Self-Assembled Monolayers of Molybdenum Sulfide Clusters on Au Electrode as Hydrogen Evolution Catalyst for Solar Water Splitting

 ,
,  , , and
, , and 
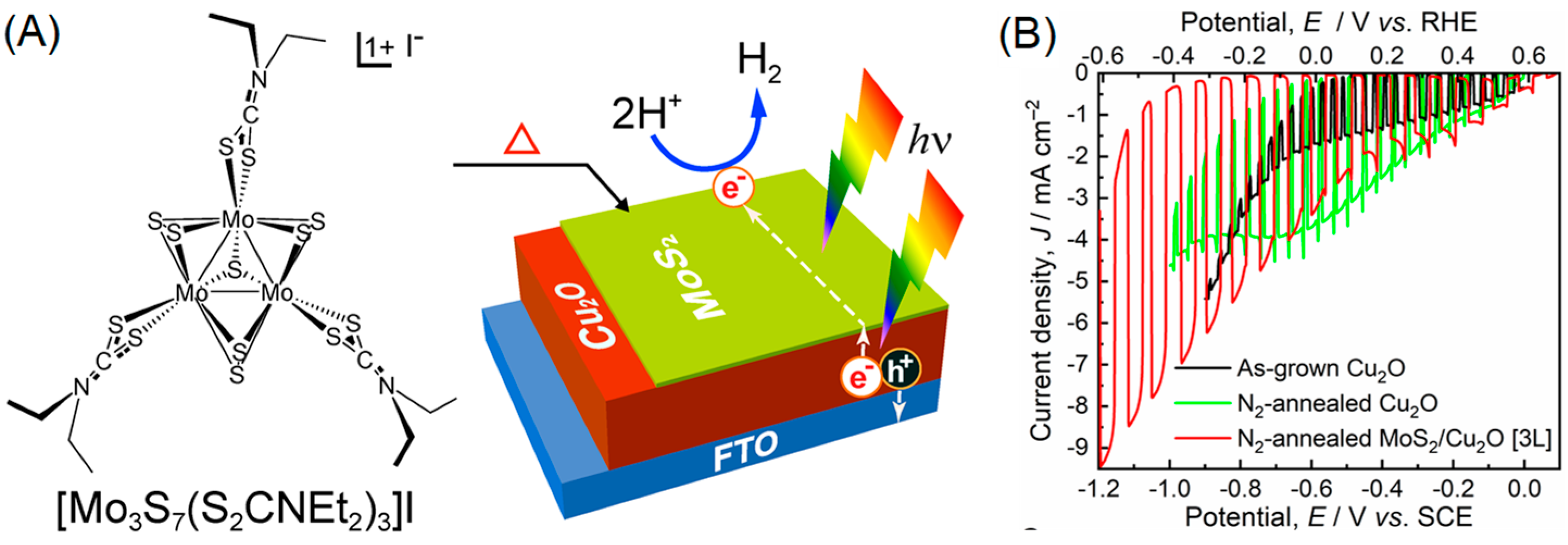{kind=link}
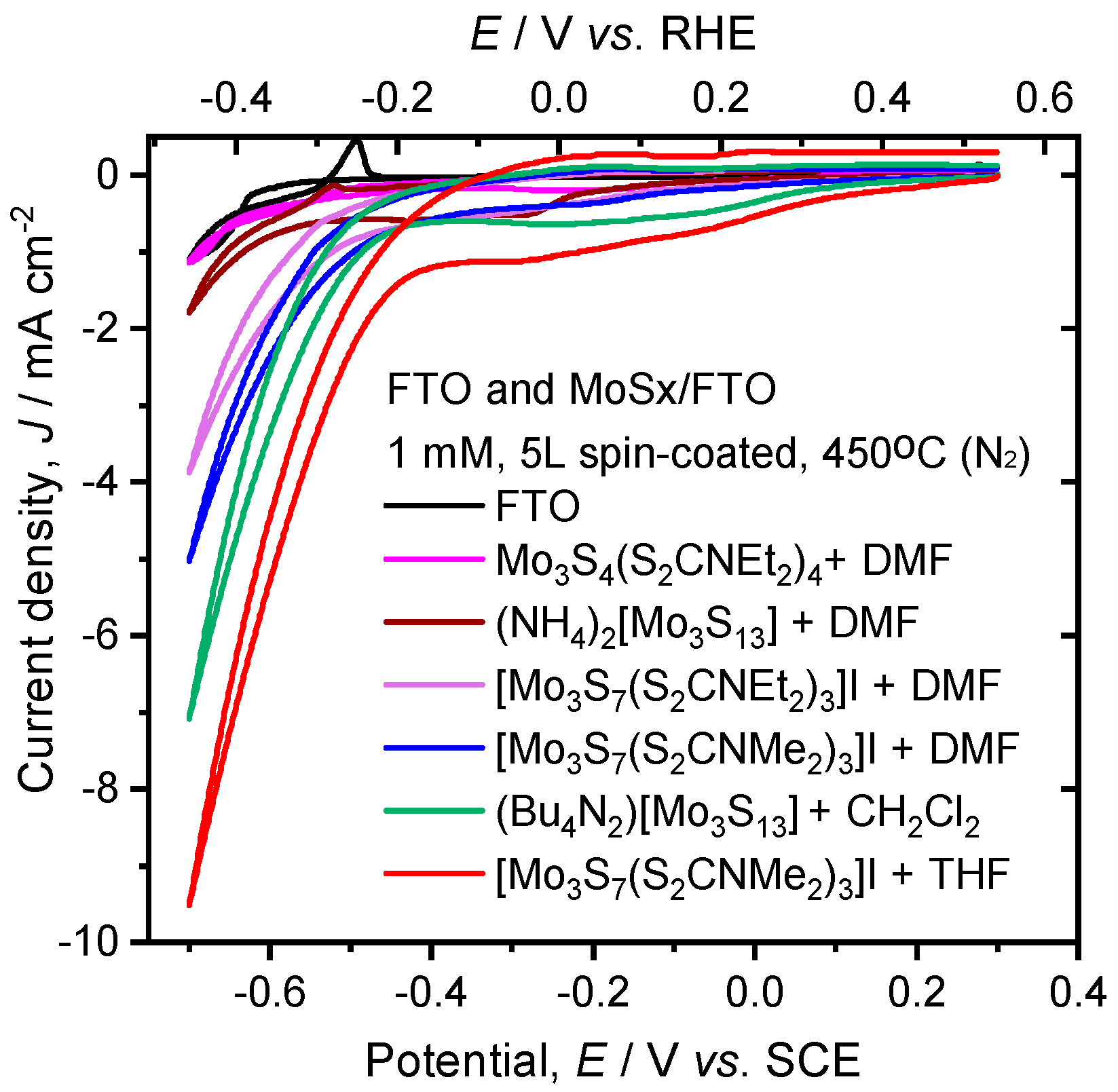{kind=link}
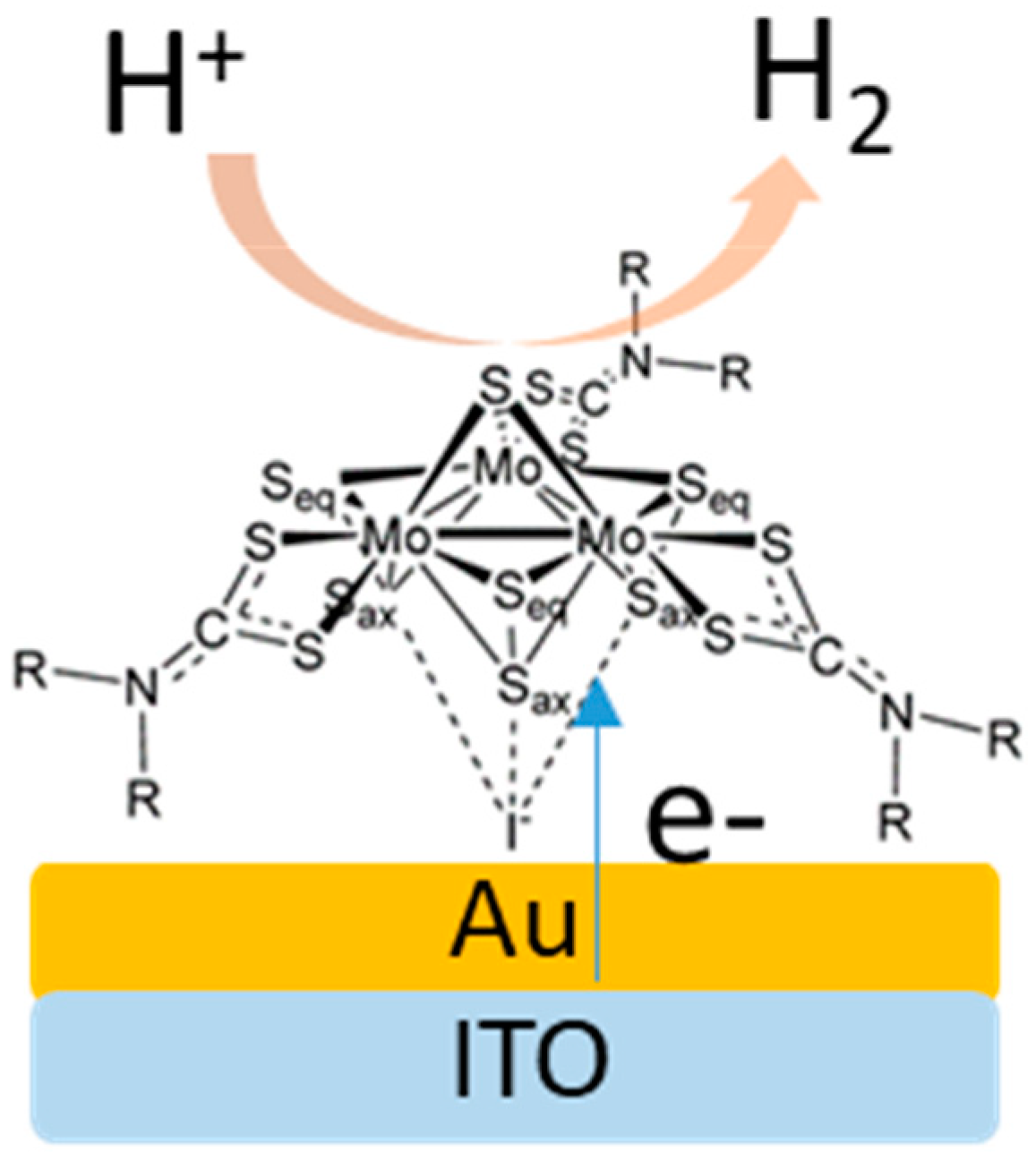{kind=link}
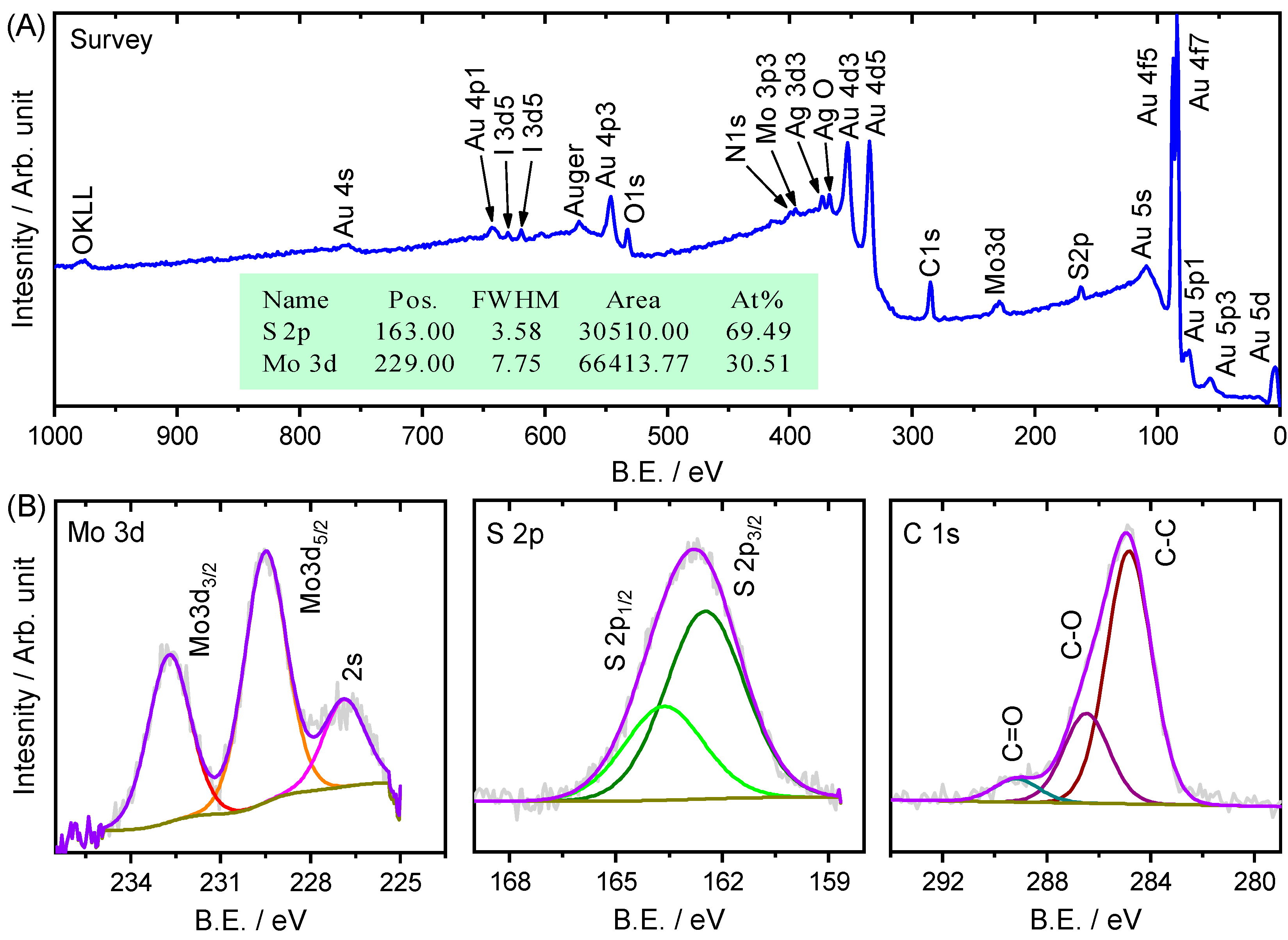{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
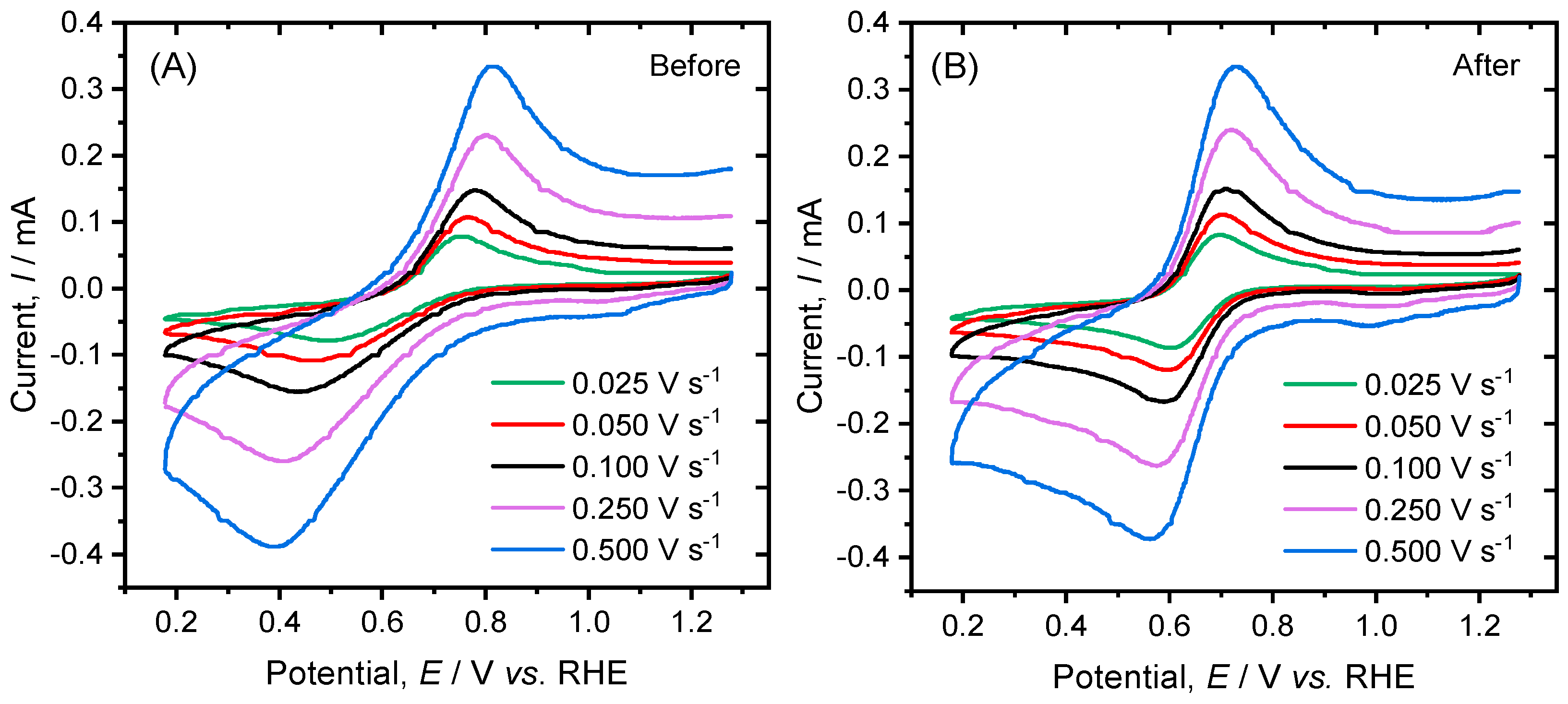
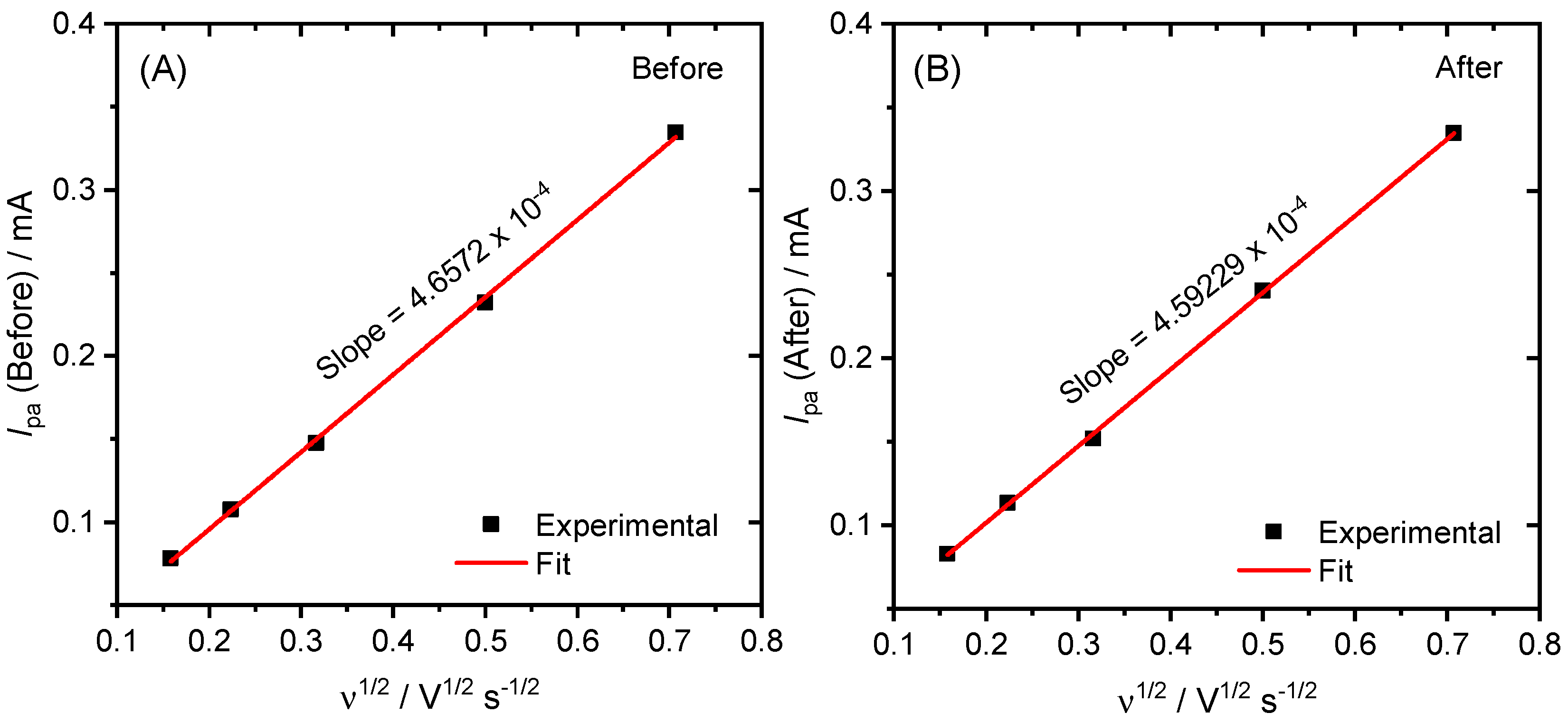
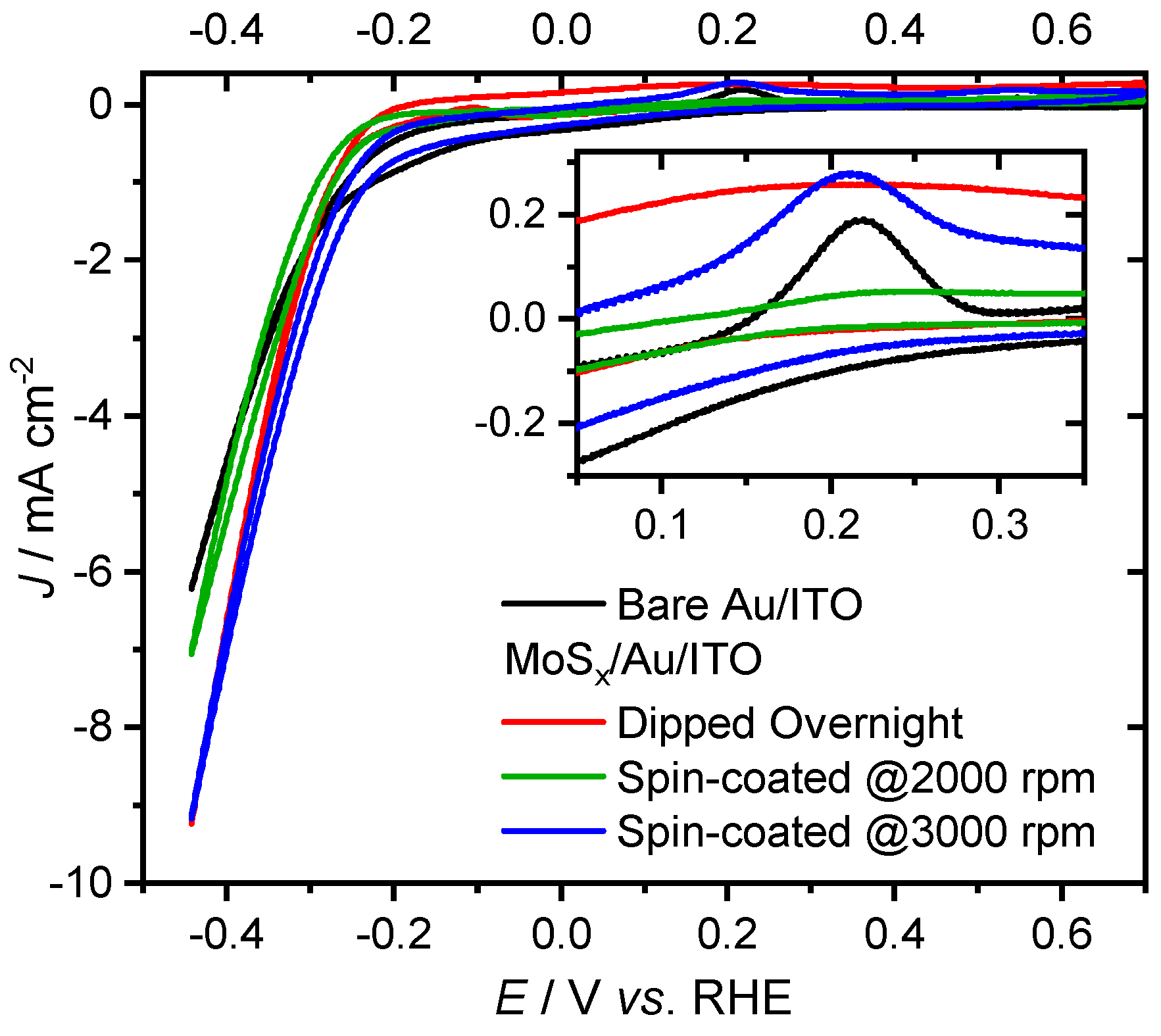
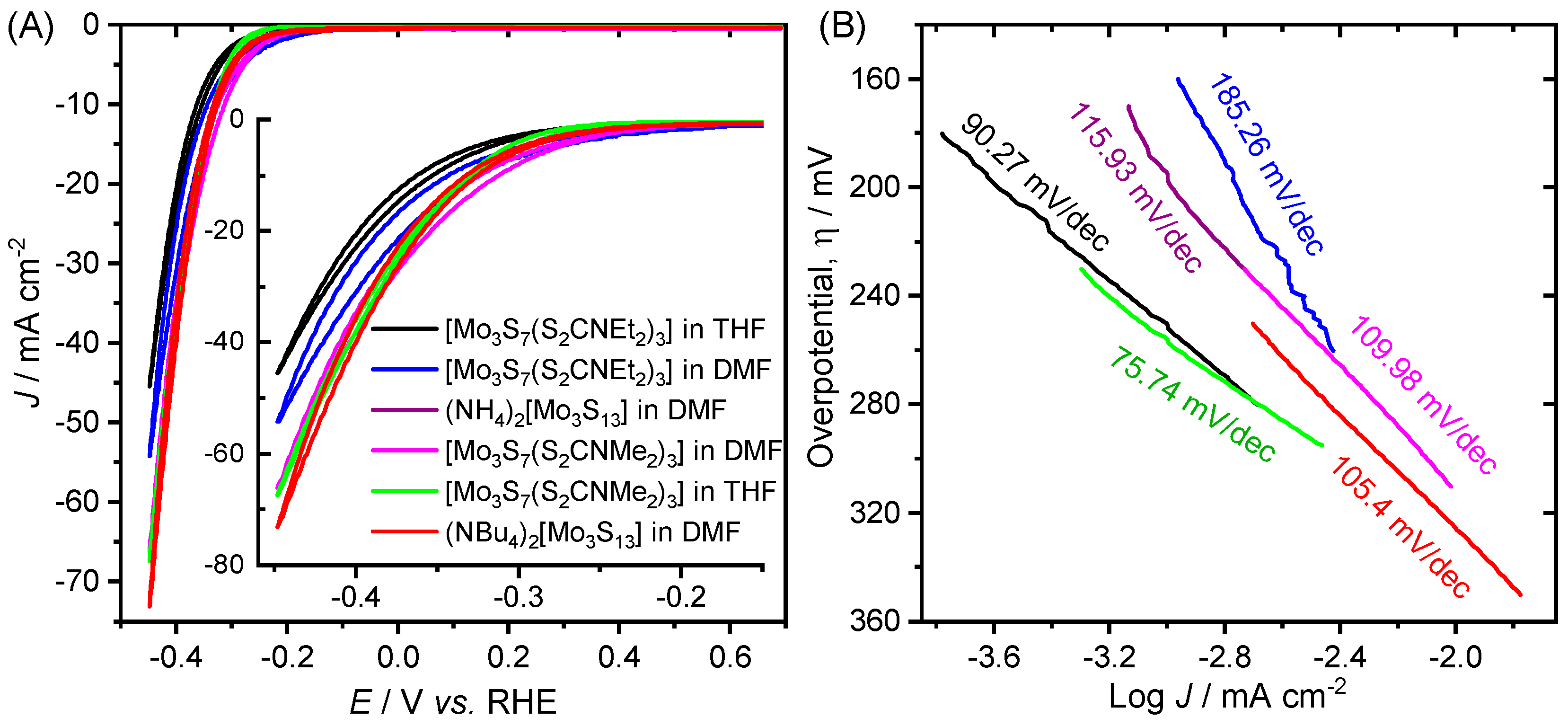
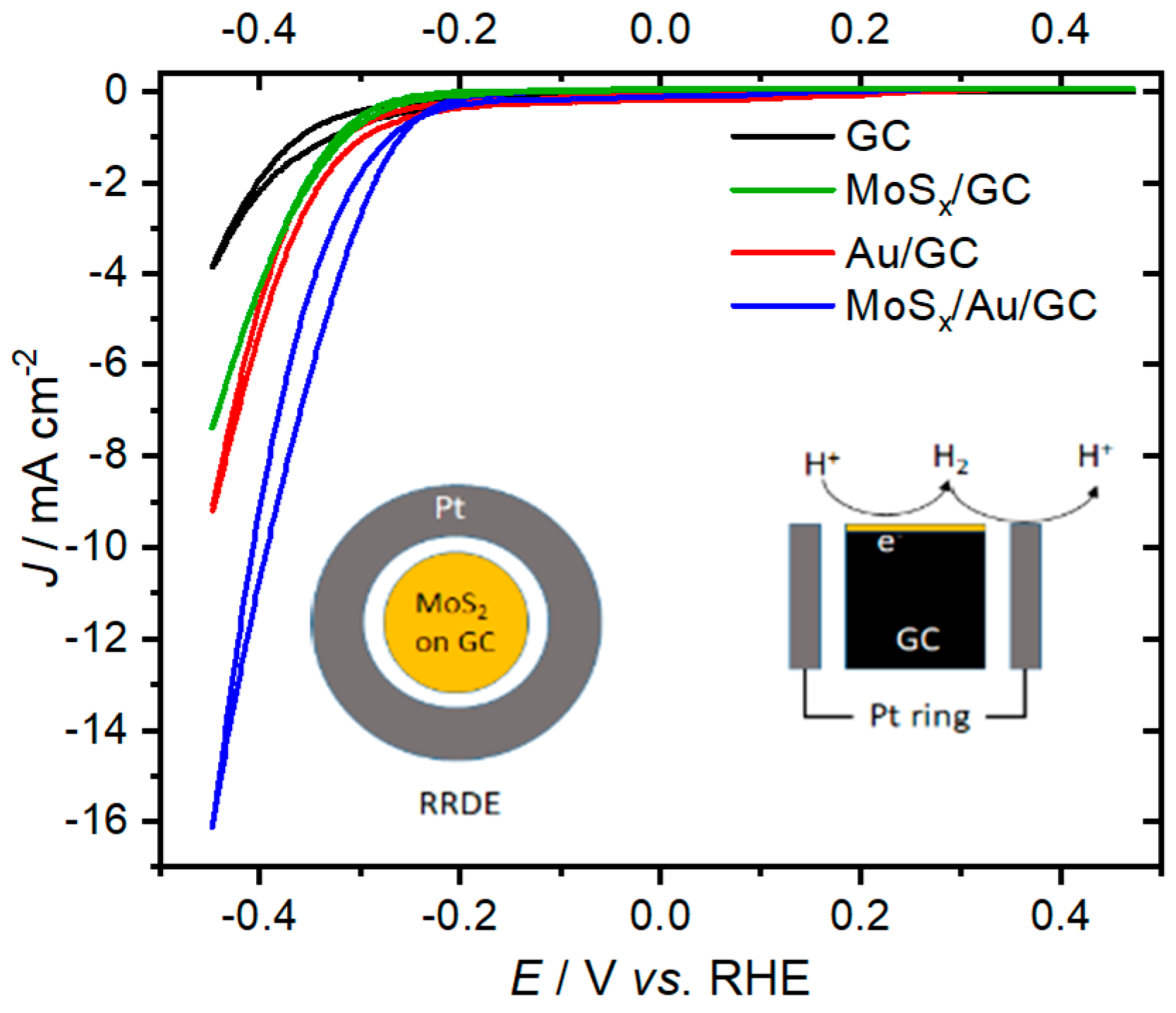
2. Results and Discussion
- (i)
- Electrochemical hydrogen adsorption, H3O+ + e− → Hads + H2O (Tafel slope: 120 mV dec−1)
- (ii)
- Electrochemical desorption, Hads + H3O+ + e− → H2 + H2O (Tafel slope: 40–120 mV dec−1)
- (iii)
- Chemical desorption, Hads + Hads → H2 (Tafel slope: 30–40 mV dec−1)
3. Materials and Methods
3.1. Materials
3.2. Preparation and Assembling of Monolayer on Au-Disc Electrode
3.3. Preparation and Assembling of Monolayer on Au/ITO Electrode
3.4. RRDE Study
3.5. Proton Reduction Measurement
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Blankenship, R.E.; Tiede, D.M.; Barber, J.; Brudvig, G.W.; Fleming, G.; Ghirardi, M.; Gunner, M.R.; Junge, W.; Kramer, D.M.; Melis, A.; et al. Comparing Photosynthetic and Photovoltaic Efficiencies and Recognizing the Potential for Improvement. Science 2011, 332, 805–809. [Google Scholar] [CrossRef] [Green Version]
- Armaroli, N.; Balzani, V. Solar Electricity and Solar Fuels: Status and Perspectives in the Context of the Energy Transition. Chem. Eur. J. 2016, 22, 32–57. [Google Scholar] [CrossRef] [PubMed]
- Chouhan, N.; Liu, R.-S.; Zhang, J. Photochemical Water Splitting: Materials and Applications; CRC Press: Boca Raton, FL, USA, 2017. [Google Scholar]
- Sarina, S.; Waclawik, E.R.; Zhu, H. Photocatalysis on Supported Gold and Silver Nanoparticles Under Ultraviolet and Visible Light Irradiation. Green Chem. 2013, 15, 1814–1833. [Google Scholar] [CrossRef]
- Yang, L.; Guo, S.; Li, X. Au Nanoparticles@MoS2 Core–Shell Structures with Moderate MoS2 Coverage for Efficient Photocatalytic Water Splitting. J. Alloys Compd. 2017, 706, 82–88. [Google Scholar] [CrossRef]
- Roger, I.; Shipman, M.A.; Symes, M.D. Earth-Abundant Catalysts for Electrochemical and Photoelectrochemical Water Splitting. Nat. Rev. Chem. 2017, 1. [Google Scholar] [CrossRef]
- Li, H.; Tsai, C.; Koh, A.L.; Cai, L.; Contryman, A.W.; Fragapane, A.H.; Zhao, J.; Han, H.S.; Manoharan, H.C.; Abild-Pedersen, F.; et al. Activating and optimizing MoS2 basal planes for hydrogen evolution through the formation of strained sulphur vacancies. Nat. Mater. 2015, 15, 48. [Google Scholar] [CrossRef]
- Sun, Y.J.; Liu, C.; Grauer, D.C.; Yano, J.K.; Long, J.R.; Yang, P.D.; Chang, C.J. Electrodeposited Cobalt-Sulfide Catalyst for Electrochemical and Photoelectrochemical Hydrogen Generation from Water. J. Am. Chem. Soc. 2013, 135, 17699–17702. [Google Scholar] [CrossRef] [Green Version]
- Voiry, D.; Yamaguchi, H.; Li, J.W.; Silva, R.; Alves, D.C.B.; Fujita, T.; Chen, M.W.; Asefa, T.; Shenoy, V.B.; Eda, G.; et al. Enhanced Catalytic Activity in Strained Chemically Exfoliated WS2 Nanosheets for Hydrogen Evolution. Nat. Mater. 2013, 12, 850–855. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhao, G.; Rui, K.; Chen, Y.; Xu, X.; Dou, S.X.; Sun, W. Engineering Additional Edge Sites on Molybdenum Dichalcogenides Toward Accelerated Alkaline Hydrogen Evolution Kinetics. Nanoscale 2019, 11, 717–724. [Google Scholar] [CrossRef]
- Zhou, H.Q.; Wang, Y.M.; He, R.; Yu, F.; Sun, J.Y.; Wang, F.; Lan, Y.C.; Ren, Z.F.; Chen, S. One-Step Synthesis of Self-Supported Porous NiSe2/Ni Hybrid Foam: An Efficient 3d Electrode for Hydrogen Evolution Reaction. Nano Energy 2016, 20, 29–36. [Google Scholar] [CrossRef]
- Xu, Y.F.; Gao, M.R.; Zheng, Y.R.; Jiang, J.; Yu, S.H. Nickel/Nickel(II) Oxide Nanoparticles Anchored onto Cobalt(IV) Diselenide Nanobelts for the Electrochemical Production of Hydrogen. Angew. Chem. Int. Ed. 2013, 52, 8546–8550. [Google Scholar] [CrossRef]
- Popczun, E.J.; McKone, J.R.; Read, C.G.; Biacchi, A.J.; Wiltrout, A.M.; Lewis, N.S.; Schaak, R.E. Nanostructured Nickel Phosphide as an Electrocatalyst for the Hydrogen Evolution Reaction. J. Am. Chem. Soc. 2013, 135, 9267–9270. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Liu, Q.; Liang, Y.H.; Tian, J.Q.; Asiri, A.M.; Sun, X.P. A Cost-Effective 3D Hydrogen Evolution Cathode with High Catalytic Activity: FeP Nanowire Array as the Active Phase. Angew. Chem. Int. Ed. 2014, 53, 12855–12859. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.L.; Liu, P.F.; Yan, X.C.; Gu, L.; Yang, Z.Z.; Yang, H.G.; Qiu, S.L.; Yao, X.D. Atomically Isolated Nickel Species Anchored on Graphitized Carbon for Efficient Hydrogen Evolution Electrocatalysis. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.P.; Wang, Y.X.; Lin, H.L.; Zhang, H.B.; Shen, M.K.; Xie, S.H.; Zhang, Y.H.; Gao, Q.S.; Tang, Y. Porous Nanomoc@Graphite Shell Derived from a MOFs-Directed Strategy: An Efficient Electrocatalyst for the Hydrogen Evolution Reaction. J. Mater. Chem. A 2016, 4, 6006–6013. [Google Scholar] [CrossRef]
- Jacques, P.-A.; Artero, V.; Pécaut, J.; Fontecave, M. Cobalt and Nickel Diimine-Dioxime Complexes as Molecular Electrocatalysts for Hydrogen Evolution with Low Overvoltages. Proc. Natl. Acad. Sci. USA 2009, 106, 20627–20632. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.-G.; Zhang, H.-L.; Tao, Y.-W.; Wu, Q.-Y.; Tian, D.; Zhang, Q. Recent Progress in Ligand-Centered Homogeneous Electrocatalysts for Hydrogen Evolution Reaction. Inorg. Chem. Front. 2019, 6, 343–354. [Google Scholar] [CrossRef]
- Uchida, T.; Mogami, H.; Yamakata, A.; Sasaki, Y.; Osawa, M. Hydrogen Evolution Reaction Catalyzed by Proton-Coupled Redox Cycle of 4,4′-Bipyridine Monolayer Adsorbed on Silver Electrodes. J. Am. Chem. Soc. 2008, 130, 10862–10863. [Google Scholar] [CrossRef]
- McAllister, J.; Bandeira, N.A.G.; McGlynn, J.C.; Ganin, A.Y.; Song, Y.-F.; Bo, C.; Miras, H.N. Tuning and Mechanistic Insights of Metal Chalcogenide Molecular Catalysts for the Hydrogen-Evolution Reaction. Nat. Commun. 2019, 10, 370. [Google Scholar] [CrossRef]
- Benck, J.D.; Chen, Z.; Kuritzky, L.Y.; Forman, A.J.; Jaramillo, T.F. Amorphous Molybdenum Sulfide Catalysts for Electrochemical Hydrogen Production: Insights into the Origin of their Catalytic Activity. ACS Catal. 2012, 2, 1916–1923. [Google Scholar] [CrossRef]
- Vrubel, H.; Hu, X. Growth and Activation of an Amorphous Molybdenum Sulfide Hydrogen Evolving Catalyst. ACS Catal. 2013, 3, 2002–2011. [Google Scholar] [CrossRef] [Green Version]
- Kibsgaard, J.; Chen, Z.; Reinecke, B.N.; Jaramillo, T.F. Engineering the Surface Structure of MoS2 to Preferentially Expose Active Edge Sites for Electrocatalysis. Nat. Mater. 2012, 11, 963. [Google Scholar] [CrossRef] [PubMed]
- Shinde, P.S.; Fontenot, P.R.; Donahue, J.P.; Waters, J.L.; Kung, P.; McNamara, L.E.; Hammer, N.I.; Gupta, A.; Pan, S. Synthesis of MoS2 from [Mo3S7(S2CNEt2)3]I for Enhancing Photoelectrochemical Performance and Stability of Cu2O Photocathode toward Efficient Solar Water Splitting. J. Mater. Chem. A 2018, 6, 9569–9582. [Google Scholar] [CrossRef]
- Merki, D.; Fierro, S.; Vrubel, H.; Hu, X.L. Amorphous Molybdenum Sulfide Films as Catalysts for Electrochemical Hydrogen Production in Water. Chem. Sci. 2011, 2, 1262–1267. [Google Scholar] [CrossRef]
- Vrubel, H.; Merki, D.; Hu, X. Hydrogen Evolution Catalyzed by MoS3 and MoS2 Particles. Energy Environ. Sci. 2012, 5, 6136–6144. [Google Scholar] [CrossRef]
- Lin, L.; Miao, N.; Wen, Y.; Zhang, S.; Ghosez, P.; Sun, Z.; Allwood, D.A. Sulfur-Depleted Monolayered Molybdenum Disulfide Nanocrystals for Superelectrochemical Hydrogen Evolution Reaction. ACS Nano 2016, 10, 8929–8937. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.F.; Zhang, H.; Li, S.; Wang, R.X.; Sun, X.; Zhou, M.; Zhou, J.F.; Lou, X.W.; Xie, Y. Defect-Rich MoS2 Ultrathin Nanosheets with Additional Active Edge Sites for Enhanced Electrocatalytic Hydrogen Evolution. Adv. Mater. 2013, 25, 5807–5813. [Google Scholar] [CrossRef] [PubMed]
- Merki, D.; Vrubel, H.; Rovelli, L.; Fierro, S.; Hu, X. Fe, Co, and Ni Ions Promote the Catalytic Activity of Amorphous Molybdenum Sulfide Films for Hydrogen Evolution. Chem. Sci. 2012, 3, 2515–2525. [Google Scholar] [CrossRef]
- Roger, I.; Moca, R.; Miras, H.N.; Crawford, K.G.; Moran, D.A.J.; Ganin, A.Y.; Symes, M.D. The Direct Hydrothermal Deposition of Cobalt-Doped MoS2 onto Fluorine-Doped SnO2 Substrates for Catalysis of the Electrochemical Hydrogen Evolution Reaction. J. Mater. Chem. A 2017, 5, 1472–1480. [Google Scholar] [CrossRef]
- Gao, D.; Si, M.; Li, J.; Zhang, J.; Zhang, Z.; Yang, Z.; Xue, D. Ferromagnetism in Freestanding MoS2 Nanosheets. Nanoscale Res. Lett. 2013, 8, 129. [Google Scholar] [CrossRef]
- Li, G.; Zhang, D.; Qiao, Q.; Yu, Y.; Peterson, D.; Zafar, A.; Kumar, R.; Curtarolo, S.; Hunte, F.; Shannon, S.; et al. All the Catalytic Active Sites of MoS2 for Hydrogen Evolution. J. Am. Chem. Soc. 2016, 138, 16632–16638. [Google Scholar] [CrossRef] [PubMed]
- Gan, X.; Zhao, H.; Quan, X. Two-Dimensional MoS2: A Promising Building Block for Biosensors. Biosens. Bioelectron. 2017, 89, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Sengupta, K.; Bandyopadhyay, S.; Dey, A. Ammonium Tetrathiomolybdate as a Novel Electrode Material for Convenient Tuning of the Kinetics of Electrochemical O2 Reduction by using Iron–Porphyrin Catalysts. J. Mater. Chem. A 2016, 4, 6819–6823. [Google Scholar] [CrossRef]
- Zheng, D.; Zhang, G.; Hou, Y.; Wang, X. Layering MoS2 on Soft Hollow g-C3N4 Nanostructures for Photocatalytic Hydrogen Evolution. Appl. Catal. A 2016, 521, 2–8. [Google Scholar] [CrossRef]
- Eda, G.; Yamaguchi, H.; Voiry, D.; Fujita, T.; Chen, M.; Chhowalla, M. Photoluminescence from Chemically Exfoliated MoS2. Nano Lett. 2011, 11, 5111–5116. [Google Scholar] [CrossRef] [PubMed]
- Hamzah, I.H.; Manaf, A.A.; Sidek, O. A Study on Characteristic and Reliability of Fabricated Microfluidic Three Electrodes Sensor Based on Randle-Sevcik Equation. In Proceedings of the 2010 IEEE Asia Pacific Conference on Circuits and Systems, Kuala Lumpur, Malaysia, 6–9 December 2010; pp. 816–819. [Google Scholar]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spring, S.; Shinde, P.S.; Fontenot, P.R.; Donahue, J.P.; Pan, S. Self-Assembled Monolayers of Molybdenum Sulfide Clusters on Au Electrode as Hydrogen Evolution Catalyst for Solar Water Splitting. Inorganics 2019, 7, 79. https://doi.org/10.3390/inorganics7060079
Spring S, Shinde PS, Fontenot PR, Donahue JP, Pan S. Self-Assembled Monolayers of Molybdenum Sulfide Clusters on Au Electrode as Hydrogen Evolution Catalyst for Solar Water Splitting. Inorganics. 2019; 7(6):79. https://doi.org/10.3390/inorganics7060079
Chicago/Turabian StyleSpring, Stephanie, Pravin S. Shinde, Patricia R. Fontenot, James P. Donahue, and Shanlin Pan. 2019. "Self-Assembled Monolayers of Molybdenum Sulfide Clusters on Au Electrode as Hydrogen Evolution Catalyst for Solar Water Splitting" Inorganics 7, no. 6: 79. https://doi.org/10.3390/inorganics7060079
APA StyleSpring, S., Shinde, P. S., Fontenot, P. R., Donahue, J. P., & Pan, S. (2019). Self-Assembled Monolayers of Molybdenum Sulfide Clusters on Au Electrode as Hydrogen Evolution Catalyst for Solar Water Splitting. Inorganics, 7(6), 79. https://doi.org/10.3390/inorganics7060079