studies with cis-diammine-1,1-cyclobutane dicarboxylate platinum II. Cancer Chemother. Pharmacol. 1982, 9, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Sharma, H.; Thatcher, N.; Baer, J.; Zaki, A.; Smith, A.; McAucliffe, C.A.; Crowther, D.; Owens, S.; Fox, B.W. Blood clearance of radioactively labelled cis-diammine 1,1-cyclobutane dicarboxylate platinum(II) (CBDCA) in cancer patients. Cancer Chemother. Pharmacol. 1983, 11, 5–7. [Google Scholar] [CrossRef]
- Tashiro, T.; Kawada, Y.; Sakurai, Y.; Kidani, Y. Antitumor activity of a new platinum complex, oxalato (trans-l-1,2-diaminocyclohexane)platinum(II): New experimental data. Biomed. Pharmacother. 1989, 43, 251–260. [Google Scholar] [CrossRef]
- Cvitkovic, E. Ongoing and unsaid on oxaliplatin: The hope. Br. J. Cancer 1998, 77 (Suppl. 4), 8–11. [Google Scholar] [CrossRef]
- Seeber, S.; Osieka, R.; Schmidt, C.G.; Achterrath, W.; Crooke, S.T. In vivo resistance towards anthracyclines, etoposide, and cis-diamminedichloroplatinum(II). Cancer Res. 1982, 42, 4719–4725. [Google Scholar]
- Eastman, A.; Illenye, S. Murine leukemia L1210 cell lines with different patterns of resistance to platinum coordination complexes. Cancer Treat. Rep. 1984, 68, 1189–1190. [Google Scholar]
- Eastman, A.; Bresnick, E. Studies on the resistance of a murine leukemia L1210 cell line cis-diamminedichloroplatinum(II). Biochem. Pharmacol. 1981, 30, 2721–2723. [Google Scholar] [CrossRef]
- Komeda, S.; Ohishi, H.; Yamane, H.; Harikawa, M.; Sakaguchi, K.-I.; Chikuma, M. An NMR study and crystal structure of [{cis-Pt(NH3)2(9EtG-kN7)2(m-pz)][NO3]3 (9EtG = 9-ethylguanine) as a model compound for the 1,2-intrastrand GG crosslink. J. Chem. Soc. Dalton Trans. 1999, 17, 2959–2962. [Google Scholar] [CrossRef]
- Komeda, S.; Lutz, M.; Spek, A.L.; Chikuma, M.; Reedijk, J. New antitumor-active azole-bridged dinuclear platinum(II) complexes: Synthesis, characterization, crystal structures, and cytotoxic studies. Inorg. Chem. 2000, 39, 4230–4236. [Google Scholar] [CrossRef] [PubMed]
- Komeda, S.; Lin, Y.L.; Chikuma, M. A Tetrazolato-Bridged Dinuclear Platinum(II) Complex Exhibits Markedly High in vivo Antitumor Activity against Pancreatic Cancer. ChemMedChem 2011, 6, 987–990. [Google Scholar] [CrossRef] [PubMed]
- Farrell, N.; Ha, T.T.; Souchard, J.P.; Wimmer, F.L.; Cros, S.; Johnson, N.P. Cytostatic trans-platinum(II) complexes. J. Med. Chem. 1989, 32, 2240–2241. [Google Scholar] [CrossRef] [PubMed]
- Farrell, N.; Qu, Y. Chemistry of Bis(Platinum) Complexes–Formation of Trans Derivatives from Tetraamine Complexes. Inorg. Chem. 1989, 28, 3416–3420. [Google Scholar] [CrossRef]
- Farrell, N.; Kiley, D.M.; Schmidt, W.; Hacker, M.P. Chemical-Properties and Antitumor-Activity of Complexes of Platinum Containing Substituted Sulfoxides [PtCl(R′R″SO)(Diamine)]NO3. Chirality and Leaving-Group Ability of Sulfoxide Affecting Biological Activity. Inorg. Chem. 1990, 29, 397–403. [Google Scholar] [CrossRef]
- Farrell, N.; Qu, Y.; Feng, L.; Van Houten, B. Comparison of chemical reactivity, cytotoxicity, interstrand cross-linking and DNA sequence specificity of bis(platinum) complexes containing monodentate or bidentate coordination spheres with their monomeric analogues. Biochemistry 1990, 29, 9522–9531. [Google Scholar] [CrossRef]
- Farrell, N.; Qu, Y.; Hacker, M.P. Cytotoxicity and antitumor activity of bis(platinum) complexes. A novel class of platinum complexes active in cell lines resistant to both cisplatin and 1,2-diaminocyclohexane complexes. J. Med. Chem. 1990, 33, 2179–2184. [Google Scholar] [CrossRef]
- Yang, D.; van Boom, S.S.G.E.; Reedijk, J.; van Boom, J.H.; Wang, A.H.J. Structure and isomerization of an intrastrand cisplatin-cross-linked octamer DNA duplex by NMR analysis. Biochemistry 1995, 34, 12912–12920. [Google Scholar] [CrossRef]
- van Boom, S.S.; Yang, D.; Reedijk, J.; van der Marel, G.A.; Wang, A.H. Structural effect of intra-strand cisplatin-crosslink on palindromic DNA sequences. J. Biomol. Struct. Dyn. 1996, 13, 989–998. [Google Scholar] [CrossRef]
- Shellard, S.A.; Fichtinger-Schepman, A.M.; Lazo, J.S.; Hill, B.T. Evidence of differential cisplatin-DNA adduct formation, removal and tolerance of DNA damage in three human lung carcinoma cell lines. Anticancer Drugs 1993, 4, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Takahara, P.M.; Rosenzweig, A.C.; Frederick, C.A.; Lippard, S.J. Crystal structure of double-stranded DNA containing the major adduct of the anticancer drug cisplatin. Nature 1995, 377, 649–652. [Google Scholar] [CrossRef] [PubMed]
- Coste, F.; Malinge, J.M.; Serre, L.; Shepard, W.; Roth, M.; Leng, M.; Zelwer, C. Crystal structure of a double-stranded DNA containing a cisplatin interstrand cross-link at 1.63 A resolution: Hydration at the platinated site. Nucleic Acids Res. 1999, 27, 1837–1846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamieson, E.R.; Lippard, S.J. Structure, Recognition, and Processing of Cisplatin-DNA Adducts. Chem. Rev. 1999, 99, 2467–2498. [Google Scholar] [CrossRef] [PubMed]
- Teletchea, S.; Komeda, S.; Teuben, J.M.; Elizondo-Riojas, M.A.; Reedijk, J.; Kozelka, J. A pyrazolato-bridged dinuclear platinum(II) complex induces only minor distortions upon DNA-binding. Chemistry 2006, 12, 3741–3753. [Google Scholar] [CrossRef]
- Magistrato, A.; Ruggerone, P.; Spiegel, K.; Carloni, P.; Reedijk, J. Binding of novel azole-bridged dinuclear platinum(II) anticancer drugs to DNA: Insights from hybrid QM/MM molecular dynamics simulations. J. Phys. Chem. B 2006, 110, 3604–3613. [Google Scholar] [CrossRef]
- Mlcouskova, J.; Kasparkova, J.; Suchankova, T.; Komeda, S.; Brabec, V. DNA conformation and repair of polymeric natural DNA damaged by antitumor azolato-bridged dinuclear Pt(II) complex. J. Inorg. Biochem. 2012, 114, 15–23. [Google Scholar] [CrossRef]
- Mlcouskova, J.; Malina, J.; Novohradsky, V.; Kasparkova, J.; Komeda, S.; Brabec, V. Energetics, conformation, and recognition of DNA duplexes containing a major adduct of an anticancer azolato-bridged dinuclear Pt(II) complex. Biochim. Biophys. Acta 2012, 1820, 1502–1511. [Google Scholar] [CrossRef]
- Imai, R.; Komeda, S.; Shimura, M.; Tamura, S.; Matsuyama, S.; Nishimura, K.; Rogge, R.; Matsunaga, A.; Hiratani, I.; Takata, H.; et al. Chromatin folding and DNA replication inhibition mediated by a highly antitumor-active tetrazolato-bridged dinuclear platinum(II) complex. Sci. Rep. 2016, 6, 24712. [Google Scholar] [CrossRef]
- Uemura, M.; Yoshikawa, Y.; Yoshikawa, K.; Sato, T.; Mino, Y.; Chikuma, M.; Komeda, S. Second- and higher-order structural changes of DNA induced by antitumor-active tetrazolato-bridged dinuclear platinum(II) complexes with different types of 5-substituent. J. Inorg. Biochem. 2013, 127, 169–174. [Google Scholar] [CrossRef]
- Komeda, S.; Takayama, H.; Suzuki, T.; Odani, A.; Yamori, T.; Chikuma, M. Synthesis of antitumor azolato-bridged dinuclear platinum(II) complexes with in vivo antitumor efficacy and unique in vitro cytotoxicity profiles. Metallomics 2013, 5, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Uemura, M.; Suzuki, T.; Nishio, K.; Chikuma, M.; Komeda, S. An in vivo highly antitumor-active tetrazolato-bridged dinuclear platinum(II) complex largely circumvents in vitro cisplatin resistance: Two linkage isomers yield the same product upon reaction with 9-ethylguanine but exhibit different cytotoxic profiles. Metallomics 2012, 4, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Uemura, M.; Hoshiyama, M.; Furukawa, A.; Sato, T.; Higuchi, Y.; Komeda, S. Highly efficient uptake into cisplatin-resistant cells and the isomerization upon coordinative DNA binding of anticancer tetrazolato-bridged dinuclear platinum(II) complexes. Metallomics 2015, 7, 1488–1496. [Google Scholar] [CrossRef]
- Komeda, S.; Yoneyama, H.; Uemura, M.; Muramatsu, A.; Okamoto, N.; Konishi, H.; Takahashi, H.; Takagi, A.; Fukuda, W.; Imanaka, T.; et al. Specific Conformational Change in Giant DNA Caused by Anticancer Tetrazolato-Bridged Dinuclear Platinum(II) Complexes: Middle-Length Alkyl Substituents Exhibit Minimum Effect. Inorg. Chem. 2017, 56, 802–811. [Google Scholar] [CrossRef] [PubMed]
- Komeda, S.; Yoneyama, H.; Uemura, M.; Tsuchiya, T.; Hoshiyama, M.; Sakazaki, T.; Hiramoto, K.; Harusawa, S. Synthesis and Structure–Activity Relationships of Tetrazolato-Bridged Dinuclear Platinum(II) Complexes: A Small Modification at Tetrazole C5 Markedly Influences the In Vivo Antitumor Efficacy. J. Inorg. Biochem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Hanna, M.W.; Ashbaugh, A.L. Nuclear Magnetic Resonance Study of Molecular Complexes of 7,7,8,8-Tetracyanoquinodimethane and Aromatic Donors1, 2. J. Phys. Chem. 1964, 68, 811–816. [Google Scholar] [CrossRef]
- Benesi, H.A.; Hildebrand, J. A spectrophotometric investigation of the interaction of iodine with aromatic hydrocarbons. J. Am. Chem. Soc. 1949, 71, 2703–2707. [Google Scholar] [CrossRef]
- Mathur, R.; Becker, E.D.; Bradley, R.B.; Li, N.C. Proton magnetic resonance studies of hydrogen bonding of benzenethiol with several hydrogen acceptors. J. Phys. Chem. 1963, 67, 2190–2194. [Google Scholar] [CrossRef]
- Eftink, M.R.; Andy, M.L.; Bystrom, K.; Perlmutter, H.D.; Kristol, D.S. Cyclodextrin inclusion complexes: Studies of the variation in the size of alicyclic guests. J. Am. Chem. Soc. 1989, 111, 6765–6772. [Google Scholar] [CrossRef]
- Leong, N.J.; Prankerd, R.J.; Shackleford, D.M.; Mcintosh, M.P. The Effect of Intravenous Sulfobutylether7-β-Cyclodextrin on the Pharmacokinetics of a Series of Adamantane-Containing Compounds. J. Pharm. Sci. 2015, 104, 1492–1498. [Google Scholar] [CrossRef]
- Carlstedt, J.; Bilalov, A.; Krivtsova, E.; Olsson, U.; Lindman, B.R. Cyclodextrin–surfactant coassembly depends on the cyclodextrin ability to crystallize. Langmuir 2012, 28, 2387–2394. [Google Scholar] [CrossRef] [PubMed]
- Trofymchuk, I.; Belyakova, L.; Grebenyuk, A. Study of complex formation between β-cyclodextrin and benzene. J. Incl. Phenom. Macrocycl. Chem. 2011, 69, 371–375. [Google Scholar] [CrossRef]
- Kahle, C.; Holzgrabe, U. Determination of binding constants of cyclodextrin inclusion complexes with amino acids and dipeptides by potentiometric titration. Chirality 2004, 16, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Jansook, P.; Ogawa, N.; Loftsson, T. Cyclodextrins: Structure, physicochemical properties and pharmaceutical applications. Int. J. Pharm. 2018, 535, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Uemura, M.; Yoshikawa, Y.; Chikuma, M.; Komeda, S. A circular dichroism study uncovers a two-step interaction of antitumor azolato-bridged dinuclear platinum(II) complexes with calf thymus DNA. Metallomics 2012, 4, 641–644. [Google Scholar] [CrossRef]




{kind=link}
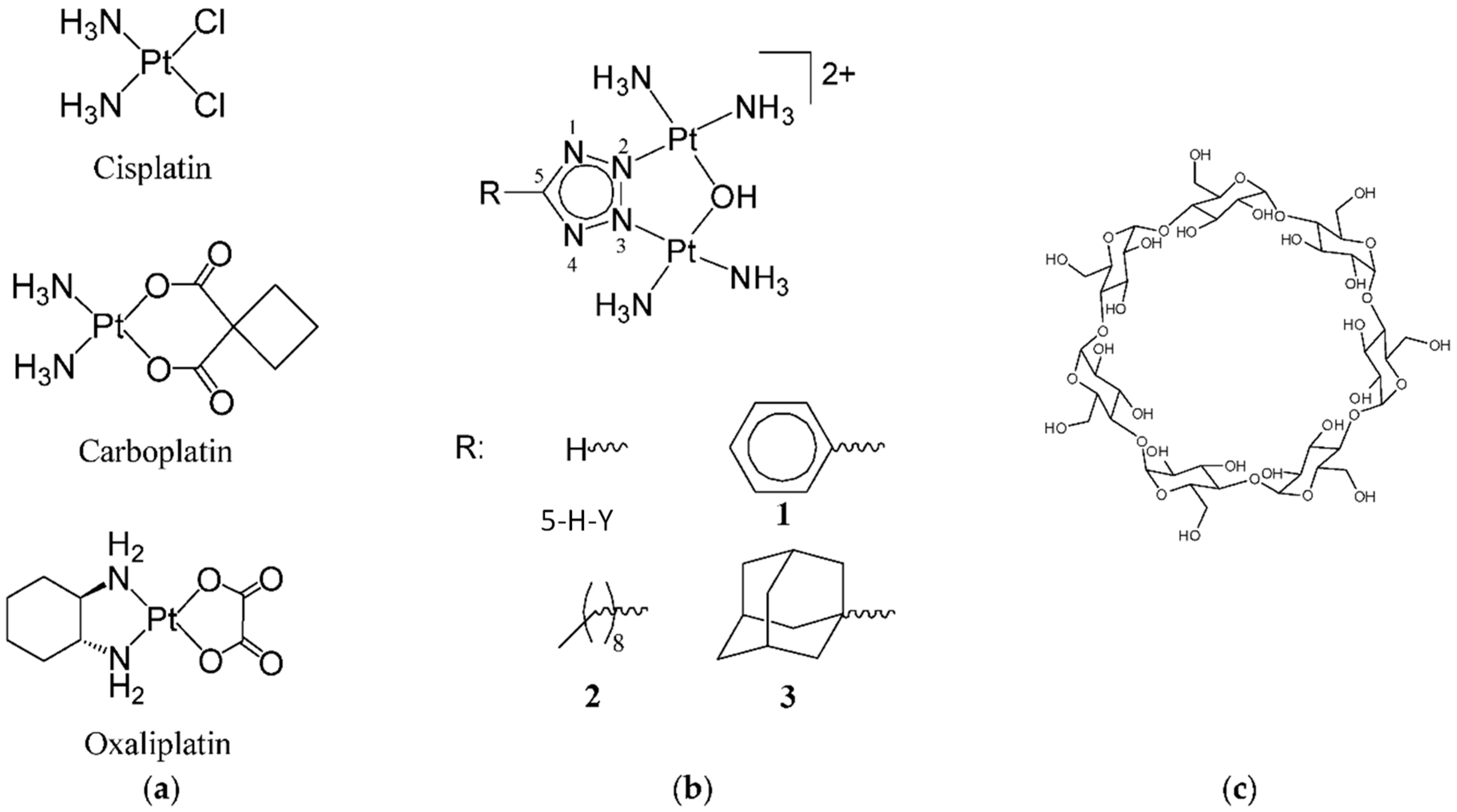{kind=link}
{kind=link}
{kind=link}
| Pt(II) Complex | Ks/M−1 |
|---|---|
| Oxaliplatin | n. d. a |
| 5-H-Y | n. d. a |
| 1 | (1.81 ± 1.28) × 103 |
| 2 | (1.30 ± 0.26) × 104 |
| 3 | (1.27 ± 0.03) × 105 |
| Pt(II) Complex | Mean IC50 ± SD/μM (n = 6) | ||
|---|---|---|---|
| −β-CD | +β-CD | +β-CD/−β-CD | |
| Oxaliplatin (24 h) a | 11 ± 3 | 28 ± 3 | 2.5 |
| Oxaliplatin (48 h) b | 5.7 ± 1.2 | 7.9 ± 0.3 | 1.4 |
| 5-H-Y (24 h) a | 0.59 ± 0.21 | 0.74 ± 0.03 | 1.3 |
| 5-H-Y (48 h) b | 0.23 ± 0.09 | 0.25 ± 0.02 | 1.1 |
| 1 (24 h) a | >360 | >270 | - |
| 1 (48 h) b | 43 ± 1 | >270 | >6.3 |
| 2 (24 h) a | 6.3 ± 0.7 | 5.6 ± 0.5 | 0.9 |
| 2 (48 h) b | 4.9 ± 0.6 | 7.4 ± 0.2 | 1.5 |
| 3 (24 h) a | 113 ± 8 | >270 | >2.4 |
| 3 (48 h) b | 109 ± 10 | >270 | >2.5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Komeda, S.; Uemura, M.; Yoneyama, H.; Harusawa, S.; Hiramoto, K. In Vitro Cytotoxicity and In Vivo Antitumor Efficacy of Tetrazolato-Bridged Dinuclear Platinum(II) Complexes with a Bulky Substituent at Tetrazole C5. Inorganics 2019, 7, 5. https://doi.org/10.3390/inorganics7010005
Komeda S, Uemura M, Yoneyama H, Harusawa S, Hiramoto K. In Vitro Cytotoxicity and In Vivo Antitumor Efficacy of Tetrazolato-Bridged Dinuclear Platinum(II) Complexes with a Bulky Substituent at Tetrazole C5. Inorganics. 2019; 7(1):5. https://doi.org/10.3390/inorganics7010005
Chicago/Turabian StyleKomeda, Seiji, Masako Uemura, Hiroki Yoneyama, Shinya Harusawa, and Keiichi Hiramoto. 2019. "In Vitro Cytotoxicity and In Vivo Antitumor Efficacy of Tetrazolato-Bridged Dinuclear Platinum(II) Complexes with a Bulky Substituent at Tetrazole C5" Inorganics 7, no. 1: 5. https://doi.org/10.3390/inorganics7010005
APA StyleKomeda, S., Uemura, M., Yoneyama, H., Harusawa, S., & Hiramoto, K. (2019). In Vitro Cytotoxicity and In Vivo Antitumor Efficacy of Tetrazolato-Bridged Dinuclear Platinum(II) Complexes with a Bulky Substituent at Tetrazole C5. Inorganics, 7(1), 5. https://doi.org/10.3390/inorganics7010005