Dimethyloxonium and Methoxy Derivatives of nido-Carborane and Metal Complexes Thereof




Abstract
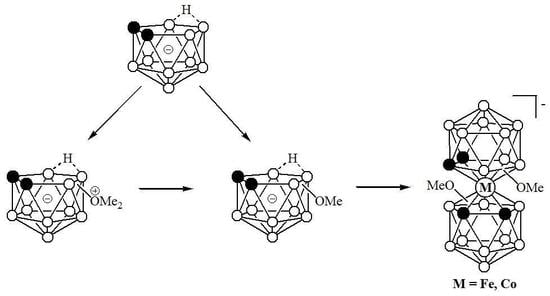
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
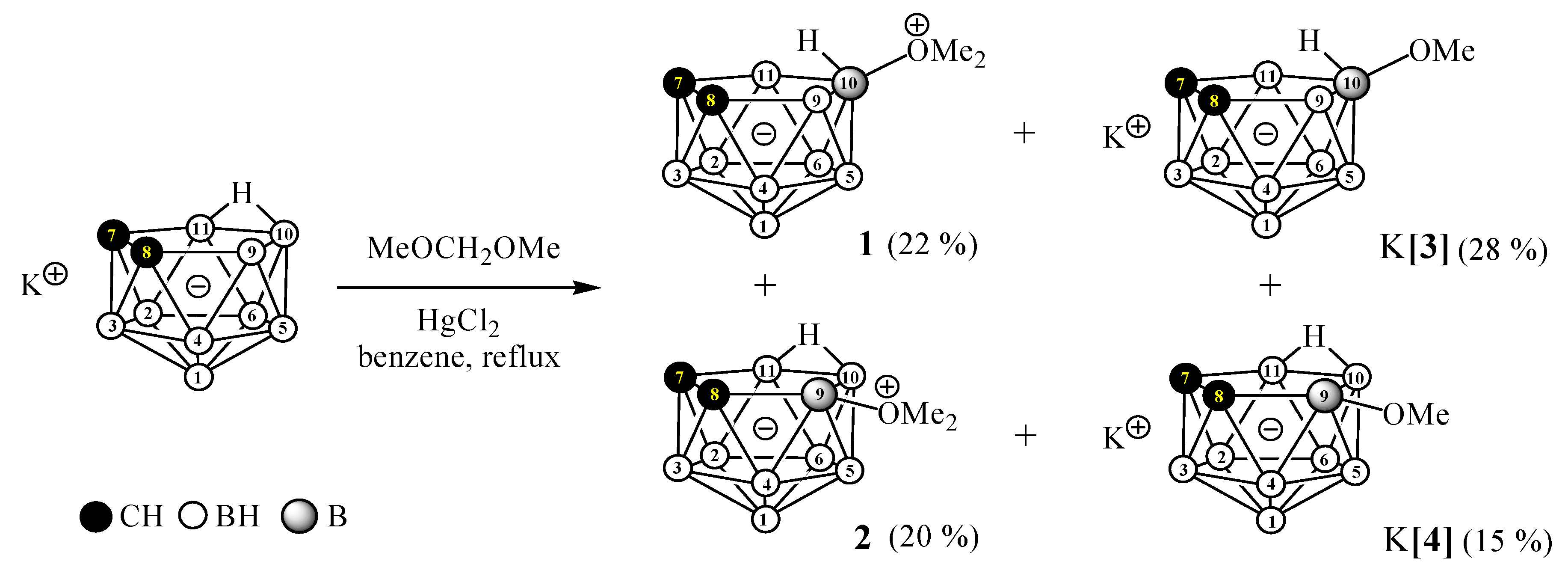
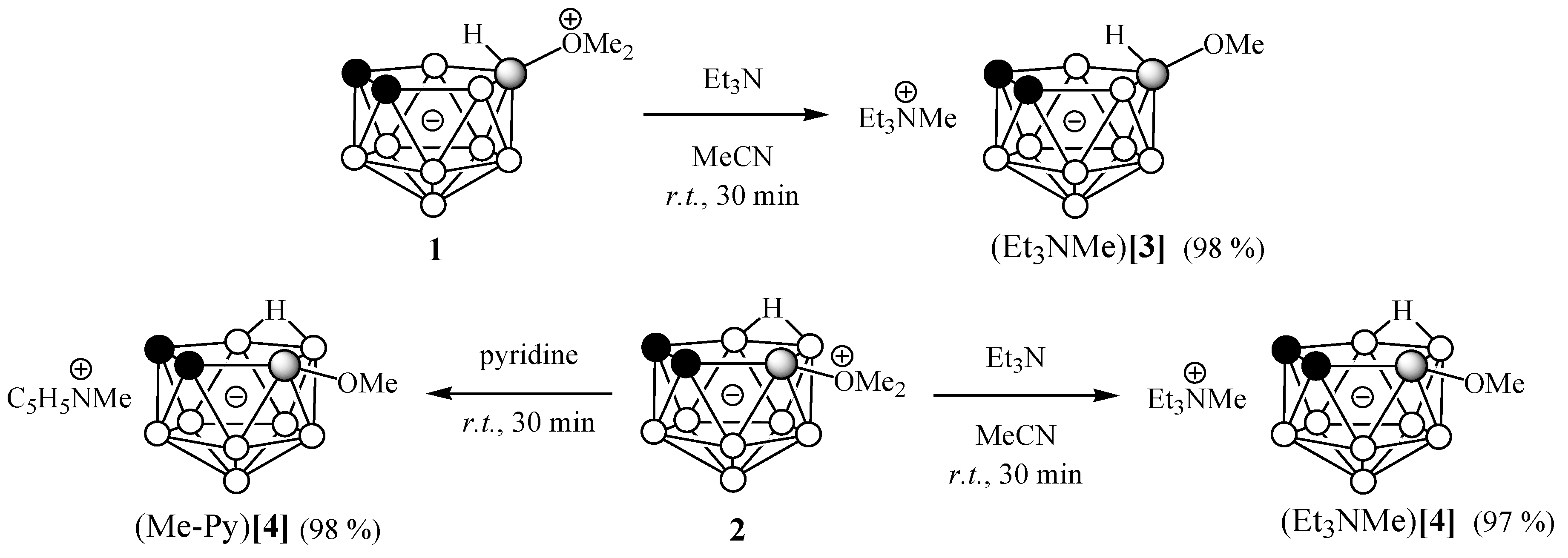
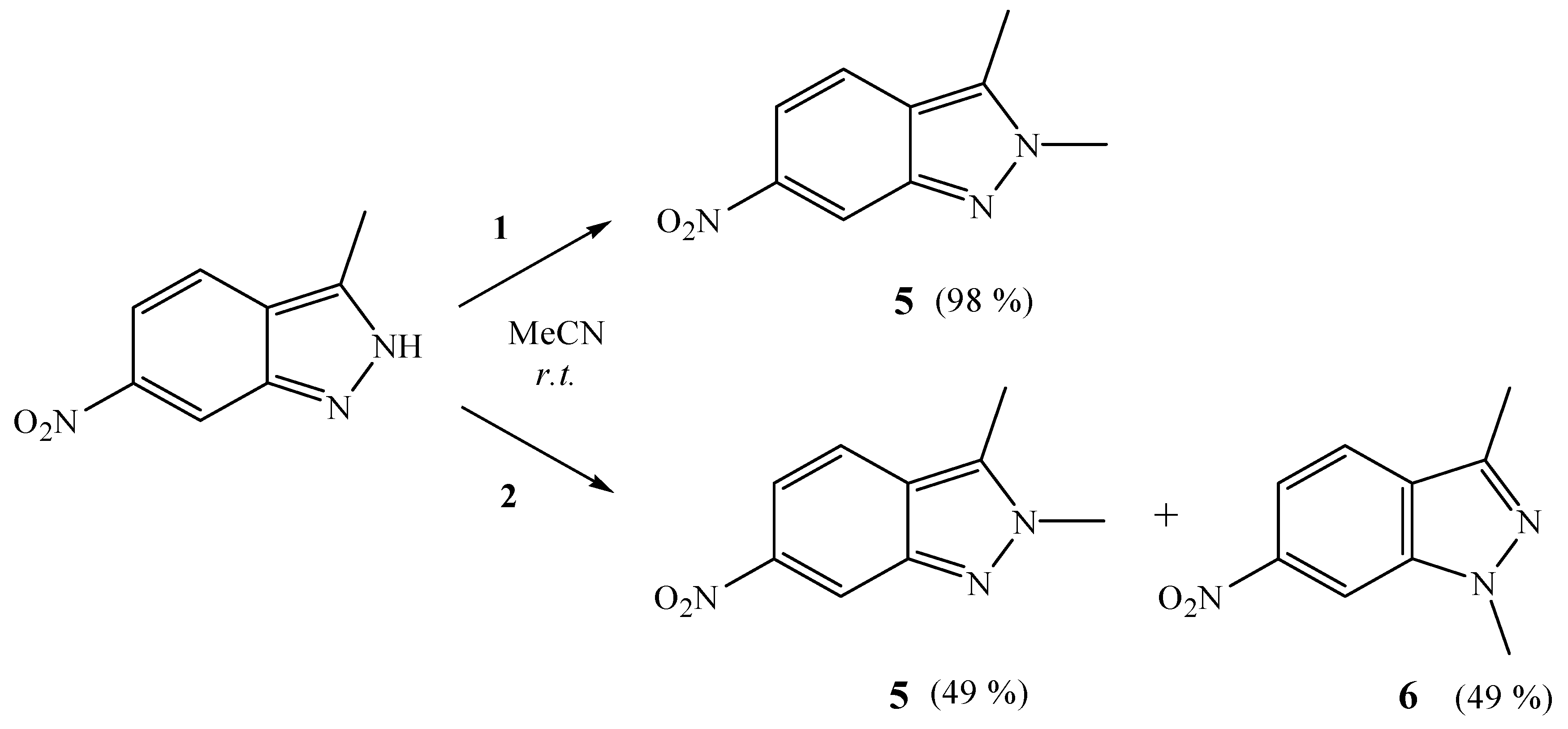
2. Results and Discussion
3. Materials and Methods
3.1. General Procedures and Instrumentation
3.2. Synthesis
3.2.1. Preparation of 10-Me2O-7,8-C2B9H11 (1), 9-Me2O-7,8-C2B9H11 (2), K[10-MeO-7,8-C2B9H11] (K[3]), and K[9-MeO-7,8-C2B9H11] (K[4])
3.2.2. Reactions of 10-Me2O-7,8-C2B9H11 and 9-Me2O-7,8-C2B9H11 with Triethylamine
3.2.3. Reaction of 9-Me2O-7,8-C2B9H11 with Pyridine
3.2.4. Reactions of 10-Me2O-7,8-C2B9H11 and 9-Me2O-7,8-C2B9H11 with 3-Methyl-6-nitro-1H-indazole
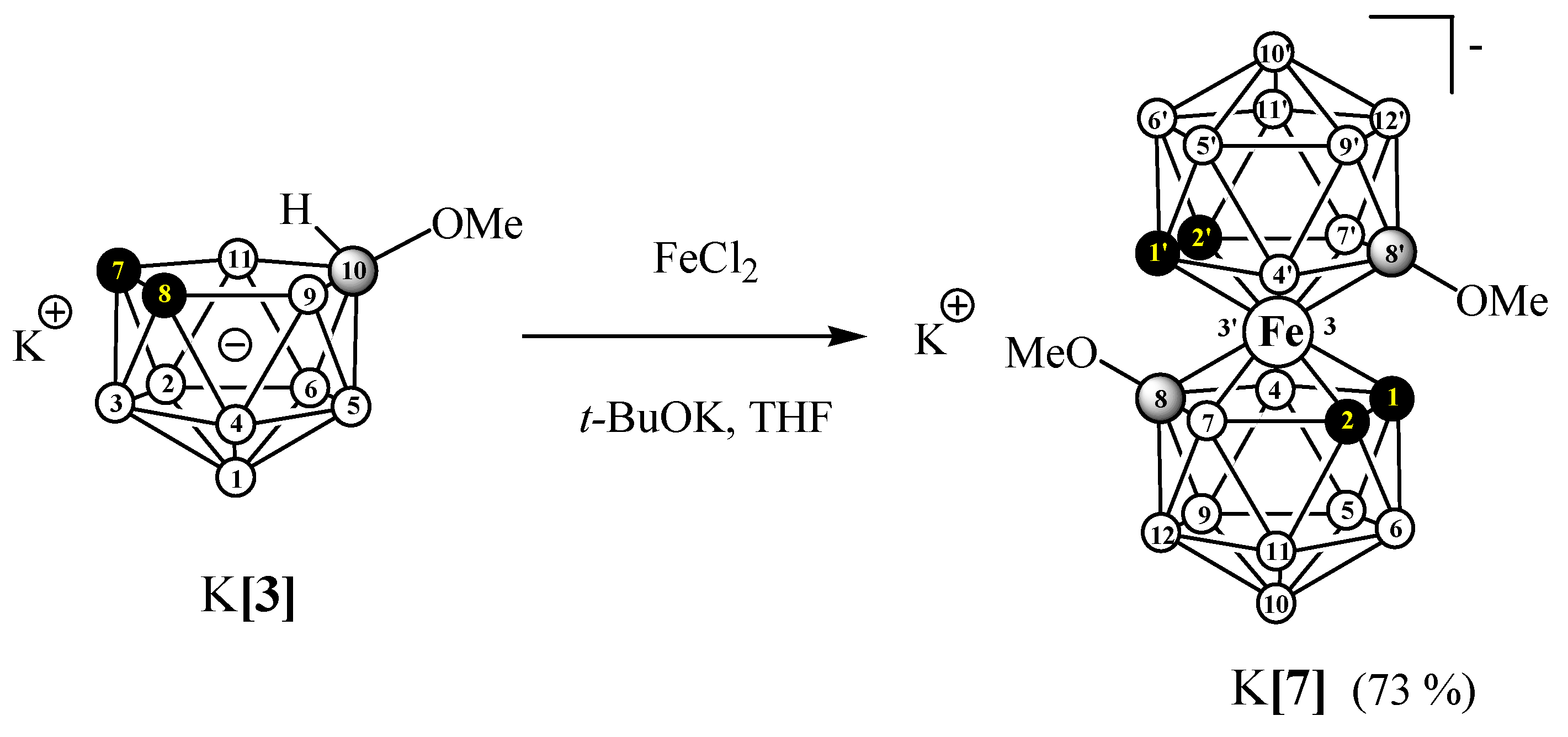
3.2.5. Synthesis of K[8,8′-(MeO)2-3,3′-Fe(1,2-C2B9H10)2] (K[7])
3.2.6. Synthesis of (Bu4N)[4,7′-(MeO)2-3,3′-Fe(1,2-C2B9H10)2] ((Bu4N)[8])
3.2.7. Synthesis of (Bu4N)[4,7′-(MeO)2-3,3′-Co(1,2-C2B9H10)2] ((Bu4N)[9])
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Semioshkin, A.A.; Sivaev, I.B.; Bregadze, V.I. Cyclic oxonium derivatives of polyhedral boron hydrides and their synthetic applications. Dalton Trans. 2008, 8, 977–992. [Google Scholar] [CrossRef] [PubMed]
- Sivaev, I.B.; Bregadze, V.I. Cyclic oxonium derivatives as an efficient synthetic tool for the modification of polyhedral boron hydrides. In Boron Science: New Technologies and Applications; Hosmane, N.S., Ed.; CRC Press: Boca Raton, FL, USA, 2012; pp. 623–637. [Google Scholar]
- Wojtczak, B.A.; Andrysiak, A.; Grüner, B.; Lesnikowski, Z.J. “Chemical Ligation”: A versatile method for nucleoside modification with boron cluster. Chem. Eur. J. 2008, 14, 10675–10682. [Google Scholar] [CrossRef]
- Bednarska, K.; Olejniczak, A.B.; Wojtczak, B.A.; Sulowska, Z.; Lesnikowski, Z.J. Adenosine and 2′-deoxyadenosine modified with boron cluster pharmacophores as new classes of human blood platelet function modulators. ChemMedChem 2010, 5, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Řezačova, P.; Pokorna, J.; Brynda, J.; Kohišek, M.; Cigler, P.; Lepšik, M.; Fanfrlik, J.; Řezač, J.; Šaškova, K.G.; Sieglova, I.; et al. Design of HIV protease inhibitors based on inorganic polyhedral metallacarboranes. J. Med. Chem. 2009, 52, 7132–7141. [Google Scholar] [CrossRef] [PubMed]
- Stogniy, M.Y.; Kazakov, G.S.; Sivaev, I.B.; Bregadze, V.I. Synthesis of podands with nido-carboranyl groups as a basis for construction of crown ethers with an incorporated metallacarborane moiety. Russ. Chem. Bull. 2013, 62, 699–704. [Google Scholar] [CrossRef]
- Kazakov, G.S.; Stogniy, M.Y.; Sivaev, I.B.; Suponitsky, K.Y.; Godovikov, I.A.; Kirilin, A.D.; Bregadze, V.I. Synthesis of crown ethers with the incorporated cobalt bis(dicarbollide) fragment. J. Organomet. Chem. 2015, 798, 196–203. [Google Scholar] [CrossRef]
- Sivaev, I.B.; Semioshkin, A.A.; Brellochs, B.; Sjöberg, S.; Bregadze, V.I. Synthesis of oxonium derivatives of the dodecahydro-closo-dodecaborate anion [B12H12]2−. Tetramethylene oxonium derivative of [B12H12]2− as a convenient precursor for the synthesis of functional compounds for boron neutron capture therapy. Polyhedron 2000, 19, 627–632. [Google Scholar] [CrossRef]
- Klyukin, I.N.; Voinova, V.V.; Selivanov, N.A.; Zhdanov, A.P.; Zhizhin, K.Y.; Kuznetsov, N.T. New methods for the synthesis of alkoxy derivatives of the closo-decaborate anion [2-B10H9(OR)]2−, where R = C2H5, iso-C3H7, C4H9. Russ. J. Inorg. Chem. 2018, 63, 1546–1551. [Google Scholar] [CrossRef]
- Plešek, J.; Jelinek, T.; Mareš, F.; Heřmanek, S. Unique dialkylsulfonio-methylation of the 7,8-C2B9H12− ion to the 9-R2S-CH2-7,8-C2B9H11 zwitterions by formaldehyde and dialkyl sulfides. General synthesis of the compounds 10-R2E-7,8-C2B9H11 (E = O, S). Collect. Czech. Chem. Commun. 1993, 58, 1534–1547. [Google Scholar] [CrossRef]
- Shmalko, A.V.; Anufriev, S.A.; Anisimov, A.A.; Stogniy, M.Y.; Sivaev, I.B.; Bregadze, V.I. On the synthesis of 6,6’-diphenyl cobalt and nickel bis(dicarbollides). Russ. Chem. Bull. 2018. submitted. [Google Scholar]
- Mullica, D.F.; Sappenfield, E.L.; Stone, F.G.A.; Woollam, S.F. Allyl Carborane complexes of molybdenum and tungsten: Cage-hydride abstraction reactions in the presence of donor molecules. Organometallics 1994, 13, 157–166. [Google Scholar] [CrossRef]
- Du, S.; Franken, A.; Jellis, P.A.; Kautz, J.A.; Stone, F.G.A.; Yu, P.-Y. Monocarbollide complexes of molybdenum and tungsten: Functionalization through reactions at a cage boron centre. J. Chem. Soc. Dalton Trans. 2001, 1846–1856. [Google Scholar] [CrossRef]
- Ma, P.; Smith Pellizzeri, T.M.; Zubieta, J.; Spencer, J.T. Synthesis and characterization of oxonium functionalized rhenium metallaborane. J. Chem. Cryst. 2019, 49. [Google Scholar] [CrossRef]
- Stogniy, M.Y.; Abramova, E.N.; Lobanova, I.A.; Sivaev, I.B.; Bragin, V.I.; Petrovskii, P.V.; Tsupreva, V.N.; Sorokina, O.V.; Bregadze, V.I. Synthesis of functional derivatives of 7,8-dicarba-nido-undecaborate anion by ring-opening of its cyclic oxonium derivatives. Collect. Czech. Chem. Commun. 2007, 72, 1676–1688. [Google Scholar] [CrossRef]
- Zakharkin, L.I.; Kalinin, V.N.; Zhigareva, G.G. Oxidation of dicarbadodecahydro-nido-undecaborate anions by mercuric chloride in tetrahydrofuran and pyridine. Bull. Acad. Sci. USSR Div. Chem. Sci. 1979, 28, 2198–2199. [Google Scholar] [CrossRef]
- Stogniy, M.Y.; Sivaev, I.B.; Malysheva, Y.B.; Bregadze, V.I. Synthesis of tetrahydropyran oxonium derivative of 7,8-dicarba-nido-undecaborate anion [10-C5H10O-7,8-C2B9H11]. Vestn. Lobachevsky State Univ. Nizhni Novgorod 2013, 4, 115–117. [Google Scholar]
- Stogniy, M.Y.; Erokhina, S.A.; Suponitsky, K.Y.; Anisimov, A.A.; Sivaev, I.B.; Bregadze, V.I. Nucleophilic addition reactions to the ethylnitrilium derivative of nido-carborane 10-EtCRN-7,8-C2B9H11. New J. Chem. 2018, 42, 17958–17967. [Google Scholar] [CrossRef]
- Colquhoun, H.M.; Greenhough, T.J.; Wallbridge, M.G.H. Carbaborane derivatives of the late- and post-transition elements. Part 2. Dicarbaundecaboranyl compounds of copper(I), gold(I), and mercury(II); the crystal and molecular tructure of 3-triphenylphosphine-3-mercura-1,2-dicarbadodecaborane(II), a pseudo-σ-bonded metallacarbaborane. J. Chem. Soc. Dalton Trans. 1979, 4, 619–628. [Google Scholar]
- Zakharkin, L.I.; Ol’shevskaya, V.A. Simple method of mercuration of nido-7-R-7,8-dicarbaundecaborate anions with formation of 10,10′-bis(7-R-7,8-dicarbaundecaborate) mercury dianions. Russ. J. Gen. Chem. 1992, 62, 114–116. [Google Scholar]
- Sivaev, I.B.; Bregadze, V.I. Lewis acidity of boron compounds. Coord. Chem. Rev. 2014, 270–271, 75–88. [Google Scholar] [CrossRef]
- Sivaev, I.B.; Bregadze, V.I. Polyhedral boron hydrides as Lewis acids. In Proceedings of the Third EuCheMS Inorganic Chemistry Conference: “Chemistry over the Horizon”, Wroclaw, Poland, 28 June–1 July 2015; p. 73. [Google Scholar]
- Yadav, J.S.; Ganganna, D.; Bhunia, D.C.; Srihari, P. NbCl5 mediated deprotection of methoxy methyl ether. Tetrahedron Lett. 2009, 50, 4318–4320. [Google Scholar] [CrossRef]
- Marchetti, F.; Pampaloni, G.; Zacchini, S. The reactivity of 1,1-dialkoxyalkanes with niobium and tantalum pentahalides. Formation of coordination compounds, C–H and C–C bond activation and the X-ray structure of the stable carboxonium species [Me2C=CHC(=OMe)Me][NbCl5(OMe)]. Dalton Trans. 2009, 38, 8096–8106. [Google Scholar] [CrossRef]
- Bini, R.; Chiappe, C.; Marchetti, F.; Pampaloni, G.; Zacchini, S. Structures and unusual rearrangements of coordination adducts of MX5 (M = Nb, Ta; X = F, Cl) with simple diethers. A crystallographic, spectroscopic, and computational study. Inorg. Chem. 2010, 49, 339–351. [Google Scholar] [CrossRef]
- Earle, M.J.; Fairhurst, R.A.; Giles, R.G.; Heaney, H. Detailed procedures for the preparation of dimethoxycarbenium and trimethyloxonium tetrafluoroborate. Synlett 1991, 10, 728. [Google Scholar] [CrossRef]
- Plešek, J.; Janoušek, Z.; Heřmanek, S. Four new (CH3)2SC2B9H11 isomers. Collect. Czech. Chem. Commun. 1978, 43, 2862–2868. [Google Scholar] [CrossRef]
- Lyssenko, K.A.; Golovanov, D.G.; Meshcheryakov, V.I.; Kudinov, A.R.; Antipin, M.Y. Nature of weak inter- and intramolecular interactions in crystals. 5. Interactions Na···H–B in a crystal of sodium salt of charge-compensated nido-carborane [9-SMe2-7,8-C2B9H10]−. Russ. Chem. Bull. 2005, 54, 933–941. [Google Scholar] [CrossRef]
- Ryschkewitsh, G.E.; Rademaker, W.J. Long-range B–H coupling and quadrupole relaxation. J. Magn. Reson. 1969, 1, 584–588. [Google Scholar] [CrossRef]
- Allerhand, A.; Moll, R.E. Indirect determination of boron-proton coupling in trimethyl borate by proton spin-echo NMR. J. Magn. Reson. 1969, 1, 488–493. [Google Scholar] [CrossRef]
- Bogdanov, V.S.; Kessenikh, A.V.; Negrebetsky, V.V. The indirect measurement of 11B–H coupling constants in some organoboron compounds. J. Magn. Reson. 1971, 5, 145–150. [Google Scholar] [CrossRef]
- Zozulin, A.J.; Jakobsen, H.J.; Moore, T.F.; Garber, A.R.; Odom, J.D. 13C-{1H,11B} triple-resonance experiments. Sign determination of 1J(11B-11B), J(13C-11B), and 2J(1H-11B) in some organoboron compounds. J. Magn. Reson. 1980, 41, 458–466. [Google Scholar] [CrossRef]
- Kultyshev, R.G.; Liu, J.; Meyers, E.A.; Shore, S.G. Synthesis and characterization of sulfide, sulfide-sulfonium, and bissulfide derivatives of [B12H12]2−. Additivity of Me2S and MeS-substituent effects in 11B NMR spectra of disubstituted icosahedral boron clusters. Inorg. Chem. 2000, 39, 3333–3341. [Google Scholar] [CrossRef]
- Hamilton, E.J.M.; Leung, H.T.; Kultyshev, R.G.; Chen, X.; Meyers, E.A.; Shore, S.G. Unusual cationic tris(dimethylsulfide)-substituted closo-boranes: Preparation and characterization of [1,7,9-(Me2S)3-B12H9]BF4 and [1,2,10-(Me2S)3-B10H7]BF4. Inorg. Chem. 2012, 51, 2374–2380. [Google Scholar] [CrossRef]
- Anufriev, S.A.; Erokhina, S.A.; Suponitsky, K.Y.; Godovikov, I.A.; Filippov, O.A.; Fabrizi de Biani, F.; Corsini, M.; Chizhov, A.O.; Sivaev, I.B. Methylsulfanyl-stabilized rotamers of cobalt bis(dicarbollide). Eur. J. Inorg. Chem. 2017, 2017, 4444–4451. [Google Scholar] [CrossRef]
- Bukowski, R.M.; Yasothan, U.; Kirkpatrick, P. Pazopanib. Nat. Rev. Drug Discov. 2010, 9, 17–18. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Chen, L.; Liu, B.; Wang, X.; Long, L.; Liu, D. Synthesis and biological evaluation of novel pazopanib derivatives as antitumor agents. Bioorg. Med. Chem. Lett. 2014, 24, 1108–1110. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.C.; Yang, B.W.; Chen, W.; Huang, D.D.; Li, Y.; Deng, X.; Liu, B.M.; Wang, J.J.; Qian, H.; Huang, W.L. A novel practical synthesis of pazopanib: An anticancer drug. Lett. Org. Chem. 2012, 9, 276–279. [Google Scholar]
- Baddam, S.R.; Kumar, N.U.; Reddy, A.P.; Bandichhor, R. Regioselective methylation of indazoles using methyl 2,2,2-trichloromethylacetamide. Tetrahedron Lett. 2013, 54, 1661–1663. [Google Scholar] [CrossRef]
- Romanovskiy, V.N.; Smirnov, I.V.; Babain, V.A.; Shadrin, A.Y. Combined processes for high level radioactive waste separations: UNEX and other extraction processes. In Advanced Separation Techniques for Nuclear Fuel Reprocessing and Radioactive Waste Treatment; Nash, K.L., Lumetta, G.J., Eds.; Woodhead Publishing: Cambridge, UK, 2011; pp. 229–265. [Google Scholar]
- Grüner, B.; Rais, J.; Selucky, P.; Lučaníkova, M. Recent progress in extraction agents based on cobalt bis(dicarbollides) for partitioning of radionuclides from high-level nuclear waste. In Boron Science: New Technologies and Applications; Hosmane, N.S., Ed.; CRC Press: Boca Raton, FL, USA, 2012; pp. 463–490. [Google Scholar]
- Gozzi, M.; Schwarze, B.; Hey-Hawkins, E. Half- and mixed-sandwich metallacarboranes in catalysis. In Handbook of Boron Science with Applications in Organometallics, Catalysis, Materials and Medicine. Volume 2: Boron in Catalysis; Hosmane, N.S., Eagling, R., Eds.; World Scientific Publishing Europe: London, UK, 2018; pp. 27–80. [Google Scholar]
- Spokoyny, A.M.; Li, T.C.; Fahra, O.K.; Machan, C.W.; She, C.; Stern, C.L.; Marks, T.J.; Hupp, J.T.; Mirkin, C.A. Electronic tuning of nickel-based bis(dicarbollide) redox shuttles in dye-sensitized solar cells. Angew. Chem. Int. Ed. 2010, 49, 5339–5343. [Google Scholar] [CrossRef]
- Bregadze, V.I.; Dyachenko, O.A.; Kazheva, O.N.; Kravchenko, A.V.; Sivaev, I.B.; Starodub, V.A. Tetrathiafulvalene-based radical cation salts with transition metal bis(dicarbollide) anions. CrystEngComm 2015, 17, 4754–4767. [Google Scholar] [CrossRef]
- Ruiz-Rosas, R.; Fuentes, I.; Viñas, C.; Teixidor, F.; Morallon, E.; Cazorla-Amoros, D. Tailored metallacarboranes as mediators for boosting the stability of carbon-based aqueous supercapacitors. Sustain. Energy Fuels 2018, 2, 345–352. [Google Scholar] [CrossRef]
- Sivaev, I.B. Ferrocene and transition metal bis(dicarbollides) as platform for design of rotatory molecular switches. Molecules 2017, 22, 2201. [Google Scholar] [CrossRef]
- Hao, E.; Jensen, T.J.; Courtney, B.H.; Vicente, M.G.H. Synthesis and cellular studies of porphyrin-cobaltacarborane conjugates. Bioconjug. Chem. 2005, 16, 1495–1502. [Google Scholar] [CrossRef]
- Hao, E.; Sibrian-Vazquez, M.; Serem, W.; Garno, J.C.; Fronczek, F.R.; Vicente, M.G.H. Synthesis, aggregation and cellular investigations of porphyrin-cobaltcarborane conjugates. Chem. Eur. J. 2007, 13, 9035–9042. [Google Scholar] [CrossRef] [PubMed]
- Efremenko, A.V.; Ignatova, A.A.; Grin, M.A.; Sivaev, I.B.; Mironov, A.F.; Bregadze, V.I.; Feofanov, A.V. Chlorin e6 fused with a cobalt-bis(dicarbollide) nanoparticle provides efficient boron delivery and photoinduced cytotoxicity in cancer cells. Photochem. Photobiol. Sci. 2014, 13, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Volovetsky, A.B.; Sukhov, V.S.; Balalaeva, I.V.; Dudenkova, V.V.; Shilyagina, N.Y.; Feofanov, A.V.; Efremenko, A.V.; Grin, M.A.; Mironov, A.F.; et al. Pharmacokinetics of chlorin e6-cobalt bis(dicarbollide) conjugate in Balb/c mice with engrafted carcinoma. Int. J. Mol. Sci. 2017, 18, 2556. [Google Scholar] [CrossRef]
- Řezačova, P.; Cigler, P.; Matejiček, P.; Lepšik, M.; Pokorna, J.; Grüner, B.; Konvalinka, J. Medicinal applications of carboranes: Inhibition of HIV protease. In Boron Science: New Technologies and Applications; Hosmane, N.S., Ed.; CRC Press: Boca Raton, FL, USA, 2012; pp. 41–70. [Google Scholar]
- Zheng, Y.; Liu, W.; Chen, Y.; Jiang, H.; Yan, H.; Kosenko, I.; Chekulaeva, L.; Sivaev, I.; Bregadze, V.; Wang, X. A highly potent antibacterial agent targeting methicillin-resistant Staphylococcus aureus based on cobalt bis(1,2-dicarbollide) alkoxy derivative. Organometallics 2017, 36, 3484–3490. [Google Scholar] [CrossRef]
- Stogniy, M.Y.; Suponitsky, K.Y.; Chizhov, A.O.; Sivaev, I.B.; Bregadze, V.I. Synthesis of 8-alkoxy and 8,8’-dialkoxy derivatives of cobalt bis(dicarbollide). J. Organomet. Chem. 2018, 865, 138–144. [Google Scholar] [CrossRef]
- Anufriev, S.A.; Erokhina, S.A.; Suponitsky, K.Y.; Anisimov, A.A.; Laskova, J.N.; Godovikov, I.A.; Fabrizi de Biani, F.; Corsini, M.; Sivaev, I.B.; Bregadze, V.I. Synthesis and structure of bis(methylsulfanyl) derivatives of iron bis(dicarbollide). J. Organomet. Chem. 2018, 865, 239–246. [Google Scholar] [CrossRef]
- Hawthorne, M.F.; Young, D.C.; Garrett, P.M.; Owen, D.A.; Schwerin, S.G.; Tebbe, F.N.; Wegner, P.A. The Preparation and Characterization of the (3)-1,2-and (3)-1,7-Dicarbadodecahydroundecaborate(−1) Ion. J. Am. Chem. Soc. 1968, 90, 862–868. [Google Scholar] [CrossRef]
- Purification of Laboratory Chemicals; Butterworth-Heinemann: Burlington, NJ, USA, 2009.
- Brauer, G. (Ed.) Handbook of Preparative Inorganic Chemistry; Academic Press: London, UK, 1963. [Google Scholar]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stogniy, M.Y.; Erokhina, S.A.; Kosenko, I.D.; Semioshkin, A.A.; Sivaev, I.B. Dimethyloxonium and Methoxy Derivatives of nido-Carborane and Metal Complexes Thereof. Inorganics 2019, 7, 46. https://doi.org/10.3390/inorganics7040046
Stogniy MY, Erokhina SA, Kosenko ID, Semioshkin AA, Sivaev IB. Dimethyloxonium and Methoxy Derivatives of nido-Carborane and Metal Complexes Thereof. Inorganics. 2019; 7(4):46. https://doi.org/10.3390/inorganics7040046
Chicago/Turabian StyleStogniy, Marina Yu., Svetlana A. Erokhina, Irina D. Kosenko, Andrey A. Semioshkin, and Igor B. Sivaev. 2019. "Dimethyloxonium and Methoxy Derivatives of nido-Carborane and Metal Complexes Thereof" Inorganics 7, no. 4: 46. https://doi.org/10.3390/inorganics7040046
APA StyleStogniy, M. Y., Erokhina, S. A., Kosenko, I. D., Semioshkin, A. A., & Sivaev, I. B. (2019). Dimethyloxonium and Methoxy Derivatives of nido-Carborane and Metal Complexes Thereof. Inorganics, 7(4), 46. https://doi.org/10.3390/inorganics7040046