1. Introduction
The bonding between group 13 (E) and group 15 (Pn) elements formulated as R
2E–PnR
2 have attracted much attention due to their relationship, including the vacant p orbital on E and the lone-pair electrons on Pn. Aminoboranes (R
2B–NR
2), for example, have an enormous number of researches describing their
−B=N
+ polar double-bond character [
1]. To contrast, the heavier analogues, λ
3,λ
3-phosphanylalumanes (R
2Al–PR
2), decrease the π-type interaction between the E and Pn atoms compared to those of aminoboranes [
2,
3,
4] and phosphanylboranes [
5,
6] due to the longer E–Pn σ-bond. These aspects can afford a characteristic reactivity reflecting the adjacent but separated Lewis acids and bases, similar to the synergetic interactions of the Lewis acid and Lewis base of frustrated Lewis pairs (FLPs) toward small-molecules [
7,
8,
9,
10].
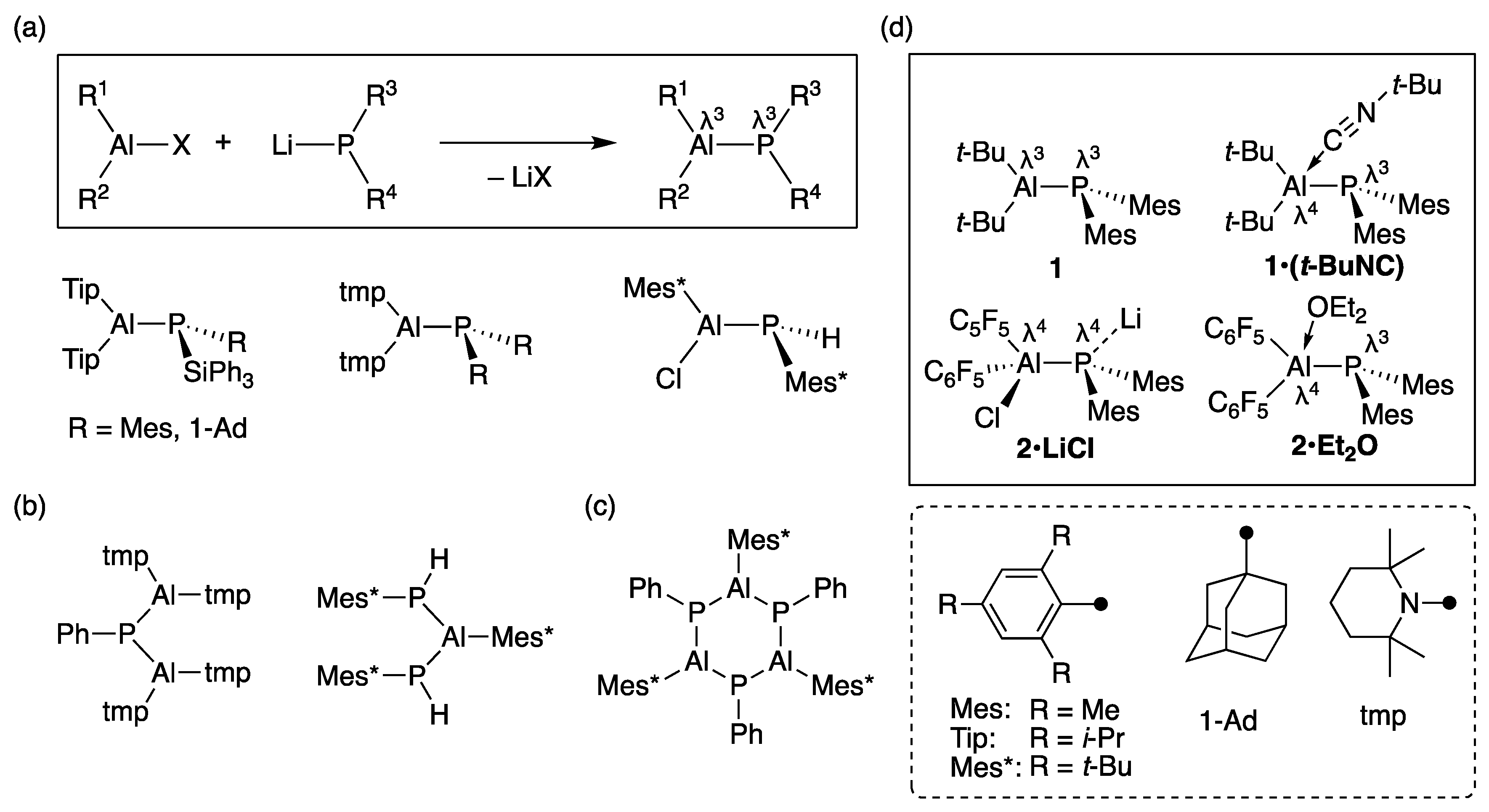
The physical properties and reactivity of λ
3,λ
3-phosphanylalumanes, however, hardly have been clarified so far. The main reason is that there are few synthetic examples due to the difficulty of protecting the vacant 3p(Al) orbital [
11,
12,
13]. Most of λ
3,λ
3-phosphanylalumanes have been synthesized by the salt elimination reactions reported by Power [
14], Nöth and Paine [
15,
16], and our group [
17] (
Figure 1a). Multi-nuclear phosphanylalumane derivatives are also known. When there are reactive substituents (H or a halogen atom) on P or Al moiety, another aluminum or phosphorus reagents react to give a λ
3,λ
3-diphosphanyl-λ
3-alumane [
16] and λ
3-phosphanyl-λ
3,λ
3-dialumane [
17], respectively (
Figure 1b). A cyclic λ
3,λ
3-phosphanylalumane (Mes*Al–PPh)
3 is synthesized by a dehydrogenation reaction, which is thought to be the trimerization of an Al–P double-bond compound (
Figure 1c) [
18]. Regarding each case, at least one substituent on an Al or P atom is a heteroatom (non-carbon atom) substituent, which will affect the nature of the Al–P bond. The Lewis acidity of aluminum, for example, is greatly impaired by mesomeric effect between the vacant 3p(Al) orbital and the one pair on aluminum-bound substituents [
19,
20]. Electropositive substituents (SiPh
3, SiMe
3, and SnMe
3) attached to the P atom also may alter the nature of the Al–P bond. Therefore, the synthesis and elucidation of the reactivity of unperturbed, all-carbon-substituted λ
3,λ
3-phosphanylalumanes might be a new challenge.
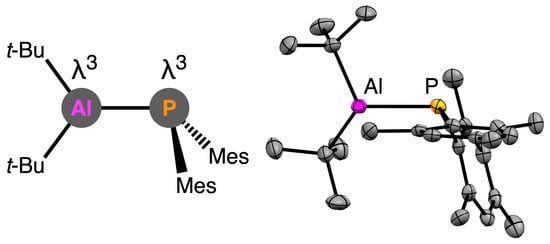
Here, we report the synthesis of λ
3,λ
3-phosphanylalumanes which are substituted fully by carbon protecting groups on the aluminum and phosphorus atoms. To understand the substituent effect on the aluminum, we chose
t-Bu and C
6F
5 groups (
Figure 1d).
2. Results and Discussion
The reaction of (
t-Bu)
2AlBr [
21] with Mes
2PLi [
22] afforded a λ
3,λ
3-phosphanylalumane
1 quantitatively, judged by
31P NMR spectroscopy (
Scheme 1a). Conversely, the use of (C
6F
5)
2AlCl·0.5(toluene) [
23] as an aluminum source in a hexane solution afforded an LiCl complex, λ
4,λ
4-phosphanylalumane
2·LiCl, in a 54% yield (
Scheme 1b). We expected that the use of (C
6F
5)
2AlBr [
23] would render a more effective salt elimination but the corresponding LiBr complex (
2·LiBr) was detected by NMR spectroscopy. No salt elimination in the case of the C
6F
5 substituent suggested the higher Lewis acidity of the aluminum atom on (C
6F
5)
2Al-moiety as compared with that on the
t-Bu
2Al moiety. The attempts of LiCl elimination from
2·LiCl by heating or the addition of silver tetrafluoroborate were not successful. Although the reaction of (C
6F
5)
2AlCl·0.5(toluene) in an Et
2O solution, rather than hexane afforded a complicated mixture, recrystallization from which gave an etherate
2·Et2O in a 38% yield. The addition of Et
2O to
2·LiCl led to the formation of compound
2·Et2O, which was evidenced in the mixture of (C
6F
5)
2AlCl·0.5(toluene) with Mes
2PLi in Et
2O. These results suggest that
2·LiCl was formed at the initial stage in both conditions and the partial decomposition occurred via Et
2O in addition to the formation of
2·Et2O.
Subsequently, the coordination of Lewis bases was examined to explore the Lewis acidity of λ
3,λ
3-phosphanylalumane
1. Although
1 did not react with carbon monoxide upon heating to 70 °C, the reaction of
1 with
t-butyl isocyanide at room temperature gave the Lewis base-coordinated λ
3,λ
4-complex
1·(t-BuNC) quantitatively (
Scheme 2). Heating of a C
6D
6 solution of
1·(t-BuNC) with the aim of further isomerization and coupling of isocyanides [
24,
25,
26] produced a mixture containing a
1,
1·(t-BuNC), Mes
2PH, and Mes
2P–PMes
2 as judged by the
31P NMR spectrum. The recrystallization from the mixture afforded a trace amount of single crystals of bis(
t-butyl)aluminium cyanide tetramer
3 [
27], as determined by NMR measurements and X-ray crystallography. The elimination of the
t-Bu group from
t-BuNC to form diorganylaluminum cyanides (R
2AlCN)
n also has been observed in the reaction of the Al–Al bond compound with
t-BuNC [
24]. Heating at elevated temperatures promotes desorption of the (
t-Bu)
2(
t-BuNC)Al- and Mes
2P-moieties to give
3, Mes
2PH, and Mes
2P–PMes
2.
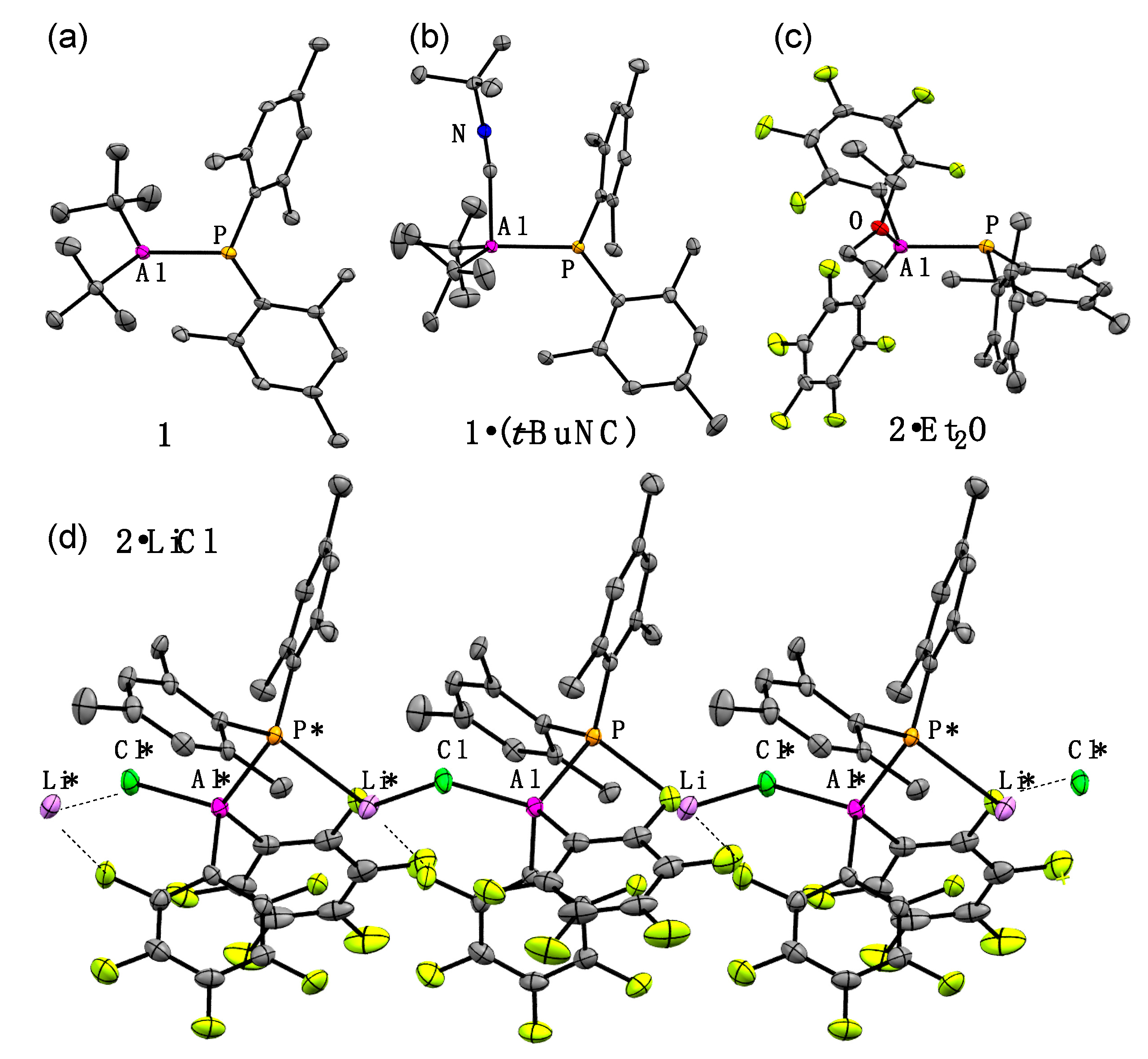
Structures of all new phosphanylalumane derivatives
1,
1·(t-BuNC),
2·LiCl, and
2·Et2O were determined finally by X-ray crystallography, and the results showed good agreement with the optimized structures calculated at the B3LYP-D3/6-31G(d) level. These structures are depicted in
Figure 2 and the structural features of these compounds are summarized in
Table 1. Particularly,
2·LiCl had a one-dimensional infinite chain structure with continuous [–Cl–Al–P–Li–] parts. One Li atom of
2·LiCl was coordinated by P and two F atoms of the C
6F
5 groups. Concerning complex
2·LiCl, its anion part (
2Cl−) without Li
+, also was used for the calculations. There was almost no structural difference between the optimized structures,
2·LiCl and
2Cl−. Lewis base-free λ
3,λ
3-phosphanylalumane
2 could not be obtained experimentally, but the optimized structural parameters of
2 also are described for the comparison.
The sum of the bond angles around the P atom (ΣP) of these compounds are 325–328°, indicating the pyramidalized structures of the phosphorus moieties despite the different coordination. Conversely, the sum of the bond angles around the Al atom (ΣAl) reflected the environment on the coordination of Al atoms. λ3,λ3-Phosphanylalumane 1 retained the planar structure with the Al center (ΣAl = 357°). The λ4-Al moieties of 1·(t-BuNC) and 2·Et2O were pyramidalized slightly (ΣAl = 354° and 347°, respectively), reflecting the weak coordination of isocyanide and diethyl ether to the λ3-Al atoms. To contrast, the λ4-Al center of 2·LiCl was pyramidalized extremely (ΣAl = 319°), suggesting its strong Al–Cl covalent bond.
The viewpoint of the Al–P bond lengths brought us the effect of electronic situations on the Al atom. The Al–P bond lengths of
1 (2.343(1) and 2.347(1) Å for two independent molecules) were comparable to that of the reported λ
3,λ
3-phosphanylalumanes (Tip
2Al–P(SiPh
3)Mes: 2.342(2) Å) [
14] or the Al
3P
3 six-membered-ring compound ((Mes*Al–PPh)
3: 2.323(3)–2.336(3) Å) [
18]. The coordination of
t-BuNC to the λ
3-Al center resulted in the large elongation of the Al–P bond (
1·(t-BuNC): 2.4120(6) Å). Alternately, the Al–P bond lengths of C
6F
5-substituted λ
4-Al-derivatives,
2·LiCl (2.348(1) Å) and
2·Et2O (2.359(2) Å), were close to those of λ
3,λ
3-phosphanylalumane
1 and much shorter than those of
1·(t-BuNC) and the reported phosphanyl-λ
4-alumanes, ((
t-Bu)
2Al–P(SiPh
3)Tip·Et
2O: 2.416(3) Å) [
12] and (Bbp(Br)Al–P(H)Mes·LiBr(Et
2O)
2: 2.4055(7) Å) [
13]. Considering the observations above, the C
6F
5-substituted λ
3-Al-derivative
2 is expected to have a quite short Al–P bond. However, the calculated Al–P bond length of
2 almost was similar to or slightly shorter than that of
1 (
1: 2.348 Å vs.
2: 2.322 Å). These results indicated that the C
6F
5 groups greatly affected the Al–P bond regardless of the coordination number to the Al center, the reasons of which will be discussed later.
The observed structural features reflect the NMR spectroscopic results. The
31P NMR spectra for λ
3,λ
3-phosphanylalumane
1 displayed a resonance at
δ = −111.7 ppm that is the most upfield among those of the synthesized phosphanylalumane derivatives (
1·(t-BuNC): −96.7 ppm,
2·Et2O: −104.8 ppm, and
2·LiCl: −89.7 ppm). Additionally, these
31P NMR signals are in much lower magnetic fields than those of tmp
2Al–P(SiMe
3)
2 and tmp
2Al–P(SnMe
3)
2 (
δ = −238 and −282 ppm, respectively) [
16] having an electron-donating substituent on λ
3-P atoms, while being more upfield than that of tmp
2Al–PPh
2 (
δ = −42.9 ppm) [
15]. These values indicate a sufficiently electron-rich environment for the λ
3-P atom with carbon protecting groups in
1. Conversely, the
27Al NMR spectra for
1 showed a single broad resonance at
δ = 261 ppm. This value is upfield as compared with those of the reported λ
3,λ
3-phosphanylalumanes, i.e., tmp
2Al–PPh
2 (
δ = 110 ppm) [
15] and tmp
2Al–P(SiMe
3)
2 (
δ = 78 ppm) [
16].
The photophysical behavior of λ
3,λ
3-phosphanylalumane
1 has been investigated for the first time to obtain further insight for the character of a λ
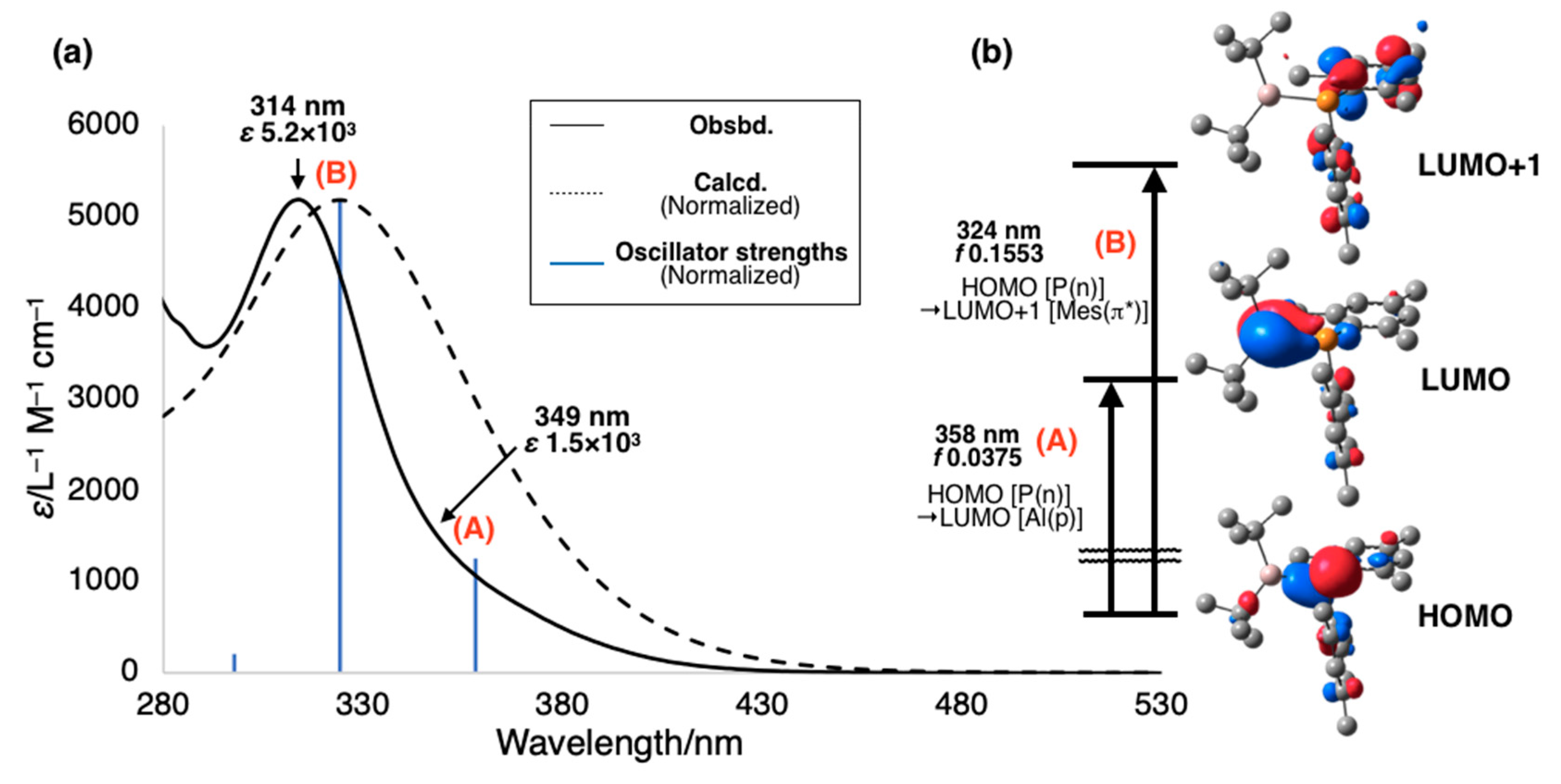
3-Al moiety. UV–vis absorption spectrum of
1 is shown in
Figure 3 with the simulated spectrum of
1 derived from time-dependent density functional theory (TD-DFT) calculations at the B3LYP-D3/6-311+G(d,p)//B3LYP-D3/6-31G(d) level of theory, which reproduced the spectrum observed experimentally. The shoulder absorption at ~350 nm should be assigned to the n-p transition from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO), which corresponds to the electron transition of a lone pair of P to a vacant p orbital of Al (
Figure 3a; (A)). The absorption maximum at 314 nm (
ε 5.2 × 10
3) was assigned to the n-π* transition from the HOMO to the LUMO+1, which corresponds to the electron transition of a lone pair of P to π* orbitals of the Mes
2P moiety (
Figure 3a; (B)). Combining the experimental and theoretical results, the vacant p orbital of λ
3,λ
3-phosphanylalumane
1 greatly contributes to the absorption behavior. Particularly, the λ
3,λ
3-phosphanylalumanes with the unperturbed vacant p orbital of Al exhibit a yellow color (
1 and Tip
2Al–P(SiPh
3)Mes (no UV–vis absorption spectrum) [
14]), whereas the λ
3,λ
3-phosphanylalumanes with the filled p orbital of Al by the Al-bound substituents are colorless (tmp
2Al–P(SiMe
3)
2 [
16] and Mes*(Cl)Al–P(H)Mes* [
17]).
The consideration of the Al–P bonds for the newly synthesized phosphanylalumane derivatives was further deepened by the natural bond orbital (NBO) analysis, as summarized in
Table 2. The π-type NBOs involving the p(Al) orbitals were not found in
1 and
2. The natural population analysis (NPA) charge on Al of
1 (1.760) is the most positive among these phosphanylalumane derivatives, indicating the substantial retention of the vacant 3p(Al) orbital. The NBO corresponding to the σ(Al–P) bond in
1 consisted mainly of 3s(Al) and 3p(P) orbitals and was polarized toward an λ
3-P moiety. The coordination of
t-BuNC to λ
3-Al increases the contribution of the hybridization of Al in σ(Al–P) bonds (
1: 18% Al vs.
1·(t-BuNC): 21% Al) and decreases the
s-character of Al (
1: Al sp
1.7 vs.
1·(t-BuNC): Al sp
2.6), as well as the experimentally observed elongation of an Al–P bond. Substitution of
t-Bu groups with C
6F
5 groups resulted in the higher
s-character of the Al atom in
2 than that of
1. These are due to the use of more
p-character in the bonding to the aryl group than that to the alkyl group and of the large inductive effect of the F atoms. This result was supported by the large Al contribution of
2,
2Cl−, and
2·LiCl compared to
1 in σ(Al–P) bonds (
1: 18% Al vs.
2,
2Cl−, and
2·LiCl: 23–26% Al) and the large
s-character of Al (
1: Al sp
1.7 vs.
2,
2Cl–, and
2·LiCl: Al sp
0.6−0.9), as well as the short Al–P bond lengths independent of the coordination-number change in the Al moieties of C
6F
5-derivatives. Additionally, compound
2 showed a WBI value greater than that of
1 (
1: 0.6595 vs.
2: 0.7781) corresponding to the short Al–P bonds of
2·LiCl and
2·Et2O, but the NPA charge of the λ
3-Al moiety of
2 was less than that of
1 (
1: 1.760 vs.
2: 1.537). These results of the theoretical calculations suggest that compound
1 is a suitable λ
3,λ
3-phosphanylalumane for reactivity studies.
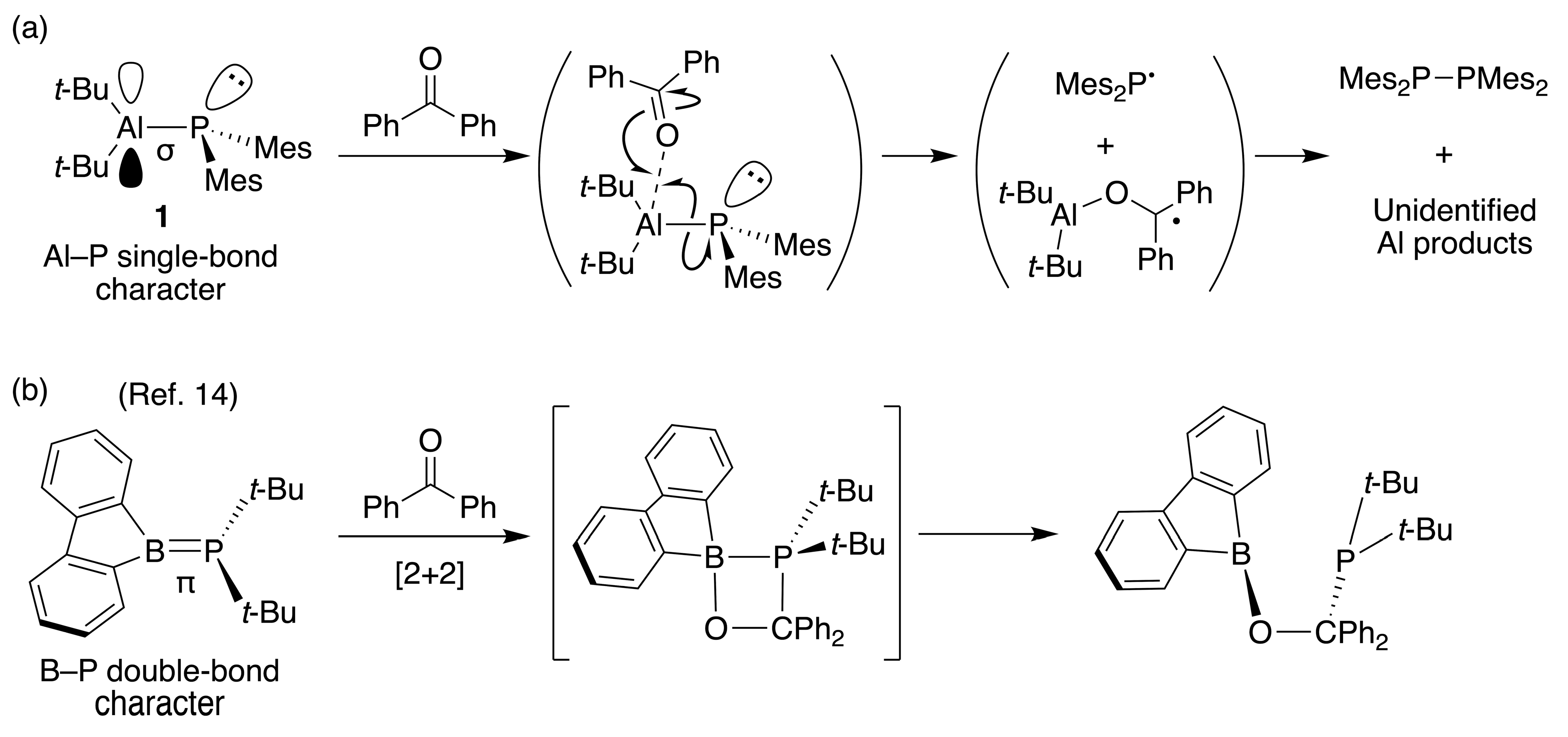
The singularity of the Al–P single-bond in
1 among the bonds between the group 13 and group 15 elements also was experimentally investigated. Treatment of
1 with benzophenone at room temperature rapidly gave Mes
2P–PMes
2 quantitatively, as judged by the
31P NMR spectrum (
Scheme 3a), but the corresponding Al moiety was not identified. This result implied that the coordination of benzophenone to Al promoted homolysis to produce an Mes
2P radical and the corresponding radical containing an Al moiety, respectively. It was difficult to clear the radical mechanism by using (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) because compound
1 reacted with TEMPO to give a mixture containing Mes
2P–PMes
2 and Mes
2PH. Conversely, a B–P bond compound with λ
3-B and P centers, phosphaboradibenzofulvene, gave a [2+2]-cycloaddition product when treated with benzophenone (
Scheme 3b) [
28]. Contrary to the large contribution of the B–P π-bond character in a phosphaboradibenzofulvene, these results suggested the Al–P single-bond character and the clear separation of the Lewis acid and Lewis base moieties due to the slight interaction between Al and P in
1.
3. Materials and Methods
3.1. General
All the manipulations were performed under a dry argon atmosphere using standard Schlenk techniques or gloveboxes. Solvents were purified by the Ultimate Solvent System, Glass Contour Company (Nikko Hansen and Co., Ltd., Osaka, Japan) (THF, toluene, and
n-hexane) [
29] or by trap-to-trap distillation from a potassium mirror prior to use (C
6D
6 and
n-hexane for UV–vis spectrum measurements). NMR spectra were measured on a JMM-ECA600 (JEOL Ltd., Tokyo, Japan) (
1H: 600 MHz,
7Li: 233 Hz,
13C: 151 MHz,
19F: 565 MHz,
27Al: 156 MHz,
31P: 243 MHz) in the Joint Usage/Research Center (JURC, Institute for Chemical Research, Kyoto University) or on a AL-300 spectrometer (JEOL Ltd., Tokyo, Japan) (
1H: 300 MHz,
13C: 75 MHz,
19F: 282 MHz,
31P: 121 MHz). Regarding the
1H NMR spectra, signals arising from residual partially hydrogenated C
6D
5H (7.15 ppm for
1H) were used as references. C
6D
6 (128.0 ppm for
13C) and Al(NO
3)
3 in D
2O (0 ppm for
27Al) were used as references.
1H and
13C NMR signals were assigned with the aid of the
1H–
1H COSY,
1H–
13C HSQC, and
1H–
13C HMBC spectra. Melting points were determined on a Yanaco micro melting point apparatus and uncorrected. Elemental analyses were carried out at the Microanalytical Laboratory, Institute for Chemical Research, Kyoto University.
AlCl
3 and AlBr
3 (Sigma-Aldrich Co., LLC., Tokyo, Japan), to prepare aluminum reagents, were purified by sublimation prior to use. The aluminum monohalides ((
t-Bu)
2AlBr [
21], and (C
6F
5)
2AlX (X = Cl, Br) or (C
6F
5)
2AlCl·0.5(toluene) [
23]) were prepared in accordance with the reported procedures. Mes
2PLi was prepared by the reaction of Mes
2PH with
n-butyllithium in
n-hexane, and the yellow solid was washed with
n-hexane carefully to remove the residual Mes
2PH [
22]. Details of theoretical calculations and XRD data are given in the
Supplementary Materials.
3.2. Synthesis of 1
To a yellow suspension of Mes2PLi (277.2 mg, 1.00 mmol) in n-hexane (3 mL), (t-Bu)2AlBr (219.5 mg, 0.998 mmol, 1.0 eq.) was slowly added at room temperature. The reaction mixture was stirred for 12 h. The almost quantitative formation of λ3,λ3-phosphanylalumane was observed, as judged by 31P NMR spectroscopy. The insoluble materials were removed by filtration through a Celite® pad using n-hexane as an eluent. The filtrate was concentrated and stored at −35 °C, presenting λ3,λ3-phosphanylalumane (1) as yellow crystals (isolated yield: 214.0 mg, 0.521 mmol, 52%). 1: Yellow crystals, mp. 54–55 °C. 1H NMR (600 MHz, C6D6, 298 K): δ = 1.09 (d, 18H, 4JHP = 1.2 Hz, C(CH3)3), 2.09 (s, 6H, Mes p-CH3), 2.36 (s, 12H, Mes o-CH3), 6.74 (m, 4H, Mes m-CH) ppm. 13C{1H} NMR (151 MHz, C6D6, 298 K): δ = 20.9 (Mes p-CH3), 24.5 (d, 3JCP = 13.6 Hz, Mes o-CH3), 29.7 (d, 3JCP = 1.7 Hz, Al-C(CH3)3), 30.53 (s, AlCMe3), 129.62 (d, 3JCP = 3.0 Hz, Mes m-C), 133.79 (d, 2JCP = 16.6 Hz, Mes o-C), 136.13 (s, Mes p-C), 141.19 (d, 1JCP = 12.1 Hz, Mes ipso-C) ppm. 27Al NMR (156 MHz, C6D6, 298 K): δ = 261.3 (br.) ppm. 31P{1H} NMR (243 MHz, C6D6, 298 K): δ = −111.7 ppm. UV–vis (hexane): λ/nm = 314 (ε 5.2 × 103), 349 (ε 1.5 × 103). Anal. Calcd. for C26H40AlP: C, 76.06; H, 9.82. Found: C, 76.07; H, 10.10.
3.3. Synthesis of 1·(t-BuNC)
To a yellow solution of 1 (40.3 mg, 0.0982 mmol) in C6D6 (0.6 mL), a small excess amount of t-BuNC (9.6 mg, 0.115 mmol, 1.2 eq.) was slowly added at room temperature. The quantitative formation of 1·(t-BuNC) was observed, as judged by 31P NMR spectroscopy. The mixture was concentrated at room temperature presenting the orange crystals. The crystals were washed with hexane to afford 1·(t-BuNC) as light orange crystals (isolated yield: 16.4 mg, 0.0332 mmol, 34%). 1·(t-BuNC): Light orange crystals, mp. 97–99 °C. 1H NMR (600 MHz, C6D6, 298 K): δ = 0.64 (s, 9H, C≡N–C(CH3)3), 1.35 (d, 4JHP = 0.6 Hz, 18H, AlC(CH3)3), 2.14 (s, 6H, Mes p-CH3), 2.55 (s, 12H, Mes o-CH3), 6.80 (m, 4H, Mes m-H) ppm. 13C{1H} NMR (151 MHz, C6D6, 298 K): δ = 18.01 (d, 2JCP = 12.1 Hz, AlCMe3), 20.95 (s, Mes p-CH3), 25.47 (d, 3JCP = 10.6 Hz, Mes o-CH3), 28.40 (s, C≡N–C(CH3)3), 32.56 (d, 3JCP = 2.4 Hz, AlC(CH3)3), 57.72 (s, C≡NCMe3), 129.04 (d, 3JCP = 4.5 Hz, Mes m-C), 132.56 (d, 2JCP = 33.2 Hz, Al←C≡N-t-Bu), 134.66 (s, Mes p-C), 137.19 (d, 2JCP = 18.1 Hz, Mes o-C), 142.19 (d, 1JCP = 10.6 Hz, Mes ipso-C) ppm. 27Al NMR (156 MHz, C6D6, 298 K): δ = +154.7 (br.) ppm. 31P{1H} NMR (121 MHz, C6D6, 298 K): δ = −154 ppm. Anal. Calcd for C31H49AlNP: C, 75.42; H, 10.00; N, 2.84. Found: C, 75.02; H, 10.10; N, 2.83.
3.4. Synthesis of 2·LiCl
To a yellow suspension of Mes2PLi (25.4 mg, 0.0939 mmol) in n-hexane (2 mL), (C6F5)2AlCl·0.5(toluene) (47.0 mg, 0.0942 mmol) was added at room temperature. The reaction mixture was stirred until the yellow color diminished (for 12 h). Following removal of the solvent, the residue was washed with the n-hexane. The insoluble materials were filtered through a Celite® pad using toluene as an eluent. n-Hexane was added to the filtrate and the mixture was scratched to give the colorless precipitate. The precipitate was washed with n-hexane carefully to afford (C6F5)2Al–PMes2·LiCl complex (2·LiCl) as a colorless solid (34.3 mg, 0.0510 mmol, 54%). 2·LiCl: Colorless solid, mp. 94 °C (dec.). 1H NMR (600 MHz, C6D6, 298 K): δ = 1.96 (s, 6H, Mes p-CH3), 2.23 (s, 12H, Mes o-CH3), 6.58 (d, 4JHP = 2.4 Hz, 4H, Mes m-H) ppm. 7Li{1H} NMR (233 MHz, C6D6, 298K): δ = −4.73 ppm. 13C{1H} NMR (151 MHz, C6D6, 298K): δ = 20.70 (s, Mes p-CH3), 24.75 (d, 3JCP = 9.1 Hz, Mes o-CH3), 117.05 (br. t, 2JCF = ~49 Hz, C6F5 ipso-C), 133.21 (d, 2JCF = 9.1 Hz, Mes o-C), 137.09 (s, Mes p-C), 137.26 (dqd, 1JCF = 252 Hz, 2JCF = ~12 Hz, 3JCF = ~4.5 Hz, C6F5 m-C), 141.53 (dm, 1JCF = 252 Hz, C6F5 p-C), 143.13 (d, 1JCP = 10.6 Hz, Mes ipso-C), 149.94 (ddm, 1JFC = 224 Hz, 2JFC = 24 Hz, C6F5 o-C) ppm. 19F NMR (282 MHz, C6D6, 298 K): δ = −123.3 (m, 4F, C6F5 o-F), –155.4 (t, J = 19.7 Hz, 2F, C6F5 p-F), −161.2 (m, 4F, C6F5 m-F) ppm. 31P{1H} NMR (121 MHz, C6D6, 298 K): δ = −89.7 (br. s) ppm. No 27Al NMR signal was observed, even after long-time measurement. Due to the flame retardancy of fluorocarbons and the extremely high air-/moisture-sensitivity, satisfactory data of the elemental analysis could not be obtained. The purity of 2·LiCl was confirmed accordingly by the 19F and 31P{1H} NMR spectra.
3.5. Synthesis of 2·LiBr
To a yellow suspension of Mes2PLi (11.9 mg, 0.044 mmol) in n-hexane (2 mL), (C6F5)2AlBr (23.3 mg, 0.0441 mmol) was added at room temperature. The solution was stirred until the yellow color diminished (for 5 h). The precipitate was washed with the n-hexane, and the residue was filtered through a Celite® pad using toluene as an eluent. Following removal of the solvent from the filtrate, the resulting materials (14.5 mg) was checked by NMR spectroscopy, suggesting the formation of (C6F5)2Al–PMes2·LiBr complex (2·LiBr). Further purification and isolation did not succeed. 2·LiBr: A colorless solid of crude materials. 1H NMR (300 MHz, C6D6, 298 K): δ = 1.96 (s, 6H, Mes p-CH3), 2.22 (s, 12H, Mes o-CH3), 6.59 (m, 4H, Mes m-H) ppm. 7Li{1H} NMR (117 MHz, C6D6, 298 K): δ = −4.38 ppm. 19F NMR (282 MHz, C6D6, 298 K): δ = −122.9 (m, 4F, C6F5 o-F), −152.9 (t, J = 19.7 Hz, 2F, C6F5 p-F), −160.8 (m, 4F, C6F5 m-F) ppm. 31P{1H} NMR (121 MHz, C6D6, r.t.): δ = −89.3 (s) ppm.
3.6. Synthesis of 2·Et2O
To a yellow suspension of Mes2PLi (24.2 mg, 0.0485 mmol) in Et2O (2 mL), (C6F5)2AlCl·0.5(toluene) (13.3 mg, 0.0492 mmol) was added at room temperature. Following removal of the solvent immediately, the residue was washed with n-hexane. The insoluble materials were separated by filtration through a Celite® pad using toluene as an eluent. Subsequent to removal of the solvent from the filtrate, the residue was washed several times with n-hexane to afford (C6F5)2Al–PMes2·Et2O (2·Et2O) as a colorless solid (14.3 mg, 0.0203 mmol, 38%). Colorless crystals, mp. 129 °C (dec.). 1H NMR (300 MHz, C6D6, 298 K): δ = 0.35 (t, 6H, OCH2CH3), 2.09 (s, 6H, Mes p-CH3), 2.35 (s, 12H, Mes o-CH3), 3.76 (q, 4H, OCH2CH3), 6.74 (m, 4H, Mes m-H). 19F NMR (282 MHz, C6D6, 298 K): δ = −119.4 (m, 4F, C6F5 o-F), −152.5 (t, J = 19.7 Hz, 2F, C6F5 p-F), −161.1 (m, 4F, C6F5 m-F) ppm. 31P{1H} NMR (121 MHz, C6D6, r.t.): δ = −104.8 ppm. No 27Al NMR signal was observed, even after long-time measurement. Satisfactory data of the 13C NMR spectrum could not be obtained due to the impurities and decomposition. Due to the flame retardancy of fluorocarbons and the extremely high air-/moisture-sensitivity, satisfactory data of the elemental analysis could not be obtained.
3.7. Reaction of 2·LiCl with Et2O
The solid of 2·LiCl (1.2 mg, 0.0018 mmol) was slowly dissolved Et2O (1 mL) at room temperature. The solvent was removed immediately and the mixture was analyzed by 1H and 19F NMR spectroscopy, presenting a mixture the same as the observation of the reaction for the (C6F5)2AlCl with Mes2PLi in Et2O solution. This result indicated that 2·LiCl is formed at the initial stage of the reaction and Et2O is coordinated toward 2·LiCl or promotes decomposition of 2·LiCl.
3.8. Thermal Isomerization of 1·(t-BuNC)
Using a J.Young NMR tube, a solution of 1·(t-BuNC) (5.6 mg, 0.011 mmol) in C6D6 (0.6 mL) was degassed by freeze-pump-thaw cycles and heated at 100 °C over 48 h. The signals of the corresponding 1, 1·(t-BuNC), Mes2PH, Mes2P–PMes2 were observed by 31P{1H} NMR spectroscopy. The recrystallization from a mixture afforded a trace amount of 3 as red crystals.
3.9. Reaction of 1 with Benzophenone
Using a J.Young NMR tube, a solution of 1 (12.7 mg, 0.0309 mmol) in C6D6 (0.6 mL) was treated with an excess amount of benzophenone (9.2 mg, 0.0505 mmol, 1.6 eq.) at room temperature. The quantitative formation of Mes2P–PMes2 was observed, as judged by 31P{1H} NMR spectroscopy.
3.10. X-Ray Crystallographic Analysis
The intensity data were collected on a Saturn 70 CCD diffractometer (Rigaku Corp., Tokyo, Japan) with a VariMax Mo optic system using Mo Kα radiation (λ = 0.71075 Å) [for
1,
1·(t-BuNC), and
2·LiCl], or a Mercury CCD diffractometer (Rigaku Corp., Tokyo, Japan) with graphite monochromated Mo Kα radiation (λ = 0.71069 Å) (for
2·Et2O). An empirical absorption correction was applied to the diffraction data using ABSPACK [
30] for
1,
1·(t-BuNC),
2·LiCl, and
2·Et2O. The structure was solved by a direct method (SHELXT [
31]) and refined by a full-matrix least-squares method on
F2 for all reflections (SHELXL-2016/4 [
32]). All hydrogen atoms were placed using AFIX instructions, while all other atoms were refined anisotropically. CCDC-1959322 (
1), CCDC-1959323 [
1·(t-BuNC)], CCDC-1959324 (
2·Et2O), and CCDC-1959325 (
2·LiCl) contain the
supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via
www.ccdc.cam.ac.uk/data_request/cif.
3.11. Theoretical Calculations
DFT calculations were performed using the Gaussian 16 (Rev. B. 01) [
33] program package (Gaussian, Inc., Wallingford, CT, USA) with B3LYP functional [
34,
35,
36] including Grimme dispersion correction (D3) [
37,
38] along with a combined basis set: 6-31G(d) level. All the geometry optimizations were performed until the residual mean force was smaller than 1.0 × 10
−5 a.u. (
tight threshold in Gaussian). The frequency calculations were carried out for each optimized structure to confirm the absence of any imaginary frequencies. The vertical excitation energy and electronic absorption spectra were simulated using time-dependent density functional theory (TD-DFT) [
39] at the B3LYP-D3/6-311G+(d,p)//B3LYP-D3/6-31G(d) level. Natural bond orbital (NBO) analyses were conducted with the NBO 6.0 program package [
40], linked to single-point calculations using Gaussian 16 (Rev. B. 01)










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}