
Anthocyanins, Carotenoids and Chlorophylls in Edible Plant Leaves Unveiled by Tandem Mass Spectrometry

Abstract
:1. Introduction
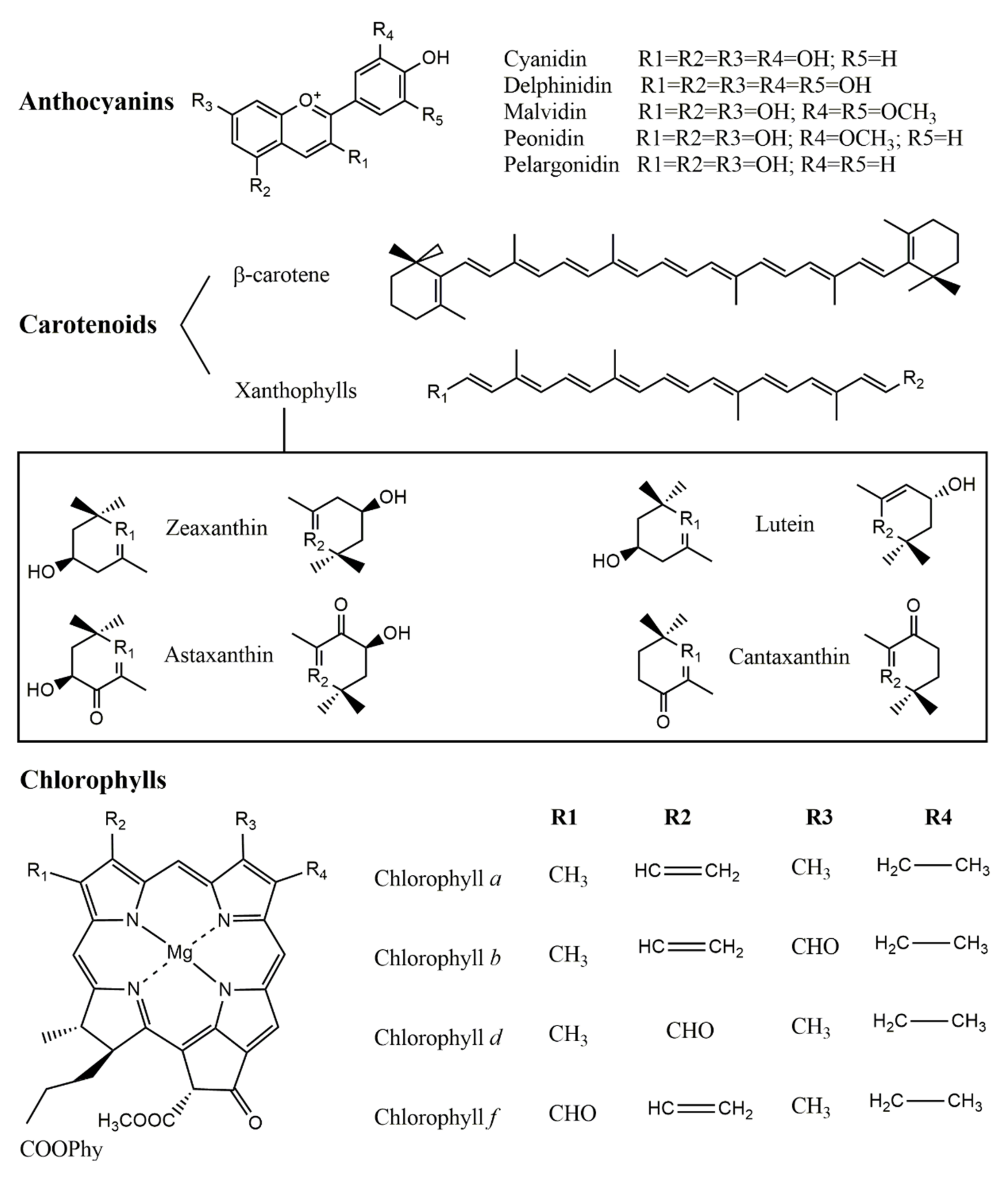
2. Natural Pigments
Mass Spectrometry: Main Principles, Advantages and Disadvantages
3. Data Analysis
4. Mass Spectrometry Analysis of Leaves Pigments
4.1. Antocyanins
4.1.1. Samples, Preprocessing and Extraction Details
4.1.2. Mass Spectra Acquisition
4.1.3. Identified and Quantified Anthocyanins
4.2. Carotenoids
4.2.1. Samples, Pre-Processing and Extraction Details
4.2.2. Mass Spectra Acquisition
4.2.3. Identified and Quantified Carotenoids
4.3. Chlorophylls
4.3.1. Samples, Pre-Processing and Extraction Details
4.3.2. Mass Spectra Acquisition
4.3.3. Identified and Quantified Chlorophylls
5. Critical Analysis and Conclusions
Funding
Informed Consent Statement
Conflicts of Interest
References
- Rao, M.P.N.; Xiao, M.; Li, W.J. Fungal and Bacterial Pigments: Secondary Metabolites with Wide Applications. Front. Microbiol. 2017, 8, 1113. [Google Scholar]
- Begum, H.; Yusoff, F.; Banerjee, S.; Khatoon, H.; Shariff, M. Availability and utilization of pigments from miocroalgae. Crit. Rev. Food Sci. Nut. 2016, 56, 2209–2222. [Google Scholar] [CrossRef] [PubMed]
- Villaró, S.; Ciardi, M.; Morillas-España, A.; Sáchez-Zurano, A.; Acién-Fernández, G.; Lafarga, T. Microalgae derived astaxanthin: Research and consumer trends and industrial use as food. Foods 2021, 10, 2303. [Google Scholar] [CrossRef]
- Andriamanantena, M.; Danthu, P.; Cardon, D.; Fawbush, F.R.; Raonizafinimanana, B.; Razafintsalama, V.E.; Rakotonandrasana, S.R.; Ethève, A.; Petit, T.; Caro, Y. Malagasy dye plant species: A promising source of novel natural colorants with potential applications- a review. Chem. Biodivers. 2019, 16, e1900442. [Google Scholar] [CrossRef] [PubMed]
- Lomax, S.Q.; Lomax, J.F.; Graham, T.K.; Moore, T.J.T.; Knapp, C.G. Historical azo pigments: Synthesis and characterization. J. Cult. Herit. 2019, 35, 218–224. [Google Scholar] [CrossRef]
- Ding, C.; Han, A.J.; Ye, M.Q.; Zhang, Y.; Yao, L.Y.; Yang, J.L. Hydrothermal synthesis and characterization of novel yellow pigments based on V5+ doped BiPO4 with high near-infrared reflectance. RSC Adv. 2018, 8, 19690–19700. [Google Scholar] [CrossRef] [Green Version]
- Zielewicz, W.; Wróbel, B.; Niedbała, G. Quantification of Chlorophyll and Carotene Pigments Content in Mountain Melick (Melica nutans L.) in Relation to Edaphic Variables. Forests 2020, 11, 1197. [Google Scholar] [CrossRef]
- Dulo, B.; Phan, K.; Githaiga, J.; Raes, K.; De Meester, S. Natural Quinone Dyes: A Review on Structure, Extraction Techniques, Analysis and Application Potential. Waste Biomass Valorization 2021, 12, 6339–6374. [Google Scholar] [CrossRef]
- Shah, A.; Smith, D.L. Flavonoids in Agriculture: Chemistry and Roles in, Biotic and Abiotic Stress Responses, and Microbial Associations. Agronomy 2020, 10, 1209. [Google Scholar] [CrossRef]
- Burmester, T.; Hankelen, T. Function and evolution of vertebrate globins. Acta Physiol. 2014, 211, 501–514. [Google Scholar] [CrossRef]
- Brenner, M.; Hearing, V.J. The protective role of melanin against UV damage in human skin. Photochem. Photobiol. 2008, 84, 539–549. [Google Scholar] [CrossRef] [Green Version]
- Khoo, H.E.; Azlan, A.; Tang, S.T.; Lim, S.M. Anthocyanidins and anthocyanins: Colored pigments as food, pharmaceutical ingredients, and the potential health benefits. Food Nut. Res. 2017, 61, 1361779. [Google Scholar] [CrossRef] [Green Version]
- Baby, B.; Antony, P.; Vijayan, R. Antioxidant and anticancer properties of berries. Crit. Rev. Food Sci. Nut. 2017, 58, 2491–2507. [Google Scholar] [CrossRef]
- Young, A.J.; Lowe, G.L. Carotenoids-antioxidant properties. Antioxidants 2018, 7, 28. [Google Scholar] [CrossRef] [Green Version]
- Tkitaoka, N.; Lu, X.; Yang, B.; Peters, R.J. The Application of Synthetic Biology to Elucidation of Plant Mono-, Sesqui-, and Diterpenoid Metabolism. Mol. Plant 2015, 8, 6–16. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Shi, Y.; Ma, L.; Yi, X.; Ruan, J. Metabolomic analysis using ultra-performance liquid chromatrography-quadrupole-time of flight mass spectrometry (UPLC-Q-TOF MS) uncovers the effects of light intensity and temperature under shading treatments on the metabolites in tea. PLoS ONE 2014, 9, e112572. [Google Scholar]
- Jayaraj, J.; Punja, Z.K. Transgenic carrot plants accumulating ketocarotenoids show tolerance to UV and oxidative stresses. Plant Physiol. Biochem. 2008, 46, 875–883. [Google Scholar] [CrossRef]
- Santos, P.M.; Batista, D.L.J.; Ribeiro, L.A.F.; Boffo, E.F.; Cerqueira, M.D.; Martins, D.; Castro, R.D.; Souza-Neta, L.C.; Pinto, E.; Zambotti-Villela, L.; et al. Identification of antioxidant and antimicrobial compounds from the oilseed crop of Ricinus communis using a multiplatform metabolite profiling approach. Ind. Crops Prod. 2018, 124, 834–844. [Google Scholar] [CrossRef]
- Wojdyło, A.; Nowicka, P.; Tkacz, K.; Turkiewicz, I.P. Fruit tree leaves as unconventional and valuable source of chlorophyll and carotenoid compounds determined by liquid chromatography-photodiode-quadrupole/time of flight-electrospray ionization-mass spectrometry (LC-PDA-qTof-ESI-MS). Food Chem. 2021, 349, 129156. [Google Scholar] [CrossRef]
- McCance, K.R.; Flanigan, P.M.; Quick, M.M.; Niemeyer, E.D. Influence of plant maturity on anthocyanin concentrations, phenolic composition, and antioxidant properties of 3 purple basil (Ocimum basilicum L.) cultivars. J. Food. Compos. Anal. 2016, 53, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Metličar, V.; Vovk, I.; Albreht, A. Japanese and bohemian knotweeds as sustainable sources of carotenoids. Plants 2019, 8, 384. [Google Scholar] [CrossRef] [Green Version]
- Castro, T.A.; Leite, B.S.; Assuncao, L.S.; Freitas, T.D.; Colauto, N.B.; Linde, G.A.; Otero, D.M.; Machado, B.A.S.; Ribeiro, C.D.F. Red tomato products as an alternative to reduce synthetic dyes in the food industry: A review. Molecules 2022, 26, 7125. [Google Scholar] [CrossRef]
- Nawaz, A.; Chaudhary, R.; Shah, Z.; Dufossé, L.; Fouillaud, M.; Mukhtar, H.; ul Haq, I. An Overview on Industrial and Medical Applications of Bio-Pigments Synthesized by Marine Bacteria. Microorganisms 2021, 9, 11. [Google Scholar] [CrossRef]
- Juric, S.; Juric, M.; Król-Kilinska, Z.; Vlahovicek-Kahlina, K.; Vincekovic, M.; Dragovic-Uzelac, V.; Donsi, F. Sources, stability, encapsulation and application of natural pigments in foods. Food Rev. Int. 2020, 1–56. [Google Scholar] [CrossRef]
- Mohd-Nasir, H.; Abd-Talib, N.; Mohd-Setapar, S.H.; Wong, L.P.; Idham, Z.; Casillas, A.C.; Ahamad, A. Natural colorants from plants for wellness industry. Int. J. Pharm. Sci. Res. 2018, 9, 836–843. [Google Scholar]
- Sigurdson, G.T.; Tang, P.P.; Giusti, M.M. Natural Colorants: Food Colorants from Natural Sources. Annu. Rev. Food Sci. Technol. 2017, 8, 261–280. [Google Scholar] [CrossRef]
- Ong, G.; Kasi, R.; Subramaniam, R. A review on plant extracts as natural additives in coating applications. Prog. Org. Coat. 2021, 151, 106091. [Google Scholar] [CrossRef]
- Alappat, B.; Alappat, J. Anthocyanin Pigments: Beyond Aesthetics. Molecules 2020, 25, 5500. [Google Scholar] [CrossRef]
- Cortez, R.; Luna-Vital, D.; Daniel Margulis, D.; Gonzalez de Mejia, E. Natural Pigments: Stabilization Methods of Anthocyanins for Food Applications. Compr. Rev. Food Sci. 2017, 16, 180–198. [Google Scholar] [CrossRef]
- Cisowska, A.; Wojnicz, D.; Hendrich, A. Anthocyanins as Antimicrobial Agents of Natural Plant Origin. Natl. Prod. Commun. 2011, 6, 149–156. [Google Scholar] [CrossRef] [Green Version]
- Hui, C.; Bin, Y.; Xiaoping, Y.; Lang, Y.; Chunye, C.; Mantian, M.; Wenhua, L. Anticancer activities of an anthocyanin-rich extract from black rice against breast cancer cells in vitro and in vivo. Nutr. Cancer 2010, 62, 1128–1136. [Google Scholar] [CrossRef] [PubMed]
- Belwal, T.; Nabavi, S.M.; Habtemariam, S. Dietary anthocyanins and insulin resistance: When food becomes a medicine. Nutrients 2017, 9, 1111. [Google Scholar] [CrossRef] [PubMed]
- Proteggente, A.; Pannala, A.; Paganga, G.; Buren, I.; Wagner, E.; Wiseman, S.; Van de put, F.; Dacombe, C.; Rice-Evans, C. The antioxidant activity of regularly consumed fruit and vegetables reflects their phenolic and vitamin C composition. Free Radic. Res. 2002, 36, 217–233. [Google Scholar] [CrossRef] [PubMed]
- Pereira, S.; Pereira, R.; Figueiredo, I.; De Freitas, V.; Dinis, T.C.P.; Almeida, L. Comparison of anti-inflammatory activities of an anthocyanin-rich fraction from Portuguese blueberries (Vaccinium corymbosum L.) and 5-aminosalicylic acid in a TNBS-induced colitis rat model. PLoS ONE 2017, 12, e0174116. [Google Scholar] [CrossRef] [Green Version]
- Cazzonelli, C.I. Carotenoids in nature: Insights from plants and beyond. Funct. Plant Biol. 2011, 38, 833–847. [Google Scholar] [CrossRef] [Green Version]
- Maoka, T. Carotenoids as natural functional pigments. J. Nat. Med. 2020, 74, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Havaux, M. Carotenoid oxidation products as stress signals in plants. Plant J. 2014, 79, 597–606. [Google Scholar] [CrossRef]
- Nisar, N.; Li, L.; Lu, S.; Khin, N.C.; Pogson, B.J. Carotenoid metabolism in plants. Mol. Plant 2015, 8, 68–82. [Google Scholar] [CrossRef] [Green Version]
- Fassett, R.G.; Coombes, J.S. Astaxanthin: A potential therapeutic agent in cardiovascular disease. Mar. Drugs 2011, 9, 447–465. [Google Scholar] [CrossRef] [Green Version]
- Stachowiak, B.; Szulc, P. Astaxanthin for the Food Industry. Molecules 2021, 26, 2666. [Google Scholar] [CrossRef]
- Chen, M. Chlorophyll modifications and their spectral extension in oxygenic photosynthesis. Annu. Rev. Biochem. 2014, 83, 317–340. [Google Scholar] [CrossRef]
- Lim. P.O.; Kim, H.J.; Nam, H.G. Leaf senescence. Annu. Rev. Plant Biol. 2007, 58, 115–136. [Google Scholar]
- Hoffman, E.; Stroobant, V. Mass Spectrometry: Principles and Applications, 3rd ed.; John Wiley & Sons: Chichester, UK, 2007. [Google Scholar]
- Li, X.; Wang, X.; Li, L.; Bai, Y.; Liu, H. Direct analysis in real time mass spectrometry: A powerful tool for fast analysis. Mass Spec. Lett. 2015, 6, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Ifa, D.R.; Wu, C.; Ouyang, Z.; Cooks, R.G. Desorption electrospray ionization and other ambient ionzation methods: Current progress and preview. Analyst 2010, 135, 669–681. [Google Scholar] [CrossRef]
- Berrueta, L.A.; Alonso-Salces, R.M.; Héberger, K. Supervised pattern recognition in food analysis. J. Chromatogr. A 2007, 1158, 196–214. [Google Scholar] [CrossRef]
- Available online: http://www.genome.jp/kegg (accessed on 19 May 2022).
- National Library of Medicine; Kim, S.; Chen, J.; Chen, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2019, 49, D1388–D1395. Available online: http://pubchem.ncbi.nlm.nih.gov (accessed on 19 May 2022). [CrossRef]
- Wishart, D.S.; Guo, A.C.; Oler, E.; Wang, F.; Anjum, A.; Peters, H.; Dizon, R.; Sayeeda, Z.; Tian, S.; Lee, B.L.; et al. HMDB 5.0: The Human Metabolome Database for 2022. Nucleic Acids Res. 2022, 50, D622–D631. Available online: http://www.hmdb.ca (accessed on 19 May 2022). [CrossRef]
- Available online: http://www.mhttp://www.kazusa.or.jp/komics/en/database-en.html (accessed on 19 May 2022).
- Available online: http://metlin.scripps.edu/ (accessed on 19 May 2022).
- Available online: https://webbook.nist.gov/chemistry/ (accessed on 19 May 2022).
- Available online: https://www.matrixscience.com/search_form_select.html (accessed on 19 May 2022).
- Available online: https://www.metaboanalyst.ca/ (accessed on 19 May 2022).
- GNPS: Global Natural Products Social Molecular Networking; Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828. Available online: https://gnps.ucsd.edu/ProteoSAFe/static/gnps-splash.jsp?redirect=auth (accessed on 19 May 2022). [CrossRef] [Green Version]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Orešič, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 395. Available online: http://mzmine.github.io/ (accessed on 19 May 2022).
- Available online: https://cytoscape.org/ (accessed on 19 May 2022).
- Jiang, L.; Shen, X.; Shoi, T.; Kanda, T.; Zhou, J.; Zhao, L. Characterization and activity of anthocyanins in Zijuan tea (Camellia sinensis var. kitamura. J. Agric. Food Chem. 2013, 61, 3306–3310. [Google Scholar] [CrossRef]
- Li, Y.; Jeyaraj, A.; Yu, H.; Wang, Y.; Ma, Q.; Chen, X.; Sun, H.; Zhang, H.; Ding, Z.; Li, X. Metabolic regulation profiling of carbon and nitrogen in tea plants [Camellia sinensis (L.) O. Kuntze] in response to shading. J. Agric. Food Chem. 2020, 68, 961–974. [Google Scholar] [CrossRef]
- Zhang, Q.; Hu, J.; Liu, M.; Shi, Y.; De Vos, R.C.H.; Ruan, J. Stimulated biosynthesis of delphinidin-related anthocyanins in tea shots reducing the quality of green tea in summer. J. Sci. Food Agric. 2020, 100, 1505–1514. [Google Scholar] [CrossRef]
- Shen, J.; Zhang, D.; Zhou, L.; Zhang, X.; Liao, J.; Duan, Y.; Wen, B.; Ma, Y.; Wang, Y.; Fang, W.; et al. Transcriptomic and metabolomics profiling of Camellia sinensis L. cv. ‘Suchazao’ exposed to temperature stresses reveals modification in protein synthesis and photosynthetic and anthocyanin biosynthetic pathways. Tree Physiol. 2019, 39, 1583–1599. [Google Scholar] [CrossRef]
- Qin, X.-X.; Zhang, M.-Y.; Han, Y.-Y.; Hao, J.-H.; Liu, C.-J.; Fan, S.-X. Beneficial phytochemicals with anti-tumor potential revealed through metabolic profiling of new red pigmented lettuces (Lactuca sativa L.). Int. J. Mol. Sci. 2018, 19, 1165. [Google Scholar] [CrossRef] [Green Version]
- Viacava, G.E.; Roura, S.I.; Berrueta, L.A.; Iriondo, C.; Gallo, B.; Alonso-Salces, R.M. Characterization of phenolic compounds in green and red oak-leaf lettuce cultivars by UHPLC-DAD-ESI-QToF/MS using MSE scan mode. J. Mass Spectrom. 2017, 52, 873–902. [Google Scholar] [CrossRef]
- Yuan, Y.; Yang, J.; Yu, X.; Huang, L.; Lin, S. Anthocyanins from buds of Lonicera japonica Thunb. var. chinensis (Wats.) Bak. Food Res. Int. 2014, 62, 812–818. [Google Scholar] [CrossRef]
- Goh, H.-H.; Khairudin, K.; Sukiran, M.N.; Normah, M.N.; Baharum, S.N. Metabolite profiling reveals temperature effects on VOCs and flavonoids of different plant populations. Plant Biol. 2016, 18, 130–139. [Google Scholar] [CrossRef]
- Simirgiotis, M.L.; Schmeda-Hirschmann, G. Determination of phenolic composition and antioxidant activity in fruits, rhizomes and leaves of the white strawberry (Fragaria chiloensis spp. chiloensis form chiloensis) using HPLC-DAD-ESI-MS and free radical quenching techniques. J. Food Compos. Anal. 2010, 23, 545–553. [Google Scholar]
- Hijaz, F.; Nehela, Y.; Jones, S.E.; Dutt, M.; Grosser, J.W.; Manthey, J.A.; Killiny, N. Metabolically engineered anthocyanin-producing lime provides additional nutricional value and antioxidant potential to juice. Plant Biotechnol. Rep. 2018, 12, 329–346. [Google Scholar] [CrossRef]
- Deng, J.; Wu, D.; Shi, J.; Balfour, K.; Wang, H.; Zhu, G.; Liu, Y.; Wang, J.; Zhu, Z. Multiple MYB activators and repressors collaboratively regulate the juvenile red fading in leaves of sweetpotato. Front. Plant Sci. 2020, 11, 941. [Google Scholar] [CrossRef]
- Gómez-Martínez, M.; Ascacio-Valdés, J.; Flores-Gallegos, A.C.; González-Domínguez, J.; Gómez-Martínez, S.; Aguillar, C.N.; Morlett-Chávez, J.A.; Rodríguez-Herrera, R. Location and tissue effects on phytochemical composition and in vitro antioxidante activity of Moringa oleifera. Ind. Crops Prod. 2020, 151, 112439. [Google Scholar] [CrossRef]
- Zhang, L.; Rocchetti, G.; Zengin, G.; Ak, G.; Saber, F.R.; Montesano, D.; Lucini, L. The UHPLC-QTOF-MS phenolic profiling and activity of Cydonia oblonga Mill. reveals a promising nutraceutical potential. Foods 2021, 10, 1230. [Google Scholar] [CrossRef] [PubMed]
- Uarrota, V.G.; Severino, R.B.; Malinowsky, C.; Oliveira, S.K.; Kuhnen, S.; Yune, R.A.; Maraschin, M. Biochemical profile of leaf, silk and grain samples of eight maize landraces (Zea mays L.) cultivated in two low-input agricultural systems. J. Food Biochem. 2014, 38, 551–562. [Google Scholar] [CrossRef]
- Bentley, J.; Olsen, E.K.; Mooe, J.P.; Farrant, J.M. The phenolic profile extracted from the desiccation-tolerant medicinal shrub Myrothamnus flabellifolia using natural deep eutectic solvents. Phytochemistry 2020, 173, 112323. [Google Scholar] [CrossRef] [PubMed]
- Saini, R.K.; Shetty, N.P.; Giridhar, P. Carotenoid content in vegetative and reproductive parts of commercially grown Moring oleifera Lam. Cultivars from India by LC-APCI-MS. Eur. Food Res. Technol. 2014, 238, 971–978. [Google Scholar] [CrossRef]
- Azevedo, C.H.; Rodriguez-Amaya, D.B. Carotenoid composition of kale as influenced by maturity, season and minimal processing. J. Sci. Food Agric. 2005, 85, 591–597. [Google Scholar] [CrossRef]
- Murillo, E.; Deli, J.; Nagy, V.; Molinar-Toribio, E.; Sándor, V.; Marton, K.; Agócs, A. Carotenoid profile of two capsorubin-rich tropical plants. J. Food Compost. Anal. 2021, 97, 103798. [Google Scholar] [CrossRef]
- Shen, J.; Zou, Z.; Zhang, X.; Zhou, L.; Fang, W.; Zhy, X. Metabolic analysis reveal different mechanisms of leaf color change I two purple-leaf tea plant (Camellia sinensis L.) cultivars. Hort. Res. 2018, 5, 7. [Google Scholar] [CrossRef] [Green Version]
- Lachowicz, S.; Oszmiański, J.; Wojdyło, A.; Cebulak, T.; Hirnle, L.; Siewiński, M. UPLC-PDA-Q/TOF-MS identification of bioactive compounds and on-line UPLC-ABTS assay in Fallopia japonica Houtt and Fallopia sachalinensis (F-Schmid) leaves and rhizomes grown in Poland. Eur. Food Res. Technol. 2019, 245, 691–706. [Google Scholar] [CrossRef] [Green Version]
- Mi, J.; Jia, K.-P.; Wang, J.Y.; Al-Babili, S. A rapid LC-MS method for qualitative and quantitative profiling plant apocarotenoids. Anal. Chim. Acta 2018, 1035, 87–95. [Google Scholar] [CrossRef]
- Delpino-Rius, A.; Cosovanu, D.; Era, J.; Vilaró, F.; Balcells, M.; Canela-Garayoa, R. A fast and reliable ultrahigh-performance liquid chromatography method to assess the fate of chlorophylls in teas and processed vegetable foodstuff. J. Chromatogr. A 2018, 1568, 69–79. [Google Scholar] [CrossRef]
- Kao, T.; Chen, C.J.; Chen, B.H. An improved high performance liquid chromatography-photodiode array detection-atmospheric pressure chemical ionization-mass spectrometry method for determination of chlorophylls and their derivatives in freeze-dried and hot-air-dried Rhinacanthus nasutus (l.) Kurz. Talanta 2011, 86, 349–355. [Google Scholar]

{kind=link}
| Plant | Sample | Solvents | Sample:Solvent | Time Extraction/Conditions | Ref |
|---|---|---|---|---|---|
| C. sinensis | Commercial tea leaves | MeOH (0.1% TFA) | 250 g:no info | Maceration (overnight, 4 °C)—complex procedure with the isolation of 4 structures | [58] |
| C. sinensis | Frozen in N2 | Cold MeOH | 100 mg:1000 mL | Chloroform+H2O (vortex 1’)—centrifuge (4000 rpm, 15’) | [59] |
| C. sinensis | Frozen in N2-lyophilized | MeOH:H2O:FA (75:24:1) | 0.1 g:1 mL | Ultrasonic bath (10’)—centrifuge (12,000 rpm, 10’) | [16] |
| C. sinensis | Frozen in N2-lyophilized | MeOH:H2O:FA (75:24:1) | 25 mg:1 mL | Ultrasonic bath (15’)— centrifuge | [60] |
| C. sinensis | Stored (−80 °C)-lyophilized | MeOH (0.1 mg L−1 lidocaine) | 100 mg:1 mL | Overnight (4 °C)—centrifuge (10’; 10,000× g) | [61] |
| L. sativa | Lyophilized | MeOH:H2O:FA (80:19:1) | 1 g:10 mL | Ultrasonic bath (12 kHz, 70′, 45 °C) | [62] |
| L. sativa | Lyophilized | MeOH:H2O:AA (30:65:5) + 2 g/L AscA | 0.1 g:5 mL | Ultrasonic bath (10′)—centrifuge (6000 rpm, 15′, 4 °C) | [63] |
| L. japonica | Lyophilized | MeOH:H2O:FA:TFA (70:27:2:1) | 500 mg:10 mL | Room temp (24 h) | [64] |
| P. minus | Lyophilized | MeOH | 500 mg:5 mL | 1 h (room temp; 3 times) | [65] |
| F. chiloensis | Lyophilized | MeOH:FA (99:1) | 5 g:50 mL | 1 h (room temp; 3 times), evaporation to dryness, H2O-Amberlite column, eluate evaporated to dryness with redissolution of MeOH:FA | [66] |
| ‘Mexican lime’ | Frozen in N2 | Acetone (80%) + ethyl acetate | 100 mg:400 + 240 mL | Dark (ice,10’; twice), H2O, centrifuge (8500× g, 5’,4 °C) | [67] |
| I. batatas | Frozen in N2 | MeOH | 0.5 g:5mL | 24 h darkness (4 °C) | [68] |
| O. basilicum | Frozen in N2 | MeOH (0.2 M HCL) | ‘known mass’:500 mL | Shaken (room temp, 40′), centrifuge (13,200 rpm; 30′), dried 3 times and reconstituted in 0.1% FA | [20] |
| M. oleifera | Dried in shade | EtOH:H2O (1:1) | 1 g:7 mL | Ultrasonic bath (20′; 60 Hz), centrifuge (419 g; 10′) | [69] |
| C. oblonga | Dried in shade | MeOH | 100 g:1000 mL | Ultrasonic bath (45′, 50 °C), evaporation dryness | [70] |
| Z. mays | Dried | MeOH:HCl 1N (85:15) | 1 g:24 mL | Ice (15′)-centrifuge(3000 rpm, 5′)-redissoluion | [71] |
| M. flabellifolia | Naturally desiccated | 4 different NaDES | 50 mg:1 mL | Diluted H2O ultrasound bath (1.5 h, 50–55 °C), diluted with H2O centrifuge (16,000 rpm, 20′) | [72] |
| Plant | Column | Eluent | Ionization Source | (LC)-MS Technique | Ref |
|---|---|---|---|---|---|
| C. sinensis | RP C18 column (250 × 4.6mm, 5 µm) | H2O:ACN (90:10; 0.1% TFA)—H2O:ACN (50:50; 0.1% TFA) | ESI + | LC-ESI-MS | [58] |
| C. sinensis | No info | H2O (0.1% FA)—ACN (0.1% FA) | ESI + | UPLC-MS | [59] |
| C. sinensis | RP ACQUITY UPLC HSS T3 C18 (100 × 2.1 mm, 1.8 µm) | H2O (0.1% FA—ACN (0.1% FA) | ESI + | UPLC-QTOF-MS | [16] |
| C. sinensis | RP Luna C18 Phenomenex (150 × 2.0 mm, 3 µm) | H2O—ACN (0.1% FA) | ESI − | HPLC-Orbitrap-MS | [60] |
| C. sinensis | RP ACQUITY UPLC HSS T3 C18 (100 × 2.1 mm, 1.8 µm) | H2O (0.04% AA)—ACN (0.04% AA) | ESI + | HPLC- ion trap-LC-MS/MS | [61] |
| L. sativa | No info | H2O (0.01% FA)—ACN | ESI + and − | UPLC-QTOF MS | [62] |
| L. sativa | RP ACQUITY UPLC BEH C18 column (100 × 2.1 mm, 1.7 µm) | H2O (0.1% AA)—MeOH (0.1% AA) | ESI + and − | UPLC-DAD-ESI-QTOF/MS | [63] |
| L. japonica | RP Zorbax Eclipse XDB-C18 (250 × 4.6 mm, 5μm) | ACN:FA(99:1)—H2O:FA (1:99:1) | ESI + | HPLC-DAD-ESI-MS/MS | [64] |
| P. minus | RP C18 (250 × 2.0 mm, 5 µm) | H2O (0.1% FA)—ACN | ESI + | LC-TOF (micrOTOF-Q) MS | [65] |
| F. chiloensis | RP Luna C18 (250 × 4.6 mm, 5 µm) | H2O (1% FA)—ACN | ESI + and − | HPLC-DAD-ESI-MS | [66] |
| ‘Mexican lime’ | RP Waters XBridge C8 (4.6 × 100 mm) | ACN:H2O (19:90, 0.5% FA)—ACN | ESI + | HPLC-MS | [67] |
| I. batatas | No info | No info | No info | UPLC-MS/MS | [68] |
| O. basilicum | nanotip in-house packed C18 (50 × 2.1 mm, 3.5 µm) | H2O (0.1% FA + 0.2% ACN)—ACN | ESI + | HPLC-QTOF MS | [20] |
| M. oleifera | RP Denali C18 (150 × 2.1 mm, 3 µm) | H2O (0.2% FA)—ACN | ESI − | HPLC-MS | [69] |
| C. oblonga | RP Zorbax eclipse plus C18 column (50 × 2.1 mm, 1.8 µm) | H2O (0.1% FA)—ACN (0.1% FA) | ESI + | UHPLC-QTOF-MS | [70] |
| Z. mays | No column | - | MALDI | TOF MS | [71] |
| M. flabellifolia | RP ACQUITY BEH C18 column (100 × 2.1 mm, 1.7 µm) | H2O (0.1% FA)—ACN (0.1% FA) | ESI − | UPLC QTOF MS | [72] |
| [58] | [59] | [16] | [60] | [61] | [62] | [63] | [64] # | [65] | [66] | [67] | [68] | [20] # | [69] | [70] | [71] | [72] | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cyanidin (Cyn) | ♦ | ♦ | ♦ | ||||||||||||||
| Delphinidin (Delp) | |||||||||||||||||
| Malvidin (Mal) | ♦ | ||||||||||||||||
| Pelargonidin (Pel) | ♦ | ♦ | |||||||||||||||
| Peonidin (Peo) | ♦ | ||||||||||||||||
| Petunidin (Pet) | |||||||||||||||||
| Cyn-3-(acetyl)-gluc | 1.08 ± 0.04 | ||||||||||||||||
| Cyn-3-o-(6″-o-acetyl)-gluc | ♦ | ||||||||||||||||
| Cyn-3-acetyl glucosamine | ♦♦ | ||||||||||||||||
| Cyn 3-(6″-caffeylgluc) | ♦ | ||||||||||||||||
| Cyn 3-p-coumaroyl gluc | ♦♦ | ||||||||||||||||
| Cyn-3-(p-coumaroyl)-rutin-5-gluc | 0.89 ± 0.04 | ||||||||||||||||
| Cyn 3-(p-coumaroyl) derivative | ♦ | ||||||||||||||||
| Cyn-3-o-(6-p-coumaroyl-60-caffeoyl)sophoroside-5- o-gluc | 0.13 ± 0.02–0.82 ± 0.23 ## | ||||||||||||||||
| Cyn-3-o-(6-p-coumaroyl-X-malonyl-60-caffeoyl)sophoroside-5-o-glucoside * | 0.05 ± 0.01–0.44 ± 0.11 ## | ||||||||||||||||
| Cyn-3-o-(6,60-di-p-coumaroyl)sophoroside-5-o-gluc | 0.41 ± 0.02–2.02 ± 0.55 ## | ||||||||||||||||
| Cyn-3-o- (6,60- di-p-coumaroyl-X-malonyl)sophoroside-5-o-gluc * | 0.10 ± 0.01–1.18 ± 0.31 ## | ||||||||||||||||
| Cyn-3-o-β-D-(6-(E)-p-coumaroyl) galpyr | ♦ | ||||||||||||||||
| Cyn 3-(p-feruloyl) derivative | ♦ | ||||||||||||||||
| Cyn 3-galact | ♦ | 0.37 ± 0.02 | |||||||||||||||
| Cyn-3-o-β-D-galact | ♦ | ||||||||||||||||
| Cyn-3-o-gluc | ♦ | ♦ | ♦ | 32.74 ± 0.71 | ♦♦ | ♦ | |||||||||||
| Cyn-3-o-gluc chloride | ♦ | ||||||||||||||||
| Cyn 3-o-glucosyl-magluc | ♦ | ||||||||||||||||
| Cyn-3,5-o-digluc | ♦ | 39.13 ± 0.87 | ♦♦ | ♦ | |||||||||||||
| Cyn-3-digluc-5-gluc | 1.56 ± 0.04 | ||||||||||||||||
| Cyn o-hexosyl-o-hexosyl-o-hexoside | ♦ | ||||||||||||||||
| Cyn 3-malgluc ** | ♦ | ||||||||||||||||
| Cyn 3-(3″-malgluc) | ♦ | ||||||||||||||||
| Cyn-3-o-(3″-o-malonyl)-gluc | ♦ | ||||||||||||||||
| Cyn-3-o-(6″-o-malonyl)-gluc | ♦ | ||||||||||||||||
| Cyn-3-o-(6″-o-malonyl-2’’-o-glucoronil) gluc | ♦ | ||||||||||||||||
| Cyn-o-malonyl-malonylhexoside | ♦ | ||||||||||||||||
| Cyn 3-rutin | 1.27 ± 0.05 | ♦ | |||||||||||||||
| Cyn 3-rutin-5-gluc | 4.18 ± 0.13 | ||||||||||||||||
| Cyn 3-o-sophoroside | ♦ | ||||||||||||||||
| Cyn o-syringic acid | ♦ | ||||||||||||||||
| Cyn 3-o-[2″-o-(xylosyl)-6″-o-(p-o-(glucosyl)-p-coumaryl)gluc]5-o-gluc | ♦ | ||||||||||||||||
| Delp 3-o-arabinose | ♦ | ||||||||||||||||
| Delp 3-coumaroyl gluc | ♦♦ | ||||||||||||||||
| Delp 3- o-β-D-(6-(E)-p-coumaroyl) galpyr | ♦ | ||||||||||||||||
| Delp 3-galac | ♦ | ||||||||||||||||
| Delp 3-o-β-D-galac | ♦ | ||||||||||||||||
| Delp gluc | ♦ | ||||||||||||||||
| Delp 3-gluc | ♦♦ | ||||||||||||||||
| Delp 3-o-gluc | ♦ | ♦ | |||||||||||||||
| Delp hexose-coumaroyl | ♦ | ||||||||||||||||
| Delp 3-(6″-malonylgluc)/6-OH-cyn 3-(6-malonylgluc) | ♦ | ||||||||||||||||
| 6-OH-delp-3-(6-malonylgluc) | ♦ | ||||||||||||||||
| Delp derivative | ♦ | ||||||||||||||||
| Mal-3-acetyl gluc | ♦♦ | ||||||||||||||||
| Mal-3-coumaroyl gluc | ♦♦ | ||||||||||||||||
| Mal-3-gluc | ♦♦ | ||||||||||||||||
| Mal 3-o-gluc | ♦ | ||||||||||||||||
| Pel gluc | ♦ | ||||||||||||||||
| Pel 3-(6″-p-coumsambubi)-5-(6″-magluc) | ♦ | ||||||||||||||||
| Peo 3-o-(6″-acetyl-gluc) | ♦ | ||||||||||||||||
| Peo 3-o-gluc | ♦ | ♦♦ | |||||||||||||||
| Peo 3-o-hexoside | ♦♦ | ||||||||||||||||
| Peo 3-malgluc | ♦ | ||||||||||||||||
| Peo 3-rutin | ♦ | ||||||||||||||||
| Peo 3-o-sophoroside-5-o-gluc | ♦♦ | ||||||||||||||||
| Pet 3-acetyl gluc | ♦♦ | ||||||||||||||||
| Pet 3-coumaroyl gluc | ♦♦ | ||||||||||||||||
| Pet 3-gluc | ♦ | ||||||||||||||||
| Pet gluc | ♦ | ||||||||||||||||
| Procyn tetramer *** | ♦ | ||||||||||||||||
| Proanthocyn III | ♦ | ||||||||||||||||
| Prodelp-O-gallate II | ♦ |
| Plant | Sample | Solvents | Sample:Solvent | Time Extraction/Conditions | Ref |
|---|---|---|---|---|---|
| Carrot leaves | Fresh leaves | Hexane:Acetone:EtOH:Toluene (10:7:6:7) | 1 g: no info | Extraction (56 °C)-mix with 10% sodium sulphate (epiphase withdrawn),evaporation to dryness, dissolution in chloroform | [17] |
| M. oleifera | Fresh leaves | Cold acetone | 5 g: 50–100 mL | repeated extractions, partition to 10% ethyl ether in PE, evaporation to dryness, dissolution in acetone | [73] |
| B. oleracea | Fresh leaves ground in household food processor | Cold acetone | 3–5 g:no info | Extraction-partition to 10% ethyl ether in PE, evaporation to dryness, dissolution in acetone | [74] |
| R. communis | Dried (25 °C) and powdered | Hexane; EtOH and ethylacetate | No info | Maceration-ethyl acetate and EtOH extraction, evaporation to dryness, dissolution in ACN | [18] |
| Z. dressleri | Brown 20 days old leaves | Acetone + 10 g NaHCO3 | 100 g:no info | Repeated extractions till colorless samples-extract diluted in ether:hexane (1:1), washed (H2O), dried (Na2SO4), evaporation to dryness, saponification | [75] |
| C. sinensis | Frozen in liquid N2 (stored −80 °C) | Cold MeOH:H2O (1:1) | 25 mg:800 µL | TissueLyser LT (5′, 60 Hz)-, centrifuged (20′, 25,000× g, 4 °C) | [76] |
| F. japonica// F. sachalinensis | Frozen (−25 °C)-lyophilized (stored −80 °C) | Hexane:Acetone:MeOH (2:1:1) (10% of MgCO3 in BHT–1%) | 500 mg:5 mL | Orbital shaker (300 rpm, 30′) dark—centrifuged (10′, 19,000× g, 4 °C), re-extraction, evaporation to dryness, dissolution in MeOH | [77] |
| S. oleracea | Lyophilized-powdered (stored at −20 °C) | n-hexane; Dichloromethane; Ethyl acetate; Acetone; MeOH; MeOH (0.1% BHT) ** | 20 mg:2 mL | 15’ ultrasound, centrifuged (8′, 3800 rpm, 4 °C), re-extraction, evaporation to dryness—dissolution in ACN | [78] |
| Fruit tree leaves * | Lyophilized-powdered | Hexane:Acetone:MeOH (2:1:1) (10% of MgCO3 in BHT–1%) | 100 mg:3 mL | Orbital shaker (300 rpm, 30′), dark—re-extraction 4 times, evaporation to dryness, dissolution in MeOH | [19] |
| Plant | Column | Eluent | Ionization Source | (LC)-MS Technique | Ref |
|---|---|---|---|---|---|
| Carrot leaves | RP YMC C30 carotenoid column | MeOH-MTBE (0–100%) | APCI | HPLC Quadrupole Ion trap MS | [17] |
| M. oleifera | RP YMC C30 carotenoid column (250 × 4.6 mm, 5 µm) | MeOH:MTBE:H2O (81:15:4)–MTBE/MeOH (91:9) | APCI | HPLC-QTOF MS | [73] |
| B. oleracea | RP C18 Spherisorb ODS2 (150 × 4.6 mm, 3 µm) | CAN (0.05% triethylamine):MeOH:ethyl acetate (95:5:0 to 60:20:20) | Thermabeam ESI + | HPLC MS | [74] |
| R. communis | RP C18 column (3.0 × 150 mm, 2.6 µm) | H2O (0.1% FA)–ACN (0.1% FA) | ESI + | LC-microTOF MS | [18] |
| Z. dressleri | RP YMC C30 carotenoid column (250 × 4.6 mm, 3 µm) | MeOH:MTBE:H2O (81:15:4)–MeOH:MTBE:H2O (6:90:4) | APCI | QTOF LC MS | [75] |
| C. sinensis | RP ACQUITY UPLC BEH C18 column (100 × 2.1 mm, 1.7 µm) | H2O (0.1% FA)–ACN (0.1% FA) | ESI+/ESI− | UPLC-QTOF MS | [76] |
| F. japonica// F. sachalinensis | RP ACQUITY UPLC BEH C18 column (100 × 2.1 mm, 1.7 µm) | ACN:MeOH (7:3)–H2O (0.1% FA) | ESI + | LC-QTOF MS | [77] |
| S. oleracea | RP ACQUITY UPLC BEH C18 column (100 × 2.1 mm, 1.7 µm) + UPLC BEH C18 guard column (5 × 2.1 mm, 1.7 µm) | H2O:ACN:FA (80:20:0.1)–ACN:IPA:FA (60:40:0.1) | ESI + | UHPLC-Q-Orbitrap MS | [78] |
| Fruit tree leaves * | RP ACQUITY UPLC BEH C18 column (100 × 2.1 mm, 1.7 µm) | H2O (0.1% FA)–ACN:MeOH (7:3) | ESI + | LC-PDA-QTOF MS | [19] |
| Carrot Leaves (Wild //Trangenic)[17] * | M. oleífera [73] | B. olerácea [74] * | R. communis [18] | Z. dressleri [75] | C. sinensis [76] | F. japonica//F. Sachalinensis [77] | Fruit Tree [19] ** | |
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 9 | |
| α-carotene | 20.1 ± 0.58//12.6 ± 0.62 | ♦ | ||||||
| β-carotene | 21.8 ± 1.15//27.1 ± 0.77 | 30.7–42.4 ## | ♦ | ♦♦ | ||||
| β-carotene-5,6-epoxide | ♦ | |||||||
| 15-Z-β-carotene | 0.40–0.69 | |||||||
| All-E-β-carotene | 11.86–20.77 | |||||||
| All-trans-β-carotene | 63.70 ± 0.13//41.41 ± 0.25 | |||||||
| 9-cis-β-carotene | ♦# | 19.08 ± 0.04//12.40 ± 0.07 | ♦♦ | |||||
| 13-cis-β-carotene | ♦# | 4.43 ± 0.01//2.88 ± 0.02 | ||||||
| 13-Z-β-carotene | ♦ | |||||||
| 9-Z-β-carotene | ♦ | |||||||
| Adonirubin | 5.2 ± 0.27 § | |||||||
| Adonixanthin | 5.0 ± 0.36 § | |||||||
| Antheraxanthin | ♦ | |||||||
| Astaxanthin | 32.4 ± 1.5 § | |||||||
| Canthaxanthin | 4.1 ± 0.21 § | ♦ | ||||||
| β-cryptoxanthin | 2.8 ± 0.42 § | ♦ | ||||||
| α-cryptoxanthin | ♦ | |||||||
| Lutein | 68.5 ± 0.87//46.0 ± 1.28 | 44.0–56.7 ## | ♦ | 45.2 ± 2.1–426.5 ± 4.2 | ||||
| lutein-5,6-epoxide | 0.84 ± 0.00//0.55 ± 0.00 | |||||||
| All-trans-lutein | 24.08 ± 0.05//15.65 ± 0.09 | |||||||
| All-E-lutein | 17.6–41.16 | |||||||
| 13-Z-lutein | 1.58–5.80 | |||||||
| All-E-luteoxanthin | 2.58–5.68 | |||||||
| Zeaxanthin | 37.7 ± 0.59//29.8 ± 1.01 | ♦ # | ♦ | 3.1 ± 0.6–212.3 ± 2.5 | ||||
| All-E-zeaxanthin | 2.26–13.54 | |||||||
| All-trans-zeaxanthin | 4.46 ± 0.01//2.90 ± 0.02 | |||||||
| Violaxanthin | 29.2–42.2 ## | |||||||
| trans-violxanthin | 0.68 ± 0.00//0.44 ± 0.01 | |||||||
| 9-cis-violaxanthin | 0.3 ± 0.0–8.4 ± 1.1 | |||||||
| Neoxanthin | 12.0–25.9 ## | ♦ | ||||||
| 9-cis-neoxanthin | 1.3 ± 0.1–33.6 ± 1.1 | |||||||
| 9-Z′-neoxanthin | ♦ | |||||||
| Capsanthin | ♦ | |||||||
| 13/13-′Z-capsanthin | ♦ | |||||||
| Capsoneoxanthin | ♦ | |||||||
| Capsorubin | ♦ | |||||||
| 13-Z-capsorubin | ♦ | |||||||
| Cryptocapsin | ♦ | |||||||
| Cryptocapsin 5,6-epoxide | ♦ | |||||||
| 9-cis-β-cryptoxanthin | 11.0 ± 1.7–504.5 ± 4.5 | |||||||
| Carotenoid compound | ♦♦♦ | |||||||
| 4,4′-diapolycopenedial | ♦ | |||||||
| 3,4-dihydroanydrorhodovibrin | ♦ | |||||||
| 3′,4′-dihydrorhodovibrin | ♦ | |||||||
| OH-spheroidene | ♦ | |||||||
| Echinenone | 2.4 ± 0.31§ | |||||||
| 3′-OH-echinenone | ♦ | |||||||
| Isorenieratene | ♦ | |||||||
| Presqualene diphosphate | ♦ | |||||||
| 8′-R-neochrome | 1.91 ± 0.00//1.24 ± 0.01 | |||||||
| 8′-S-neochrome | 2.31 ± 0.00//1.50 ± 0.01 |
| Plant | Sample | Solvents | Sample:Solvent | Time Extraction/Conditions | Ref |
|---|---|---|---|---|---|
| Fruit tree leaves * | Lyophilized, powdered | Hexane/Acetone/MeOH (2:1:1) (10% of MgCO3 in BHT–1%) | 100 mg:3 mL | Orbital shaker (300rpm, 30′) dark—re-extraction 4 times, evaporation to dryness, dissolution in MeOH | [19] |
| F. japonica// F. sachalinensis | Frozen (−25 °C), lyophilized (stored −80 °C) | Hexane:Acetone:MeOH (2:1:1) (10% of MgCO3 in BHT–1%) | 500 mg:5 mL | Orbital shaker (300 rpm, 30′) dark—centrifuged (10′, 19,000× g, 4 °C), re-extraction, evaporation to dryness, dissolution in MeOH | [77] |
| C. sinensis ** | Unprocessed samples | Cold acetone (80%) | 10 mg:2 mL | No info | [79] |
| R. nasutus | Hot air drying (60 °C, 4 h); freeze-dried; stored −20 °C | Hexane:EtOH/acetone/toluene (10:6:7:7) | 200 mg:30 mL | Shake 1 h, 15 mL hexane (shake 10′), 15 mL of 10% anhydrous sodium sulphate (shake 1′), organic layer extracted (4 times 15 mL hexane), evaporation to dryness, dissolution in acetone | [80] |
| Plant | Column | Eluent | Ionization Source | Mass Analysers | Ref |
|---|---|---|---|---|---|
| Fruit tree leaves * | RP ACQUITY UPLC BEH C18 (100 × 2.1 mm, 1.7 µm) | H2O (0.1% FA)–ACN:MeOH (7:3) | ESI + | LC-PDA-QTOF MS | [19] |
| F. japonica// F. sachalinensis | RP ACQUITY UPLC BEH C18 (100 × 2.1 mm, 1.7 µm) | ACN:MeOH (7:3)–H2O (0.1% FA) | ESI + | LC-QTOF MS | [77] |
| C. sinensis ** | RP BEH C18 (150 × 2.1 mm, 1.7 m) and RP ACQUITY UPLC HSS T3 (100 × 2.1 mm, 1.8 µm) | MeOH:iPrOH:ACN (10:15:75)–MeOH:ACN:H2O (CH3COONH4, 10 mM) (25:25:50) | APCI (apolar compounds) ESI + (polar compounds) | UHPLC tandem MS | [79] |
| R. nasutus | RP Eclipse XDB-C18 | MeOH:DMF (97:3)–ACN–Acetone | APCI | HPLC-DAD MS | [80] |
| Fruit Tree [19] * | F. japonica//F. Sachalinensis [77] | C. sinensis [79] ** | R. nasutus (Hot-Air//Freeze Drying) [80] | |
|---|---|---|---|---|
| Chlorophyll a | 0.1 ± 0.0–186.4 ± 2.5 # | 22.91 ± 0.05//14.89 ± 0.09 | 72 ± 3–1250 ± 30 | 814.1 ± 11.82//4707 ± 59 ## |
| Chlorophyll a’ | # | 1.00 ± 0.01//0.65 ± 0.00 | 90 ± 5–273.8 ± 1.2 | 131.2 ± 2.10//53.47 ± 1.30 ## |
| Chlorophyll b | 7.0 ± 0.5–80.4 ± 2.6 # | 63.19 ± 0.13//41.07 ± 0.24 | 50.6 ± 0.3–1300 ± 18 | 324.7 ± 8.83//1280 ± 17 ## |
| Chlorophyll b’ | # | 4.45 ± 0.01//2.89 ± 0.02 | 30.7 ± 2.1–410 ± 6 | 67.08 ± 1.31//ND ## |
| Chlorophyllide a | 0.1 ± 0.0–12.7 ± 1.2 | 1.52 ± 0.00//0.99 ± 0.01 | 76-6 ± 2.1–136 ± 8 | |
| Chlorophyllide a’ | 87 ± 6 | |||
| Chlorophyllide b | 8.91 ± 0.02//5.79 ± 0.03 | 70.5 ± 2.4–123 ± 16 | ||
| Chlorophyllide b’ | 85 ± 3–129 ± 13 | |||
| Pheophorbid a | 0.3 ± 0.0–40.4 ± 3.1 # | 219 ± 30–1260 ± 120 | ||
| Pheophorbid a’ | # | 68.7 ±2.5–295 ± 5 | ||
| Pheophorbid b | 0.2 ± 0.0–165.1 ± 0.3 # | 72 ± 7–321 ± 30 | ||
| Pheophorbid b’ | # | 52.0 ± 1.8–219.1 ± 1.8 | ||
| Pheophytin a | 3.3 ± 0.2–221.2 ± 2.5 # | 1.48 ± 0.01//0.96 ± 0.01 | 500 ± 50–3200 ± 320 | 440.2 ± 7.02 // 84.07 ± 1.73 ## |
| Pheophytin a’ | # | 0.68 ± 0.01//0.44 ± 0.00 | 96 ± 10–573 ± 40 | 69.68 ± 1.15//ND ## |
| Pheophytin b | 4.8 ± 0.2–311.3 ± 3.1 # | 75.13 ± 0.15//48.83 ± 0.29 | 61.1 ± 0.8–368 ± 18 | 39.65 ± 2.01//ND## |
| Pheophytin b’ | # | 11.51 ± 0.02//7.48 ± 0.04 | 58.4 ± 1.9–106 ± 10 | |
| OH-chlorophyll a | 2.34 ± 0.02//1.52 ± 0.01 | 206.4 ± 3.44//ND ## | ||
| 13-OH-chlorophyll a | 111 ± 5 | |||
| 15-OH-lactone chlorophyll a | 9.25 ± 0.45//ND ## | |||
| OH-chlorophyll b | 0.2 ± 0.0–9.2 ± 0.3 | 108.6 ± 1.58//ND ## | ||
| 13-OH-chlorophyll b | 42 ± 3–226 ± 8 | |||
| OH-pheophytin a | 0.3 ± 0.0–25.3 ± 0.4 | 88.29 ± 2.42//ND ## | ||
| 13-OH-pheophitin a | 83 ± 13–470 ± 3 | |||
| 15’-OH-lactone pheophytin a | 69 ± 6–222 ± 2 | |||
| Pyropheophytin a | 73 ± 6–327 ± 30 | |||
| OH-pheophytin a’ | 69.6 ± 2.70//ND ## | |||
| 13-OH-pheophitin a’ | 68 ± 3–391 ± 24 | |||
| OH-pheophytin b | 2.7 ± 0.3–91.3 ± 1.7 | 21.29 ± 0.04//13.84 ± 0.08 | ||
| 13-OH-pheophytin b | 52.5 ± 4–167 ± 11 | |||
| 15’-OH-lactone pheophytin b | 41 ± 4–114 ± 11 | |||
| 13-OH-pheophytin b’ | 36.1 ± 2.3–159 ± 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sousa, C. Anthocyanins, Carotenoids and Chlorophylls in Edible Plant Leaves Unveiled by Tandem Mass Spectrometry. Foods 2022, 11, 1924. https://doi.org/10.3390/foods11131924
Sousa C. Anthocyanins, Carotenoids and Chlorophylls in Edible Plant Leaves Unveiled by Tandem Mass Spectrometry. Foods. 2022; 11(13):1924. https://doi.org/10.3390/foods11131924
Chicago/Turabian StyleSousa, Clara. 2022. "Anthocyanins, Carotenoids and Chlorophylls in Edible Plant Leaves Unveiled by Tandem Mass Spectrometry" Foods 11, no. 13: 1924. https://doi.org/10.3390/foods11131924
APA StyleSousa, C. (2022). Anthocyanins, Carotenoids and Chlorophylls in Edible Plant Leaves Unveiled by Tandem Mass Spectrometry. Foods, 11(13), 1924. https://doi.org/10.3390/foods11131924