On the Possible Coordination on a 3MC State Itself? Mechanistic Investigation Using DFT-Based Methods

Abstract
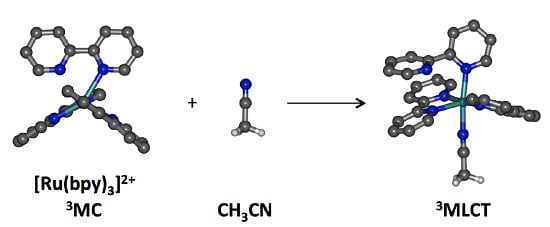
1. Introduction
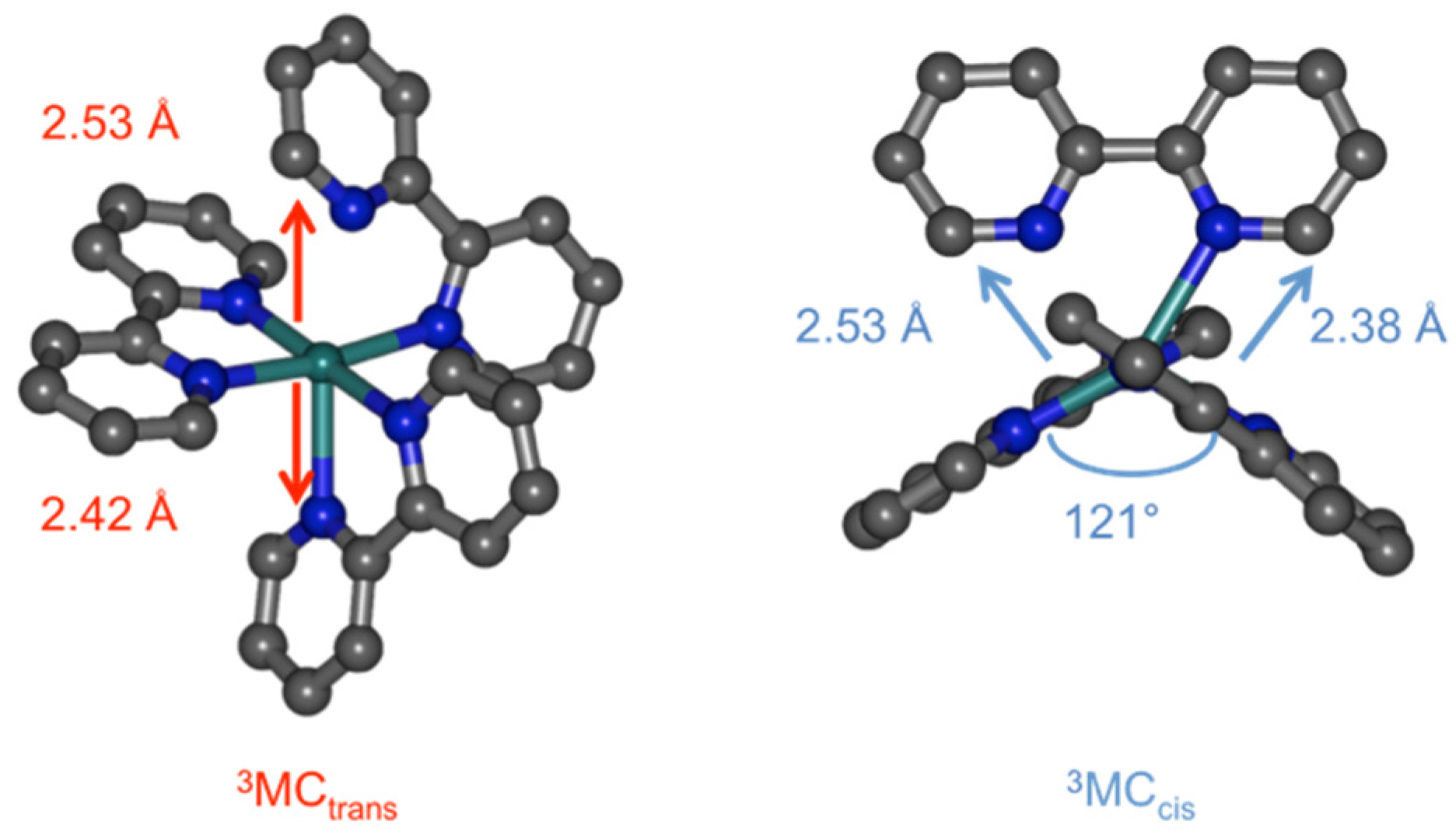
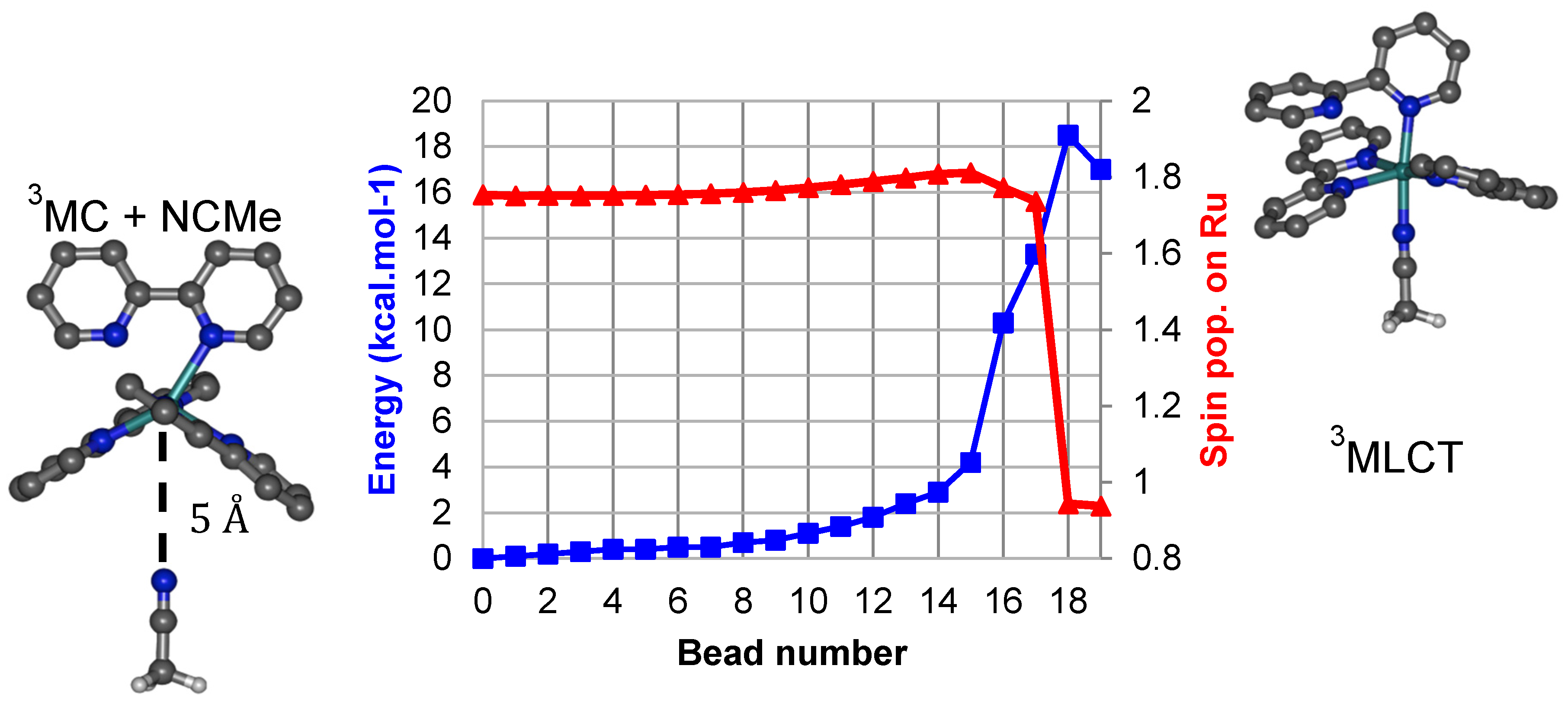
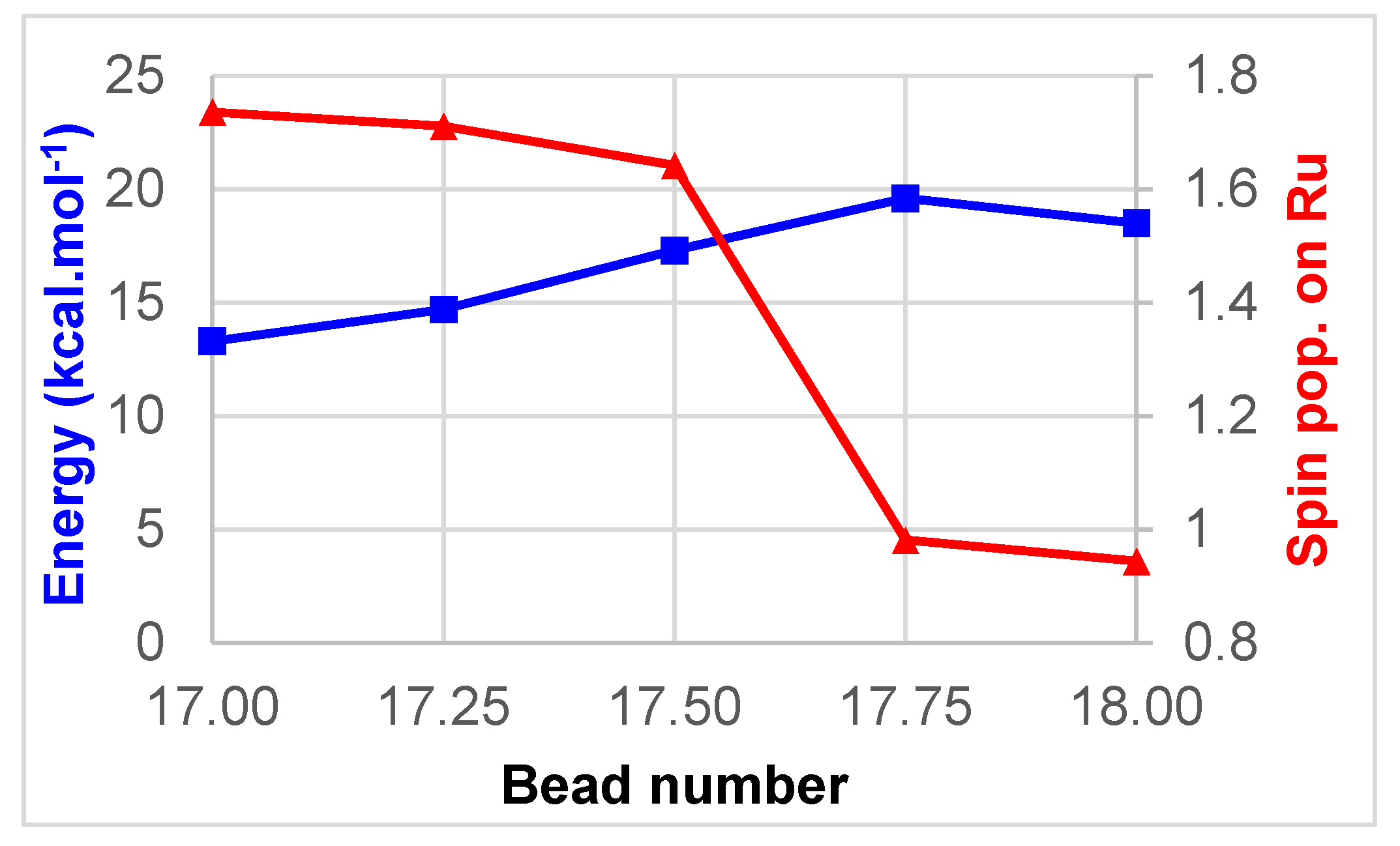
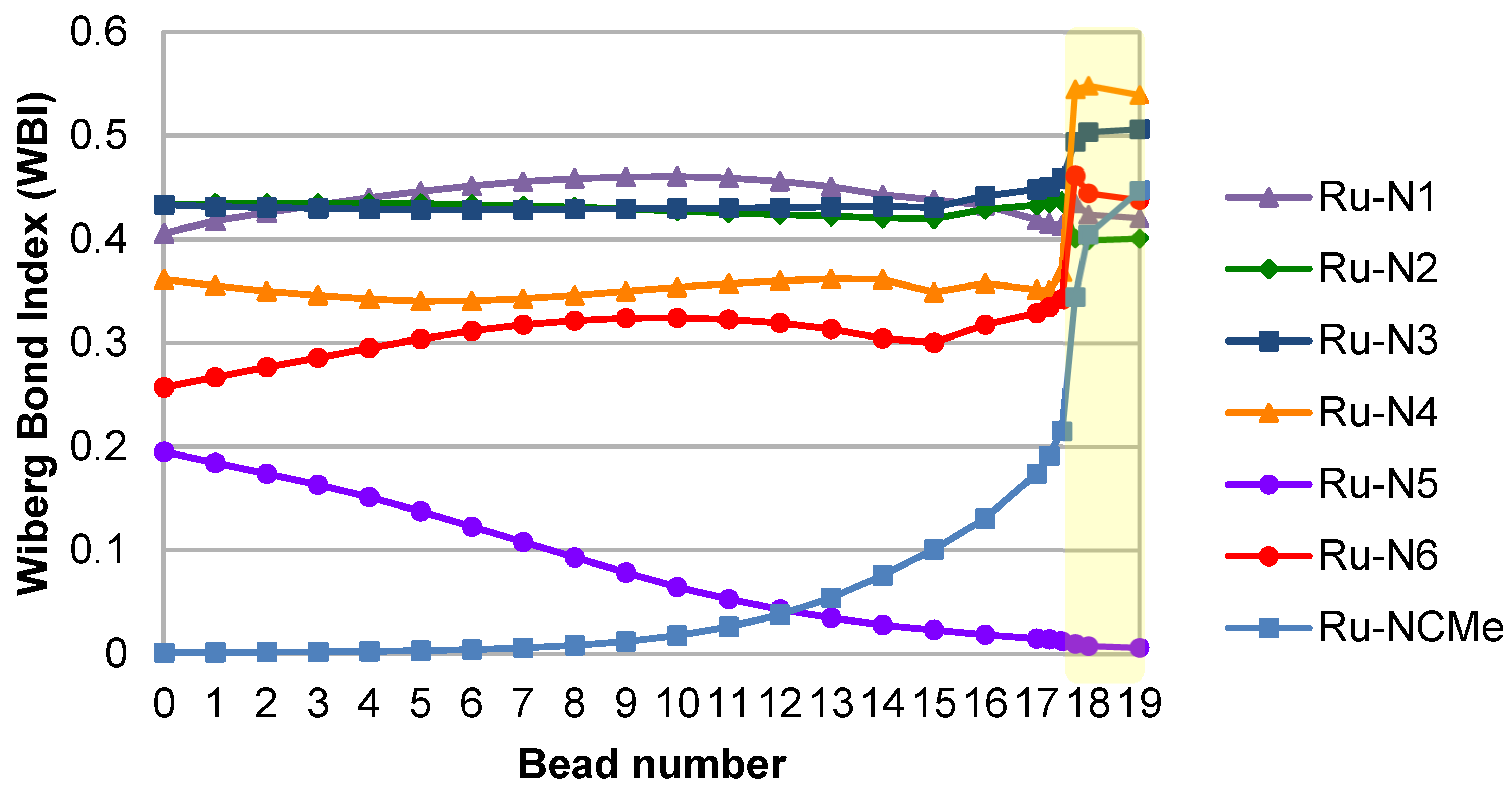
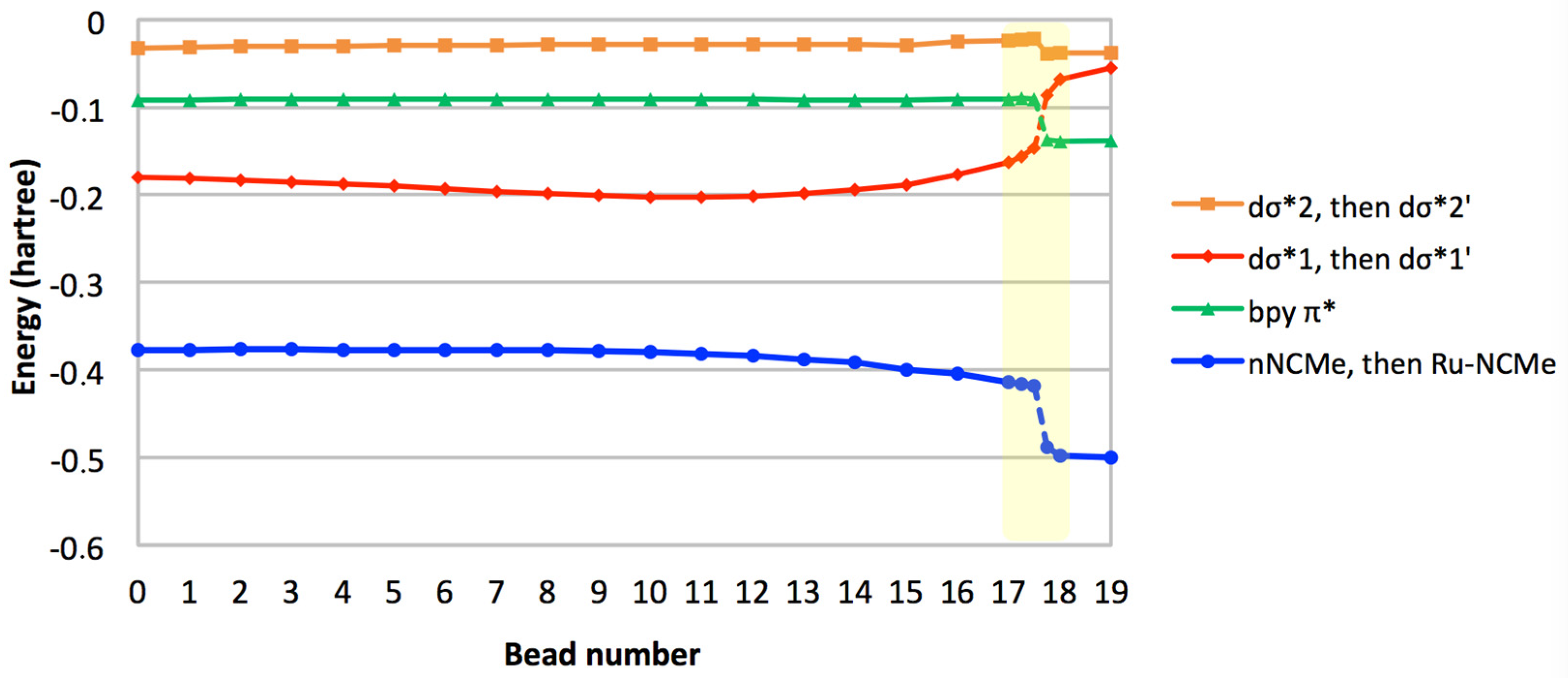
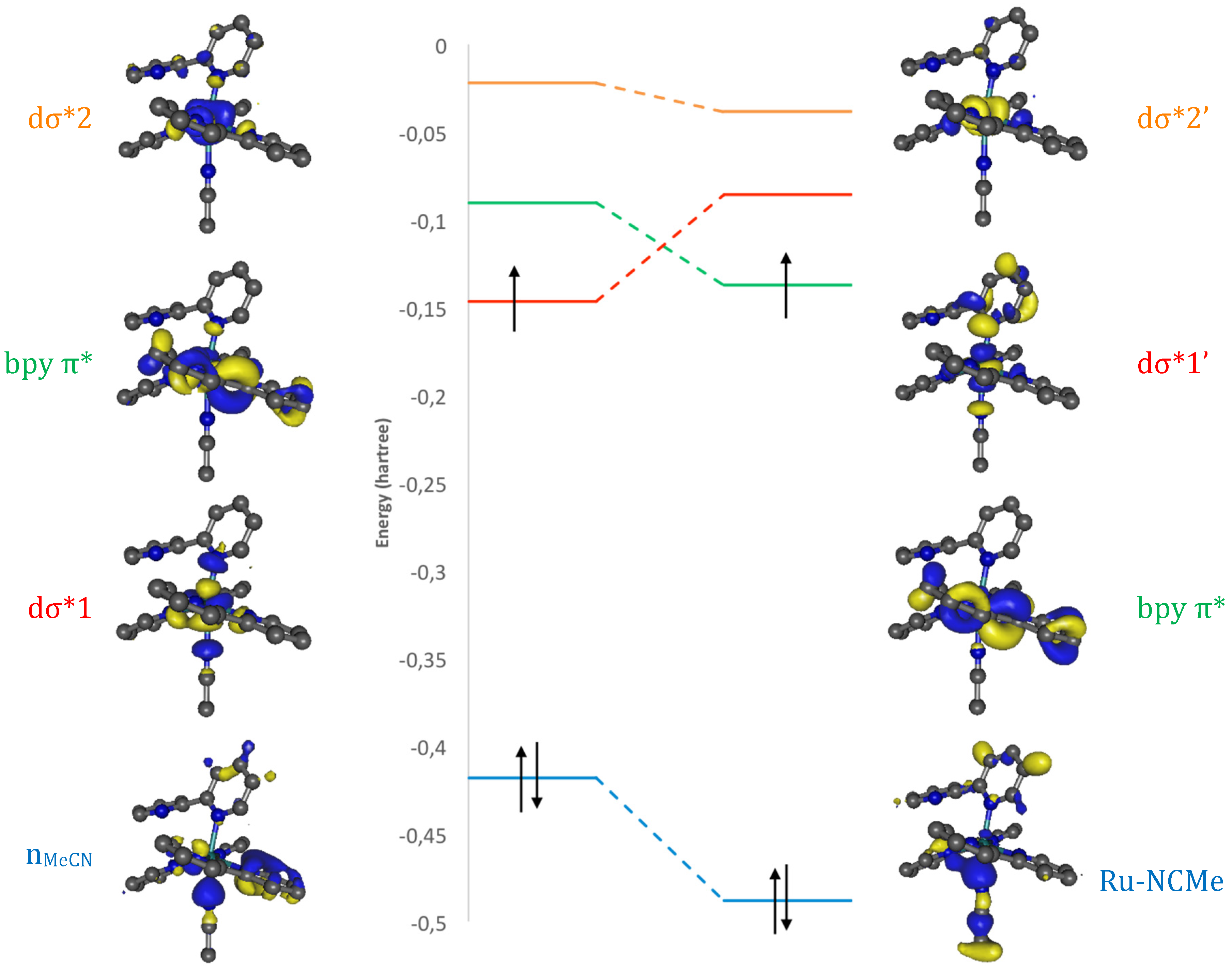
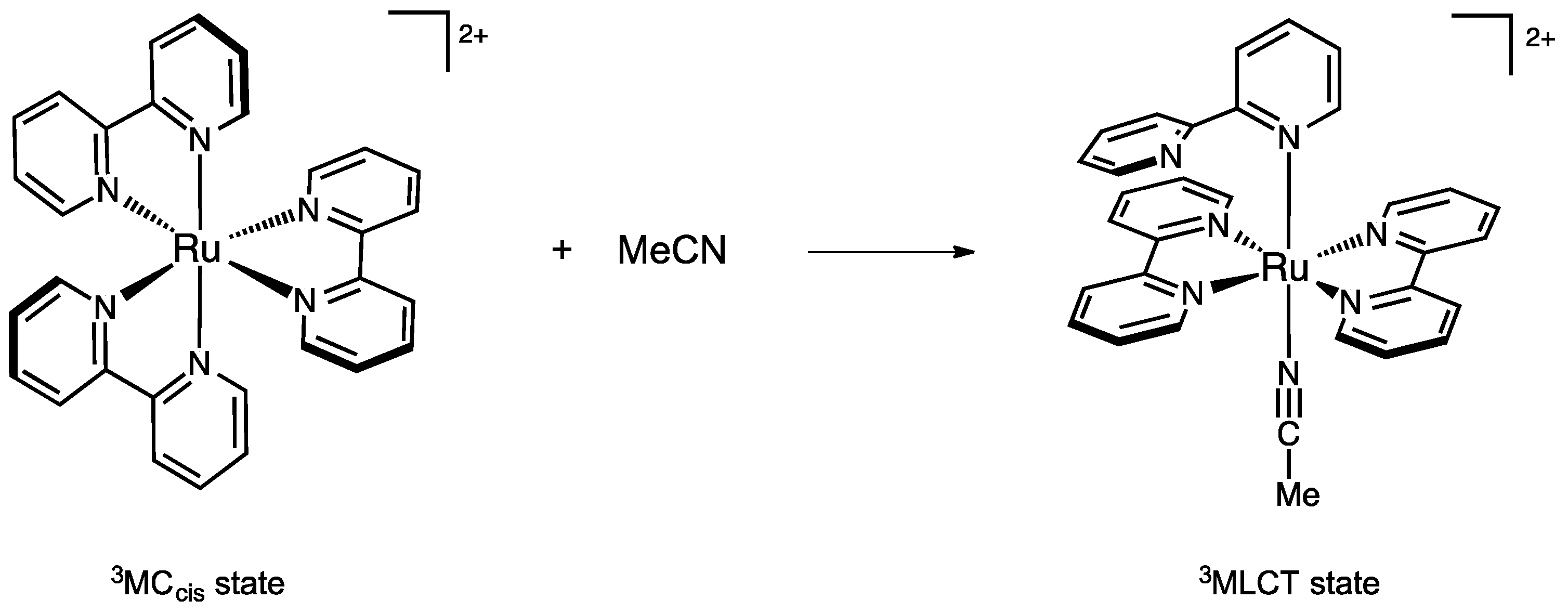
2. Results and Discussion
3. Computational Details and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Juris, A.; Balzani, V.; Barigelletti, F.; Campagna, S.; Belser, P.; von Zelewsky, A. Ru(II) Polypyridine Complexes: Photophysics, Photochemistry, Electrochemistry, and Chemiluminescence. Coord. Chem. Rev. 1988, 84, 85–277. [Google Scholar] [CrossRef]
- Van Houten, J.; Watts, R.J. Photochemistry of Tris(2,2’-bipyridyl)ruthenium(II) in Aqueous Solution. Inorg. Chem. 1978, 17, 3381–3385. [Google Scholar] [CrossRef]
- Alary, F.; Heully, J.-L.; Bijeire, L.; Vicendo, P. Is the 3MLCT the Only Photoreactive State of Polypyridyl Complexes? Inorg. Chem. 2007, 46, 3154–3165. [Google Scholar] [CrossRef]
- Alary, F.; Boggio-Pasqua, M.; Heully, J.-L.; Marsden, C.J.; Vicendo, P. Theoretical Characterization of the Lowest Triplet Excited States of the Tris-(1,4,5,8-tetraazaphenanthrene)Ruthenium Dication Complex. Inorg. Chem. 2008, 47, 5259–5266. [Google Scholar] [CrossRef]
- Heully, J.-L.; Alary, F.; Boggio-Pasqua, M. Spin-orbit effects on the photophysical properties of Ru(bpy)32+. J. Chem. Phys. 2009, 131, 184308. [Google Scholar] [CrossRef]
- Sanz García, J.; Alary, F.; Boggio-Pasqua, M.; Dixon, I.M.; Heully, J.-L. Is Photoisomerization Required for NO Photorelease in Ruthenium Nitrosyl Complexes? J. Mol. Model. 2016, 22, 284. [Google Scholar] [CrossRef]
- Göttle, A.J.; Alary, F.; Boggio-Pasqua, M.; Dixon, I.M.; Heully, J.-L.; Bahreman, A.; Askes, S.H.C.; Bonnet, S. Pivotal Role of a Pentacoordinate 3MC State on the Photocleavage Efficiency of a Thioether Ligand in Ruthenium(II) Complexes: A Theoretical Mechanistic Study. Inorg. Chem. 2016, 55, 4448–4456. [Google Scholar] [CrossRef]
- Dixon, I.M.; Heully, J.-L.; Alary, F.; Elliott, P.I.P. Theoretical illumination of highly original photoreactive 3MC states and the mechanism of the photochemistry of Ru(II) tris(bidentate) complexes. Phys. Chem. Chem. Phys. 2017, 19, 27765–27778. [Google Scholar] [CrossRef]
- Daniel, C.; Gourlaouen, C. Chemical bonding alteration upon electronic excitation in transition metal complexes. Coord. Chem. Rev. 2017, 344, 131–149. [Google Scholar] [CrossRef]
- Abrahamsson, M.; Lundqvist, M.J.; Wolpher, H.; Johansson, O.; Eriksson, L.; Bergquist, J.; Rasmussen, T.; Becker, H.-C.; Hammarström, L.; Norrby, P.-O.; et al. Steric Influence on the Excited-State Lifetimes of Ruthenium Complexes with Bipyridyl–Alkanylene–Pyridyl Ligands. Inorg. Chem. 2008, 47, 3540–3548. [Google Scholar] [CrossRef]
- Borg, O.A.; Godinho, S.S.M.C.; Lundqvist, M.J.; Lunell, S.; Persson, P. Computational Study of the Lowest Triplet State of Ruthenium Polypyridyl Complexes Used in Artificial Photosynthesis. J. Phys. Chem. A 2008, 112, 4470–4476. [Google Scholar] [CrossRef]
- Österman, T.; Abrahamsson, M.; Becker, H.-C.; Hammarström, L.; Persson, P. Influence of Triplet State Multidimensionality on Excited State Lifetimes of Bis-tridentate Ru(II) Complexes: A Computational Study. J. Phys. Chem. A 2012, 116, 1041–1050. [Google Scholar] [CrossRef]
- Fredin, L.A.; Wallenstein, J.; Sundin, E.; Jarenmark, M.; Barbosa de Mattos, D.F.; Persson, P.; Abrahamsson, M. Excited State Dynamics of Bistridentate and Trisbidentate Ru(II) Complexes of Quinoline-Pyrazole Ligands. Inorg. Chem. 2019, 58, 16354–16363. [Google Scholar] [CrossRef]
- Salassa, L.; Garino, C.; Salassa, G.; Gobetto, R.; Nervi, C. Mechanism of Ligand Photodissociation in Photoactivable [Ru(bpy)2L2]2+ Complexes: A Density Functional Theory Study. J. Am. Chem. Soc. 2008, 130, 9590–9597. [Google Scholar] [CrossRef]
- Salassa, L.; Garino, C.; Salassa, G.; Nervi, C.; Gobetto, R.; Lamberti, C.; Gianolio, D.; Bizzarri, R.; Sadler, P.J. Ligand-Selective Photodissociation from [Ru(bpy)(4AP)4]2+: A Spectroscopic and Computational Study. Inorg. Chem. 2009, 48, 1469–1481. [Google Scholar] [CrossRef]
- Borfecchia, E.; Garino, C.; Gianolio, D.; Salassa, L.; Gobetto, R.; Lamberti, C. Monitoring excited state dynamics in cis-[Ru(bpy)2(py)2]2+ by ultrafast synchrotron techniques. Catal. Today 2014, 229, 34–45. [Google Scholar] [CrossRef]
- Breivogel, A.; Meister, M.; Förster, C.; Laquai, F.; Heinze, K. Excited State Tuning of Bis(tridentate) Ruthenium(II) Polypyridine Chromophores by Push-Pull Effects and Bite Angle Optimization: A Comprehensive Experimental and Theoretical Study. Chem. Eur. J. 2013, 19, 13745–13760. [Google Scholar] [CrossRef]
- Kreitner, C.; Heinze, K. Excited state decay of cyclometalated polypyridine ruthenium complexes: Insight from theory and experiment. Dalton Trans. 2016, 45, 13631–13647. [Google Scholar] [CrossRef]
- Sun, Q.; Mosquera-Vazquez, S.; Lawson Daku, L.M.; Guénée, L.; Goodwin, H.A.; Vauthey, E.; Hauser, A. Experimental Evidence of Ultrafast Quenching of the 3MLCT Luminescence in Ruthenium(II) Tris-bipyridyl Complexes via a 3dd State. J. Am. Chem. Soc. 2013, 135, 13660–13663. [Google Scholar] [CrossRef]
- Sun, Q.; Dereka, B.; Vauthey, E.; Lawson Daku, L.M.; Hauser, A. Ultrafast Transient IR Spectroscopy and DFT Calculations of Ruthenium(II) Polypyridyl Complexes. Chem. Sci. 2017, 8, 223–230. [Google Scholar] [CrossRef]
- Ding, L.; Chung, L.W.; Morokuma, K. Excited-State Proton Transfer Controls Irreversibility of Photoisomerization in Mononuclear Ruthenium(II) Monoaquo Complexes: A DFT Study. J. Chem. Theory Comput. 2014, 10, 668–675. [Google Scholar] [CrossRef]
- Greenough, S.E.; Roberts, G.M.; Smith, N.A.; Horbury, M.D.; McKinlay, R.G.; Żurek, J.M.; Paterson, M.J.; Sadler, P.J.; Stavros, V.G. Ultrafast photo-induced ligand solvolysis of cis-[Ru(bipyridine)2(nicotinamide)2]2+: Experimental and theoretical insight into its photoactivation mechanism. Phys. Chem. Chem. Phys. 2014, 16, 19141–19155. [Google Scholar] [CrossRef]
- Camilo, M.R.; Cardoso, C.R.; Carlos, R.M.; Lever, A.B.P. Photosolvolysis of cis-[Ru(α-diimine)2(4-aminopyridine)2]2+ Complexes: Photophysical, Spectroscopic, and Density Functional Theory Analysis. Inorg. Chem. 2014, 53, 3694–3708. [Google Scholar] [CrossRef]
- Tu, Y.-J.; Mazumder, S.; Endicott, J.F.; Turro, C.; Kodanko, J.J.; Schlegel, H.B. Selective Photodissociation of Acetonitrile Ligands in Ruthenium Polypyridyl Complexes Studied by Density Functional Theory. Inorg. Chem. 2015, 54, 8003–8011. [Google Scholar] [CrossRef]
- Nisbett, K.; Tu, Y.-J.; Turro, C.; Kodanko, J.J.; Schlegel, H.B. DFT Investigation of Ligand Photodissociation in [Ru(II)(tpy)(bpy)(py)]2+ and [Ru(II)(tpy)(Me2bpy)(py)]2+ Complexes. Inorg. Chem. 2018, 57, 231–240. [Google Scholar] [CrossRef]
- Petroni, A.; Slep, L.D.; Etchenique, R. Ruthenium(II) 2,2’-Bipyridyl Tetrakis Acetonitrile Undergoes Selective Axial Photocleavage. Inorg. Chem. 2008, 47, 951–956. [Google Scholar] [CrossRef]
- Rojas Pérez, Y.; Slep, L.D.; Etchenique, R. Cis–Trans Interconversion in Ruthenium(II) Bipyridine Complexes. Inorg. Chem. 2019, 58, 11606–11613. [Google Scholar] [CrossRef]
- Feng, L.; Wang, Y.; Jia, J. Triplet Ground-State-Bridged Photochemical Process: Understanding the Photoinduced Chiral Inversion at the Metal Center of [Ru(phen)2(l-ser)]+ and Its Bipy Analogues. Inorg. Chem. 2017, 56, 14467–14476. [Google Scholar] [CrossRef]
- Jacquemin, D.; Escudero, D. The Short Device Lifetimes of Blue PhOLEDs: Insights into the Photostability of Blue Ir(III) Complexes. Chem. Sci. 2017, 8, 7844–7850. [Google Scholar] [CrossRef]
- Arroliga-Rocha, S.; Escudero, D. Facial and Meridional Isomers of Tris(bidentate) Ir(III) Complexes: Unravelling Their Different Excited State Reactivity. Inorg. Chem. 2018, 57, 12106–12112. [Google Scholar] [CrossRef]
- Escudero, D. Mer-Ir(ppy)3 to Fac-Ir(ppy)3 Photoisomerization. ChemPhotoChem 2019, 3, 697–701. [Google Scholar] [CrossRef]
- Göttle, A.J.; Dixon, I.M.; Alary, F.; Heully, J.-L.; Boggio-Pasqua, M. Adiabatic Versus Nonadiabatic Photoisomerization in Photochromic Ruthenium Sulfoxide Complexes: A Mechanistic Picture from Density Functional Theory Calculations. J. Am. Chem. Soc. 2011, 133, 9172–9174. [Google Scholar] [CrossRef]
- Göttle, A.J.; Alary, F.; Dixon, I.M.; Heully, J.-L.; Boggio-Pasqua, M. Unravelling the S→O Linkage Photoisomerization Mechanisms in cis- and trans-[Ru(bpy)2(DMSO)2]2+ Using Density Functional Theory. Inorg. Chem. 2014, 53, 6752–6760. [Google Scholar] [CrossRef]
- Sanz García, J.; Alary, F.; Boggio-Pasqua, M.; Dixon, I.M.; Malfant, I.; Heully, J.-L. Establishing the Two-Photon Linkage Isomerization Mechanism in the Nitrosyl Complex trans-[RuCl(NO)(py)4]2+ by DFT and TDDFT. Inorg. Chem. 2015, 54, 8310–8318. [Google Scholar] [CrossRef]
- Lebon, E.; Bastin, S.; Sutra, P.; Vendier, L.; Piau, R.E.; Dixon, I.M.; Boggio-Pasqua, M.; Alary, F.; Heully, J.-L.; Igau, A.; et al. Can a Functionalized Phosphine Ligand Promote Room Temperature Luminescence of the [Ru(bpy)(tpy)]2+ Core? Chem. Commun. 2012, 48, 741–743. [Google Scholar] [CrossRef]
- Triadon, A.; Grelaud, G.; Richy, N.; Mongin, O.; Moxey, G.J.; Dixon, I.M.; Yang, X.; Wang, G.; Barlow, A.; Rault-Berthelot, J.; et al. Linear and Third-Order Nonlinear Optical Properties of Fe(η5-C5Me5)(κ2-dppe)- and trans-Ru(κ2-dppe)2-Alkynyl Complexes Containing 2-Fluorenyl End Groups. Organometallics 2018, 37, 2245–2262. [Google Scholar] [CrossRef]
- Guillon, T.; Boggio-Pasqua, M.; Alary, F.; Heully, J.-L.; Lebon, E.; Sutra, P.; Igau, A. Theoretical Investigation on the Photophysical Properties of Model Ruthenium Complexes with Diazabutadiene Ligands [Ru(bpy)3−x(dab)x]2+ (x = 1–3). Inorg. Chem. 2010, 49, 8862–8872. [Google Scholar] [CrossRef]
- Vieuxmaire, O.P.J.; Piau, R.E.; Alary, F.; Heully, J.-L.; Sutra, P.; Igau, A.; Boggio-Pasqua, M. Theoretical Investigation of Phosphinidene Oxide Polypyridine Ruthenium(II) Complexes: Toward the Design of a New Class of Photochromic Compounds. J. Phys. Chem. A 2013, 117, 12821–12830. [Google Scholar] [CrossRef]
- Dixon, I.M.; Alary, F.; Boggio-Pasqua, M.; Heully, J.-L. The (N4C2)2− Donor Set as Promising Motif for Bis(tridentate) Iron(II) Photoactive Compounds. Inorg. Chem. 2013, 52, 13369–13374. [Google Scholar] [CrossRef]
- Dixon, I.M.; Khan, S.; Alary, F.; Boggio-Pasqua, M.; Heully, J.-L. Probing the photophysical capability of mono and bis(cyclometallated) Fe(II) polypyridine complexes using inexpensive ground state DFT. Dalton Trans. 2014, 43, 15898–15905. [Google Scholar] [CrossRef]
- Dixon, I.M.; Alary, F.; Boggio-Pasqua, M.; Heully, J.-L. Reversing the relative 3MLCT-3MC order in Fe(II) complexes using cyclometallating ligands: A computational study aiming at luminescent Fe(II) complexes. Dalton Trans. 2015, 44, 13498–13503. [Google Scholar] [CrossRef]
- Dixon, I.M.; Boissard, G.; Whyte, H.; Alary, F.; Heully, J.-L. Computational Estimate of the Photophysical Capabilities of Four Series of Organometallic Iron(II) Complexes. Inorg. Chem. 2016, 55, 5089–5091. [Google Scholar] [CrossRef][Green Version]
- Durham, B.; Caspar, J.V.; Nagle, J.K.; Meyer, T.J. Photochemistry of Ru(bpy)32+. J. Am. Chem. Soc. 1982, 104, 4803–4810. [Google Scholar] [CrossRef]
- Kirchhoff, J.R.; McMillin, D.R.; Marnot, P.A.; Sauvage, J.-P. Photochemistry and Photophysics of Bis(terpyridyl) Complexes of Ru(II) in Fluid Solution. Evidence for the Formation of an η2-Diphenylterpyridine Complex. J. Am. Chem. Soc. 1985, 107, 1138–1141. [Google Scholar] [CrossRef]
- Thompson, D.W.; Wishart, J.F.; Brunschwig, B.S.; Sutin, N. Efficient Generation of the Ligand Field Excited State of Tris-(2,2’-bipyridine)–ruthenium(II) through Sequential Two-Photon Capture by [Ru(bpy)3]2+ or Electron Capture by [Ru(bpy)3]3+. J. Phys. Chem. A 2001, 105, 8117–8122. [Google Scholar] [CrossRef]
- Kunnus, K.; Josefsson, I.; Rajkovic, I.; Schreck, S.; Quevedo, W.; Beye, M.; Weniger, C.; Grübel, S.; Scholz, M.; Nordlund, D.; et al. Identification of the dominant photochemical pathways and mechanistic insights to the ultrafast ligand exchange of Fe(CO)5 to Fe(CO)4EtOH. Struct. Dyn. 2016, 3, 043204. [Google Scholar] [CrossRef]
- Reinhard, M.; Auböck, G.; Besley, N.A.; Clark, I.P.; Greetham, G.M.; Hanson-Heine, M.W.D.; Horvath, R.; Murphy, T.S.; Penfold, T.J.; Towrie, M.; et al. Photoaquation Mechanism of Hexacyanoferrate(II) Ions: Ultrafast 2D UV and Transient Visible and IR Spectroscopies. J. Am. Chem. Soc. 2017, 139, 7335–7347. [Google Scholar] [CrossRef]
- Welby, C.E.; Rice, C.R.; Elliott, P.I.P. Unambiguous Characterization of a Photoreactive Ligand-Loss Intermediate. Angew. Chem. Int. Ed. 2013, 52, 10826–10829. [Google Scholar] [CrossRef]
- Welby, C.E.; Armitage, G.K.; Bartley, H.; Wilkinson, A.; Sinopoli, A.; Uppal, B.S.; Rice, C.R.; Elliott, P.I.P. Photochemistry of Ru(II) 4,4′-Bi-1,2,3-triazolyl (btz) Complexes: Crystallographic Characterization of the Photoreactive Ligand-Loss Intermediate trans-[Ru(bpy)(κ2-btz)(κ1-btz)(NCMe)]2+. Chem. Eur. J. 2014, 20, 8467–8476. [Google Scholar] [CrossRef]
- Soupart, A.; Alary, F.; Heully, J.-L.; Elliott, P.I.P.; Dixon, I.M. Exploration of Uncharted 3PES Territory for [Ru(bpy)3]2+: A New 3MC Minimum Prone to Ligand Loss Photochemistry. Inorg. Chem. 2018, 57, 3192–3196. [Google Scholar] [CrossRef]
- Jónsson, H.; Mills, G.; Jacobsen, K.W. Nudged elastic band method for finding minimum energy paths of transitions. In Classical and Quantum Dynamics in Condensed Phase Simulations; Berne, B.J., Cicotti, G., Coker, D.F., Eds.; World Scientific: Singapore, 1998; pp. 385–404. [Google Scholar]
- Henkelman, G.; Johannesson, G.; Jónsson, H. Methods for Finding Saddle Points and Minimum Energy Paths. In Progress on Theoretical Chemistry and Physics; Schwartz, S.D., Ed.; Kluwer Academic: Dordrecht, The Netherlands, 2000; pp. 269–302. [Google Scholar]
- Smidstrup, S.; Pedersen, A.; Stokbro, K.; Jónsson, H. Improved initial guess for minimum energy path calculations. J. Chem. Phys. 2014, 140, 214106. [Google Scholar] [CrossRef]
- Soupart, A.; Alary, F.; Heully, J.-L.; Elliott, P.I.P.; Dixon, I.M. Recent Progress in Ligand Photorelease Reaction Mechanisms: Theoretical Insights Focusing on Ru(II) 3MC States. Coord. Chem. Rev. 2020, 408, 213184. [Google Scholar] [CrossRef]
- Neese, F. The ORCA Program System. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Andrae, D.; Haeussermann, U.; Dolg, M.; Stoll, H.; Preuss, H. Energy-adjusted ab initio Pseudopotentials for the Second and Third Row Transition Elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate ab initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Allouche, A.-R. Gabedit-A Graphical User Interface for Computational Chemistry Softwares. J. Comput. Chem. 2011, 32, 174–182. [Google Scholar] [CrossRef]
- Herbol, H.C.; Stevenson, J.; Clancy, P. Computational Implementation of Nudged Elastic Band, Rigid Rotation, and Corresponding Force Optimization. J. Chem. Theory Comput. 2017, 13, 3250–3259. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ru–N | 3MC + MeCN | 3MLCT | |
|---|---|---|---|
 | Ru–N1 | 2.135 | 2.118 |
| Ru–N2 | 2.080 | 2.110 | |
| Ru–N3 | 2.078 | 2.049 | |
| Ru–N4 | 2.177 | 2.057 | |
| Ru–N5 | 2.530 | 3.593 | |
| Ru–N6 | 2.384 | 2.132 | |
| Ru–NCMe | 5.000 | 2.055 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soupart, A.; Alary, F.; Heully, J.-L.; Dixon, I.M. On the Possible Coordination on a 3MC State Itself? Mechanistic Investigation Using DFT-Based Methods. Inorganics 2020, 8, 15. https://doi.org/10.3390/inorganics8020015
Soupart A, Alary F, Heully J-L, Dixon IM. On the Possible Coordination on a 3MC State Itself? Mechanistic Investigation Using DFT-Based Methods. Inorganics. 2020; 8(2):15. https://doi.org/10.3390/inorganics8020015
Chicago/Turabian StyleSoupart, Adrien, Fabienne Alary, Jean-Louis Heully, and Isabelle M. Dixon. 2020. "On the Possible Coordination on a 3MC State Itself? Mechanistic Investigation Using DFT-Based Methods" Inorganics 8, no. 2: 15. https://doi.org/10.3390/inorganics8020015
APA StyleSoupart, A., Alary, F., Heully, J.-L., & Dixon, I. M. (2020). On the Possible Coordination on a 3MC State Itself? Mechanistic Investigation Using DFT-Based Methods. Inorganics, 8(2), 15. https://doi.org/10.3390/inorganics8020015