
A Novel Machine Learning Strategy for the Prediction of Antihypertensive Peptides Derived from Food with High Efficiency





Abstract
1. Introduction
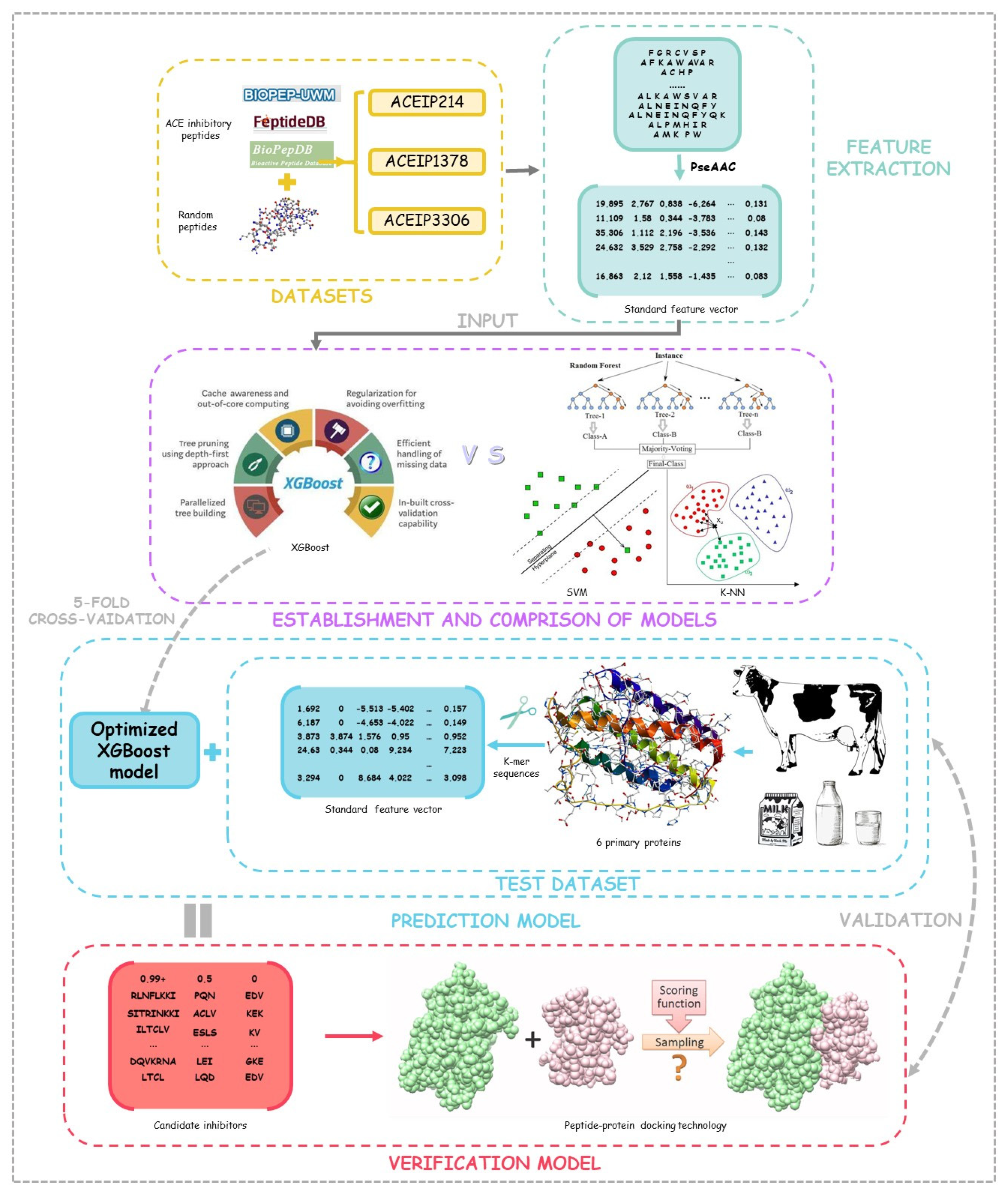
2. Method and Materials
2.1. Training Data and Test Data
2.2. Representation of Peptide Sequence Feature
2.3. Machine Learning Algorithms
2.3.1. Extreme Gradient Boosting
2.3.2. Support Vector Machine (SVM)
2.3.3. Random Forest (RF)
2.3.4. K-Nearest Neighbor (K-NN)
2.4. Performance Evaluation of Models
2.5. Prediction Model and Peptide–Protein Docking Verification
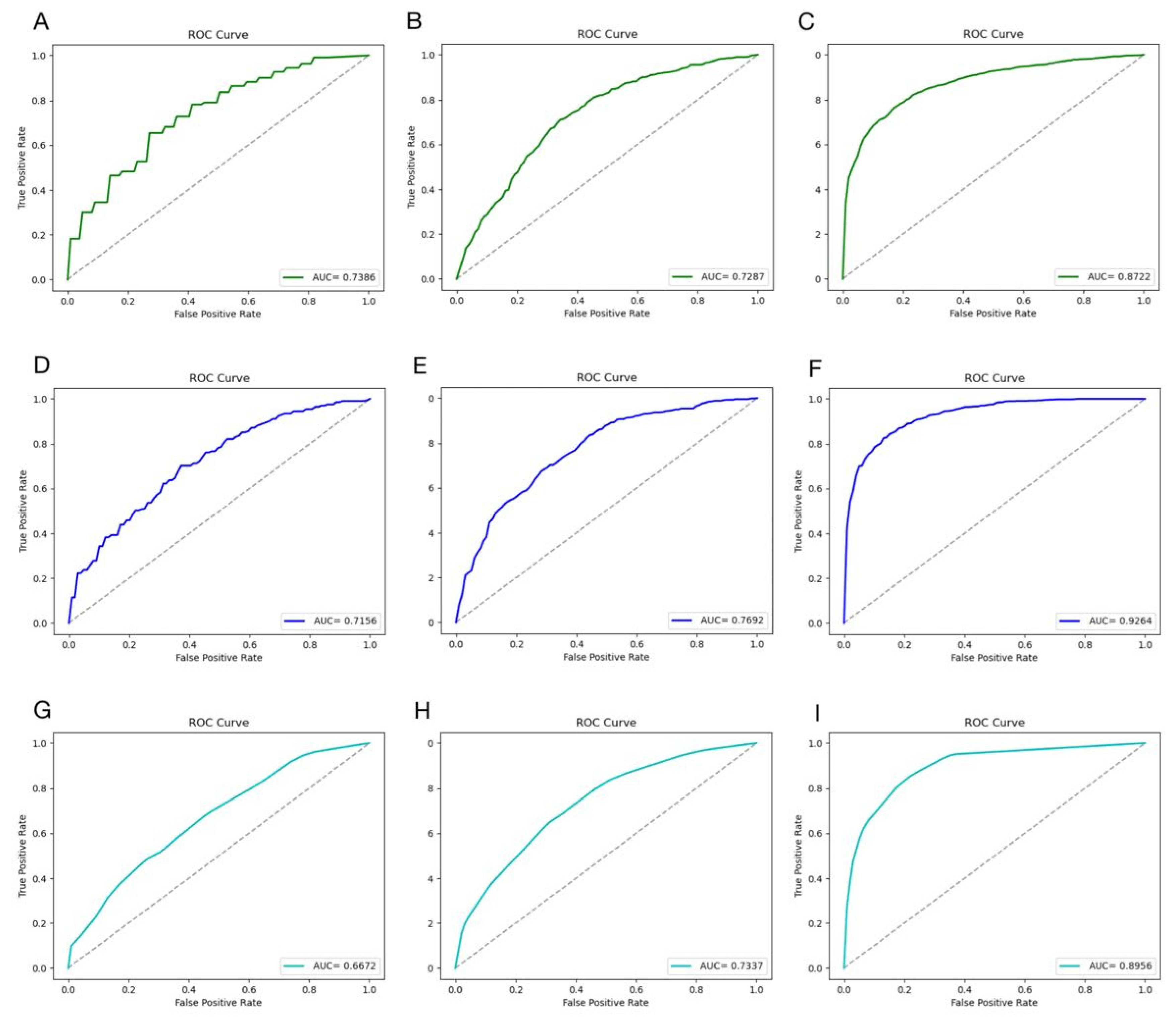
3. Results
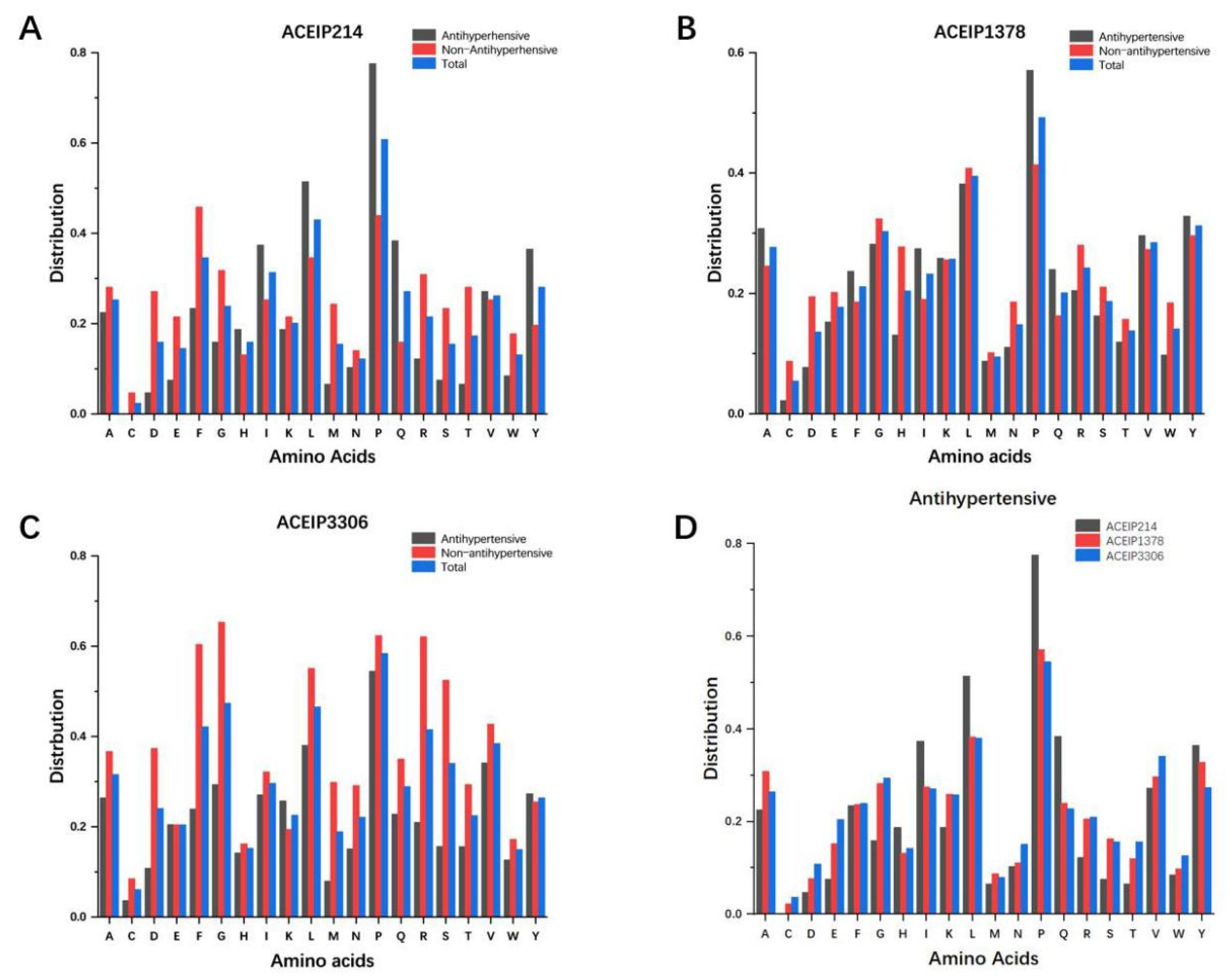
3.1. Distribution of Amino Acids in the Datasets
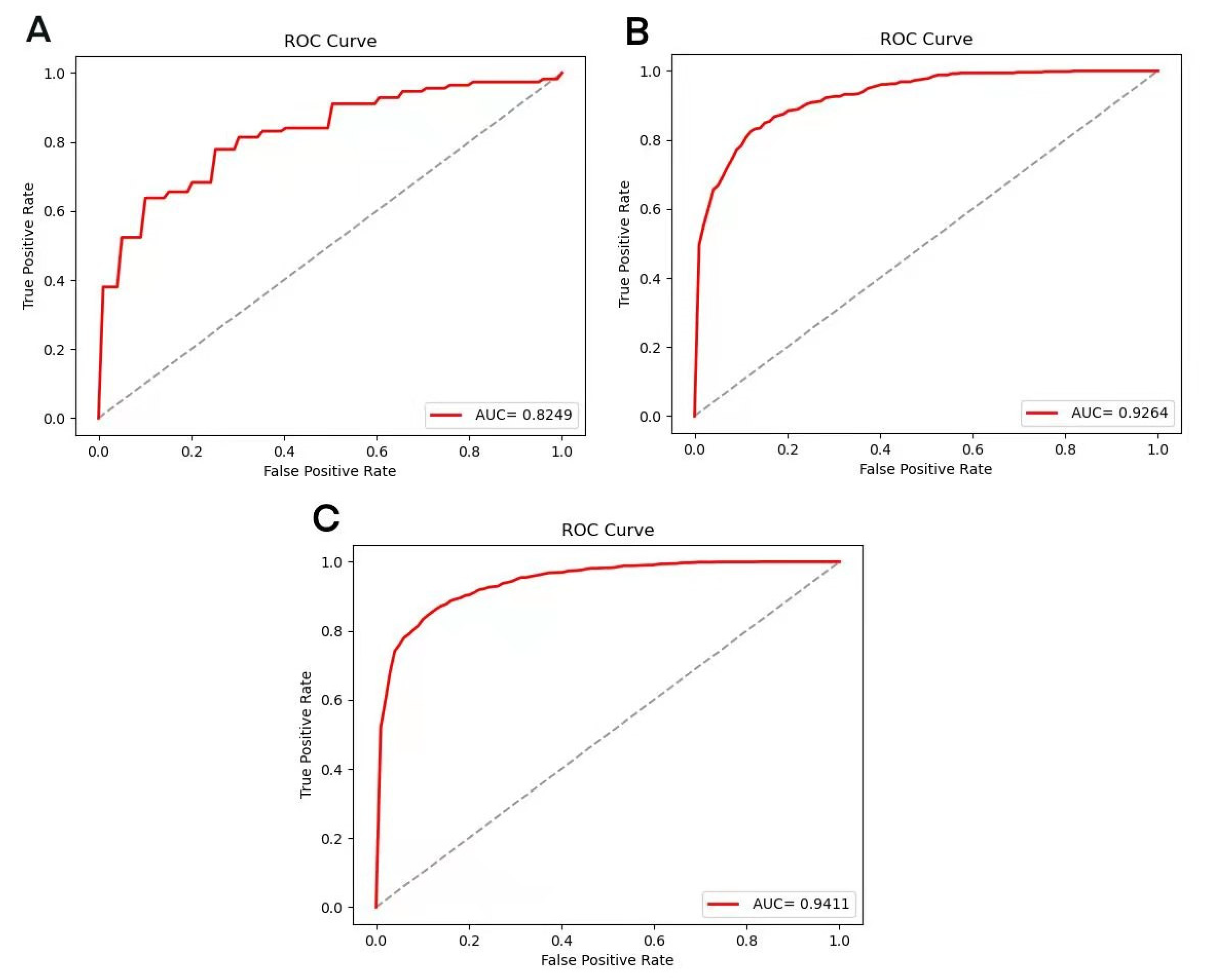
3.2. Results of XGBoost Model
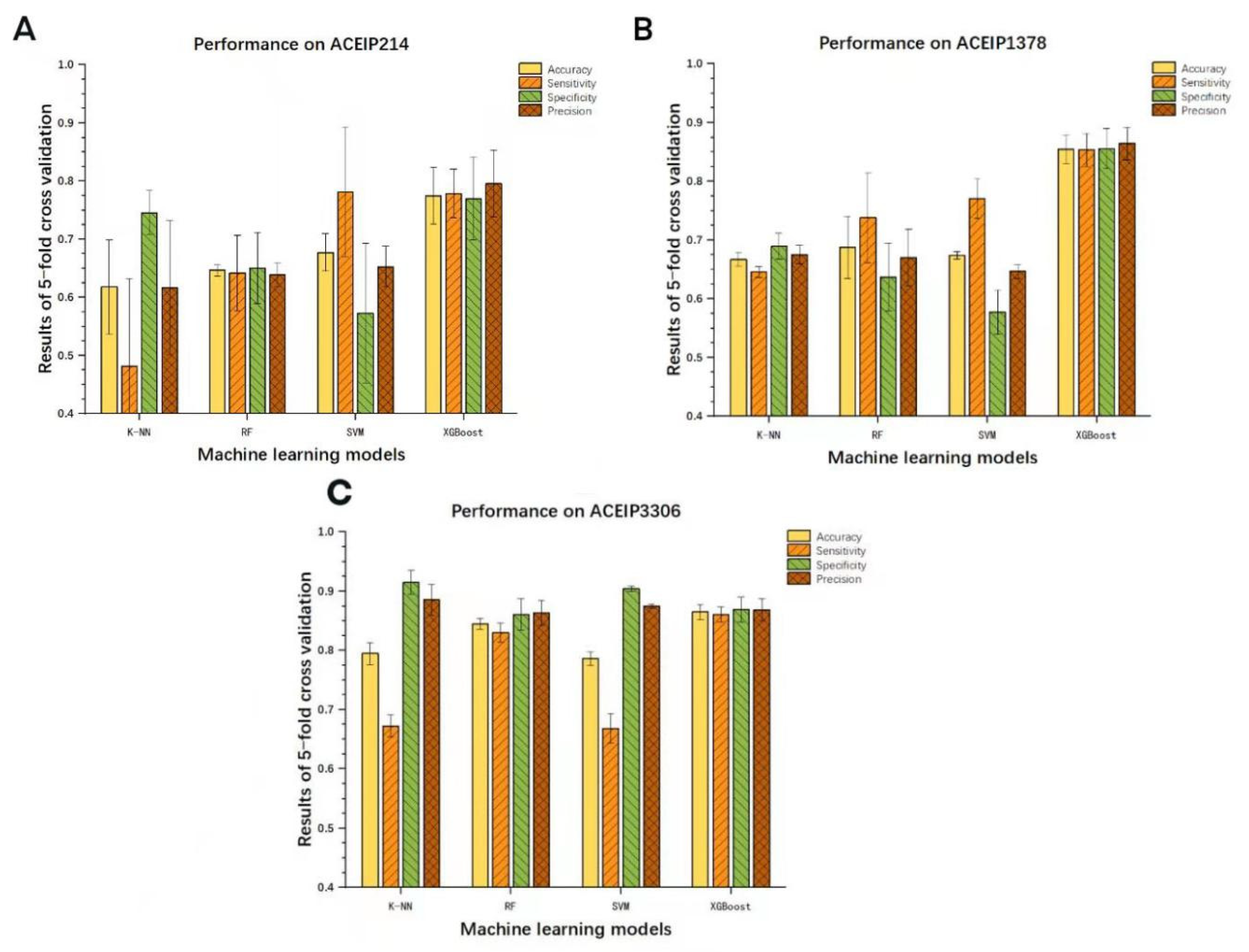
3.3. Results of Other Models
3.4. Prediction Model and Peptide–Protein Docking Verification
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Cheung, B.M.; Wat, N.M.; Man, Y.B.; Tam, S.; Cheng, C.H.; Leung, G.M.; Woo, J.; Janus, E.D.; Lau, C.P.; Lam, T.H.; et al. Relationship between the metabolic syndrome and the development of hypertension in the Hong Kong Cardiovascular risk Factor Prevalence Study-2 (CRISPS2). Am. J. Hypertens. 2008, 21, 17–22. [Google Scholar] [CrossRef]
- Huang, B.; Zhang, R.; Chen, M. Balancing the cardiovascular risk and dermatologic hazard in patients with hypertension. JAMA Dermatol. 2014, 150, 1372–1373. [Google Scholar] [CrossRef]
- Mills, K.; Bundy, J.D.; Kelly, T.N.; Reed, J.E.; Kearney, P.M.; Reynolds, K.; Chen, J.; He, J. Global burden of hypertension: Analysis of population-based studies from 89 countries. J. Hypertens. 2015, 33, e2. [Google Scholar] [CrossRef]
- Ozemek, C.; Laddu, D.R.; Arena, R.; Lavie, C.J. The role of diet for prevention and management of hypertension. Curr. Opin. Cardiol. 2018, 33, 388–393. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Luna, R.; Muñoz-Hernandez, R.; Miranda, M.L.; Costa, A.F.; Jimenez-Jimenez, L.; Vallejo-Vaz, A.J.; Muriana, F.J.; Villar, J.; Stiefel, P. Olive oil polyphenols decrease blood pressure and improve endothelial function in young women with mild hypertension. Am. J. Hypertens. 2012, 25, 1299–1304. [Google Scholar] [CrossRef]
- Bhat, Z.F.; Kumar, S.; Bhat, H.F. Antihypertensive peptides of animal origin: A review. Crit. Rev. Food Sci. Nutr. 2016, 57, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Maqueda, D.; Miralles, B.; Recio, I.; Hernández-Ledesma, B. Antihypertensive peptides from food proteins: A review. Food Funct. 2012, 3, 350–361. [Google Scholar] [CrossRef]
- Miralles, B.; Amigo, L.; Recio, I. Critical review and perspectives on food-derived antihypertensive peptides. J. Agric. Food Chem. 2018, 66, 9384–9390. [Google Scholar] [CrossRef] [PubMed]
- Ambigaipalan, P.; Al-Khalifa, A.S.; Shahidi, F. Antioxidant and angiotensin I converting enzyme (ACE) inhibitory activities of date seed protein hydrolysates prepared using Alcalase, Flavourzyme and Thermolysin. J. Funct. Foods 2015, 18, 1125–1137. [Google Scholar] [CrossRef]
- Liang, Y.; Qin, D.; Zhang, Y.; Liu, W.; Liang, G. Comprehensive interactions of ACE inhibitors with their receptor by a Support Vector Machine model and molecular docking. J. Chin. Chem. Soc. 2017, 64, 310–320. [Google Scholar] [CrossRef]
- Guan, X.; Liu, J. QSAR study of angiotensin I-converting enzyme inhibitory peptides using SVHEHS descriptor and OSC-SVM. Int. J. Pept. Res. Ther. 2019, 25, 247–256. [Google Scholar] [CrossRef]
- Cai, J.; Li, C.; Liu, Z.; Du, J.; Ye, J.; Gu, Q.; Xu, J. Predicting DPP-IV inhibitors with machine learning approaches. J. Comput. Mol. Des. 2017, 31, 393–402. [Google Scholar] [CrossRef]
- Chandra, S.; Pandey, J.; Tamrakar, A.K.; Siddiqi, M.I. SVMDLF: A novel R-based Web application for prediction of dipeptidyl peptidase 4 inhibitors. Chem. Biol. Drug Des. 2017, 90, 1173–1183. [Google Scholar] [CrossRef]
- Yi, H.-C.; You, Z.-H.; Zhou, X.; Cheng, L.; Li, X.; Jiang, T.-H.; Chen, Z.-H. ACP-DL: A deep learning long short-term memory model to predict anticancer peptides using high-efficiency feature representation. Mol. Ther. Nucleic Acids 2019, 17, 1–9. [Google Scholar] [CrossRef]
- Chou, K.-C. Using amphiphilic pseudo amino acid composition to predict enzyme subfamily classes. Bioinformatics 2004, 21, 10–19. [Google Scholar] [CrossRef]
- Chou, K.-C. Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins Struct. Funct. Bioinform. 2001, 43, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Fu, K.; Zuo, J.; Zheng, X.; Huang, T.; Ren, W. Radar emitter classification for large data set based on weighted-xgboost. IET Radar Sonar Navig. 2017, 11, 1203–1207. [Google Scholar] [CrossRef]
- Zheng, H.; Yuan, J.; Chen, L. Short-term load forecasting using EMD-LSTM neural networks with a xgboost algorithm for feature importance evaluation. Energies 2017, 10, 1168. [Google Scholar] [CrossRef]
- Lee, M.; Lee, H.; Kim, J. Dairy food consumption is associated with a lower risk of the metabolic syndrome and its components: A systematic review and meta-analysis. Br. J. Nutr. 2018, 120, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Minkiewicz, P.; Iwaniak, A.; Darewicz, M. BIOPEP-UWM database of bioactive peptides: Current opportunities. Int. J. Mol. Sci. 2019, 20, 5978. [Google Scholar] [CrossRef]
- Panyayai, T.; Ngamphiw, C.; Tongsima, S.; Mhuantong, W.; Limsripraphan, W.; Choowongkomon, K.; Sawatdichaikul, O. FeptideDB: A web application for new bioactive peptides from food protein. Heliyon 2019, 5, e02076. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhang, C.; Chen, H.; Xue, J.; Guo, X.; Liang, M.; Chen, M. BioPepDB: An integrated data platform for food-derived bioactive peptides. Int. J. Food Sci. Nutr. 2018, 69, 963–968. [Google Scholar] [CrossRef]
- Tyagi, A.; Kapoor, P.; Kumar, R.; Chaudhary, K.; Gautam, A.; Raghava, G.P.S. In silico models for designing and discovering novel anticancer peptides. Sci. Rep. 2013, 3, srep02984. [Google Scholar] [CrossRef]
- Sharma, A.; Kapoor, P.; Gautam, A.; Chaudhary, K.; Kumar, R.; Chauhan, J.S.; Tyagi, A.; Raghava, G.P.S. Computational approach for designing tumor homing peptides. Sci. Rep. 2013, 3, srep01607. [Google Scholar] [CrossRef]
- Chikhi, R.; Medvedev, P. Informed and automated k-mer size selection for genome assembly. Bioinformatics 2014, 30, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Li, F.-M.; Wang, X.-Q. Identifying anticancer peptides by using improved hybrid compositions. Sci. Rep. 2016, 6, srep33910. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zeng, D.; Wang, W.; Chen, X.; Zhang, Z.; Xu, F.; Mao, X.; Liu, X. Mutation grey wolf elite PSO balanced XGBoost for radar emitter individual identification based on measured signals. Measurement 2020, 159, 107777. [Google Scholar] [CrossRef]
- Lei, X.M.; Xie, Y.T. Improved XGBoost model based on genetic algorithm for hypertension recipe recognition. Comput. Ence 2018, 45, 476–481. [Google Scholar]
- Taherzadeh, G.; Zhou, Y.; Liew, A.W.-C.; Yang, Y. Structure-based prediction of protein– peptide binding regions using Random Forest. Bioinformatics 2017, 34, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Jia, S.; Hu, X.Z. Using random forest algorithm to predict β-hairpin motifs. Protein Pept. Lett. 2011, 18, 609–617. [Google Scholar] [CrossRef]
- Hayat, M. Discriminating outer membrane proteins with fuzzy K-nearest neighbor algorithms based on the general form of Chou’s PseAAC. Protein Pept. Lett. 2012, 19, 411–421. [Google Scholar] [CrossRef]
- Shoichet, B.K. Virtual screening of chemical libraries. Nat. Cell Biol. 2004, 432, 862–865. [Google Scholar] [CrossRef]
- Zhou, P.; Li, B.; Yan, Y.; Jin, B.; Wang, L.; Huang, S.-Y. Hierarchical flexible peptide docking by conformer generation and ensemble docking of peptides. J. Chem. Inf. Model. 2018, 58, 1292–1302. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Jin, B.; Li, H.; Huang, S.-Y. HPEPDOCK: A web server for blind peptide–protein docking based on a hierarchical algorithm. Nucleic Acids Res. 2018, 46, W443–W450. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Zhang, D.; Huang, S.-Y. Efficient conformational ensemble generation of protein-bound peptides. J. Chem. 2017, 9, 59. [Google Scholar] [CrossRef]
- Bugg, T.D.H. Appendix 2: Amino acid abbreviations [M]. In Introduction to Enzyme and Coenzyme Chemistry, 3rd ed.; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2012. [Google Scholar]
- Kim, Y.; Sidney, J.; Buus, S.; Sette, A.; Nielsen, M.; Peters, B. Dataset size and composition impact the reliability of performance benchmarks for peptide-MHC binding predictions. BMC Bioinform. 2014, 15, 241. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Fu, T.; Hao, S.; Zhang, Y.; Wang, O.; Jin, B.; Xia, M.; Liu, M.; Zhou, X.; Wu, Q.; et al. Prediction of incident hypertension within the next year: Prospective study using statewide electronic health records and machine learning. J. Med. Internet Res. 2018, 20, e22. [Google Scholar] [CrossRef]
- Carmona, P.; Climent, F.; Momparler, A. Predicting failure in the U.S. banking sector: An extreme gradient boosting approach. Int. Rev. Econ. Financ. 2019, 61, 304–323. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Entry Name | Protein Names | Gene Names | Organism | Length |
|---|---|---|---|---|---|
| P02666 | CASB_BOVIN | Beta-casein | CSN2 | Bos taurus (Bovine) | 224 |
| P02668 | CASK_BOVIN | Kappa-casein | CSN3 CSN10 CSNK | Bos taurus (Bovine) | 190 |
| P02662 | CASA1_BOVIN | Alpha-S1-casein | CSN1S1 | Bos taurus (Bovine) | 214 |
| P02663 | CASA2_BOVIN | Alpha-S2-casein | CSN1S2 | Bos taurus (Bovine) | 222 |
| P02754 | LACB_BOVIN | Beta-lactoglobulin | LGB | Bos taurus (Bovine) | 178 |
| P00711 | LALBA_BOVIN | Alpha-lactalbumin | LALBA ALACTA | Bos taurus (Bovine) | 142 |
| Name of Dataset | Acc (%) | Sens (%) | Spec (%) | Prec (%) |
|---|---|---|---|---|
| ACEIP214 | 77.49 ± 4.87 | 77.87 ± 4.20 | 77.00 ± 7.12 | 79.62 ± 5.72 |
| ACEIP1378 | 85.47 ± 2.37 | 85.35 ± 2.83 | 85.60 ± 3.42 | 86.46 ± 2.78 |
| ACEIP3306 | 86.50 ± 1.24 | 86.08 ± 1.24 | 86.92 ± 2.12 | 86.85 ± 1.87 |
| Datasets for Training | Datasets for Testing | AUC (%) |
|---|---|---|
| ACEIP3306 | ACEIP214 | 88.18 ± 1.27 |
| ACEIP3306 | ACEIP1378 | 86.33 ± 0.98 |
| 80% of total dataset | The rest of total dataset | 91.85 ± 0.82 |
| Prediction Probability of Positive Samples | Peptide Sequence | The Docking Scores |
|---|---|---|
| 0.99+ (candidate inhibitors) | RLNFLKKI | −212.37 |
| SITRINKKI | −205.44 | |
| ILTCLV | −174.48 | |
| LVVTILA | −172.51 | |
| DQVKRNA | −172.46 | |
| LTCL | −138.38 | |
| ILTC | −138.35 | |
| LILT | −136.18 | |
| 0.50 | PQN | −129.85 |
| ACLV | −115.72 | |
| ESLS | −113.95 | |
| LKK | −111.86 | |
| LEI | −100.82 | |
| LQD | −100.31 | |
| QLE | −96.19 | |
| EIV | −87.36 | |
| 0.00 | EDV | −86.31 |
| KEK | −84.55 | |
| KV | −83.00 | |
| KED | −80.58 | |
| GKE | −78.43 | |
| SEE | −62.48 | |
| EDS | −60.17 | |
| DE | −48.25 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Niu, D.; Wang, X.; Khan, J.; Shen, Q.; Xue, Y. A Novel Machine Learning Strategy for the Prediction of Antihypertensive Peptides Derived from Food with High Efficiency. Foods 2021, 10, 550. https://doi.org/10.3390/foods10030550
Wang L, Niu D, Wang X, Khan J, Shen Q, Xue Y. A Novel Machine Learning Strategy for the Prediction of Antihypertensive Peptides Derived from Food with High Efficiency. Foods. 2021; 10(3):550. https://doi.org/10.3390/foods10030550
Chicago/Turabian StyleWang, Liyang, Dantong Niu, Xiaoya Wang, Jabir Khan, Qun Shen, and Yong Xue. 2021. "A Novel Machine Learning Strategy for the Prediction of Antihypertensive Peptides Derived from Food with High Efficiency" Foods 10, no. 3: 550. https://doi.org/10.3390/foods10030550
APA StyleWang, L., Niu, D., Wang, X., Khan, J., Shen, Q., & Xue, Y. (2021). A Novel Machine Learning Strategy for the Prediction of Antihypertensive Peptides Derived from Food with High Efficiency. Foods, 10(3), 550. https://doi.org/10.3390/foods10030550