
Exploring the Successions in Microbial Community and Flavor of Daqu during Fermentation Produced by Different Pressing Patterns
 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection and Preparation of Sample
2.2. Determination of Physicochemical Properties and Enzymatic Activities
2.3. Analysis of Volatile Metabolites by HS-SPME-GC/MS
2.4. DNA Extraction, PCR Amplification and Illumina MiSeq Sequencing
2.5. Processing of Sequencing Data
2.6. Statistical Analysis and Visualization
3. Results
3.1. Temporal Changes in Physicochemical Characteristics and Enzymatic Activities of Daqu during Fermentation
3.2. Analysis of Volatile Compounds
3.3. The Succession of Microbial Community in the Two Types of Daqu
3.4. Divergence of Microbial Communities in Two Types of Daqu Samples
3.5. Interaction Network between Microbial Communities and Volatile Compounds in Daqu
3.6. Microbial Interactions and Correlation between Microbial Communities and Physicochemical Properties
3.7. Factors Contributing to the Differences in the Microbial Communities and Physicochemical Properties of the Two Daqu
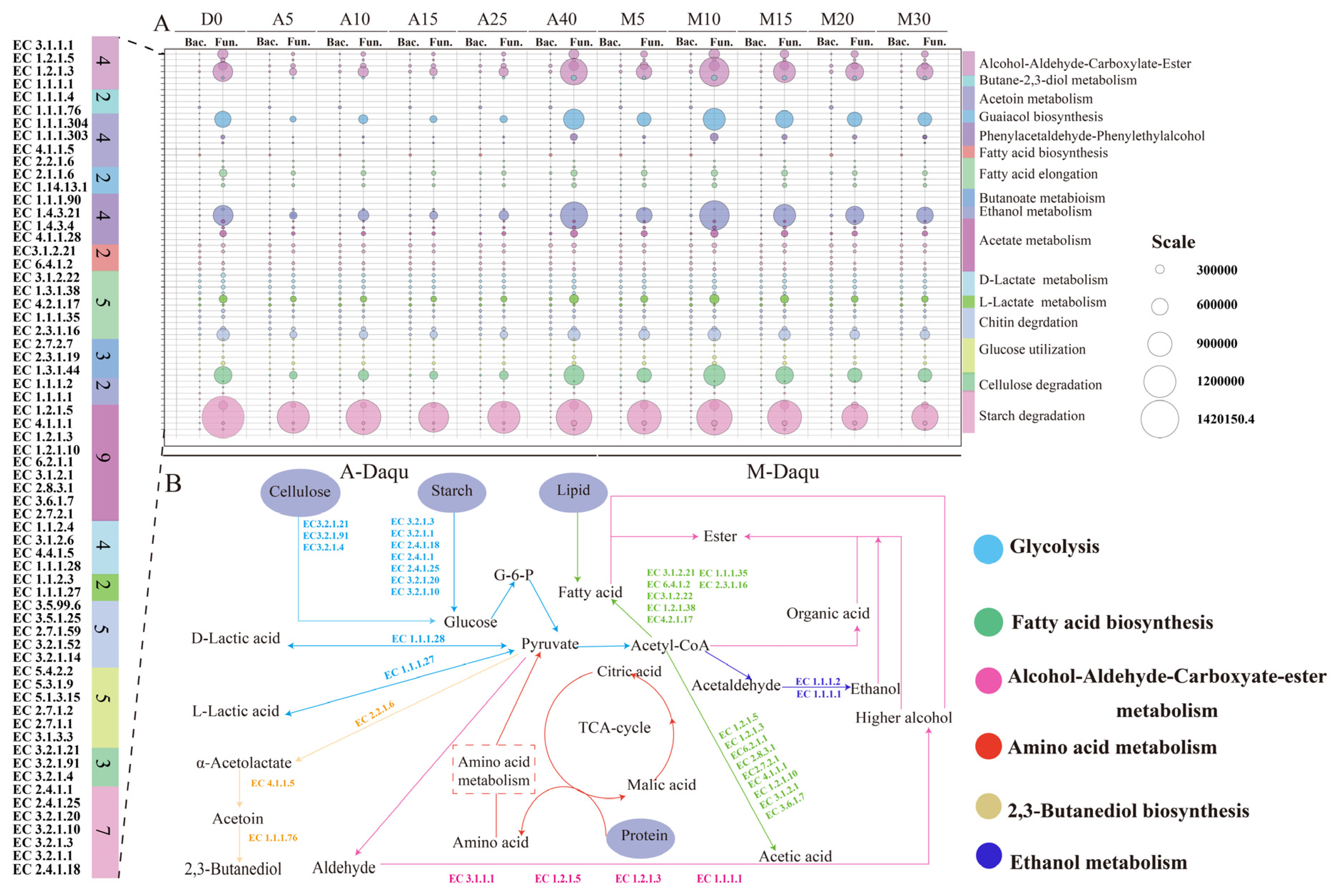
3.8. Prediction of the Microbial Functions in Daqu
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fan, G.; Fu, Z.; Sun, B.; Zhang, Y.; Wang, X.; Xia, Y.; Huang, M.; Li, X. Roles of aging in the production of light-flavored Daqu. J. Biosci. Bioeng. 2019, 127, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Deng, J.; He, G.; Zhou, R. Metaproteomic Characterization of Daqu, a Fermentation Starter Culture of Chinese Liquor. J. Proteom. Bioinform. 2016, 9, 49–52. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Chen, J.; Fan, Y.; Huang, X.; Han, B. Biochemical characterisation and dominance of different hydrolases in different types of Daqu—A Chinese industrial fermentation starter. J. Sci. Food Agric. 2018, 98, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Zhao, C.; Luo, H. Diversity and Function of Microbial Community in Chinese Strong-Flavor Baijiu Ecosystem: A Review. Front. Microbiol. 2018, 9, 671. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Li, D.; Ai, M.; Tang, Q.; Huang, J.; Ding, X.; Wu, C.; Zhou, R. Correlation between volatile profiles and microbial communities: A metabonomic approach to study Jiang-flavor liquor Daqu. Food Res. Int. 2019, 121, 422–432. [Google Scholar] [CrossRef]
- Ma, S.; Luo, H.; Zhao, D.; Qiao, Z.; Zheng, J.; An, M.; Huang, D. Environmental factors and interactions among microorganisms drive microbial community succession during fermentation of Nongxiangxing daqu. Bioresour. Technol. 2022, 345, 126549. [Google Scholar] [CrossRef]
- Zheng, X.-W.; Tabrizi, M.R.; Nout, M.J.R.; Han, B.-Z. Daqu—A Traditional Chinese Liquor Fermentation Starter. J. Inst. Brew. 2011, 117, 82–90. [Google Scholar] [CrossRef]
- Jiang, S.; Qiu, S.; Zou, J.; Chen, L.; Luo, X.; Wang, X. Preliminary studies on yeasts from sauce-flavor Daqu made by traditional and mechanical methods. China Brew. 2017, 36, 59–65. [Google Scholar]
- Guan, T.; Yang, H.; Ou, M.; Zhang, J. Storage period affecting dynamic succession of microbiota and quality changes of strong-flavor Baijiu Daqu. LWT Food Sci. Technol. 2020, 139, 110544. [Google Scholar] [CrossRef]
- Wang, J.; Zhong, Q.; Yang, Y.; Li, H.; Wang, L.; Tong, Y.; Fang, X.; Liao, Z. Comparison of Bacterial Diversity between Two Traditional Starters and the Round-Koji-Maker Starter for Traditional Cantonese Chi-Flavor Liquor Brewing. Front. Microbiol. 2018, 9, 1053. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.-D.; Qiu, S.-Y.; Li, P.; Ban, S.-D. Analysis of Microbial Community Structure in Traditional and Automated Moutai-Flavor Daqu. J. Am. Soc. Brew. Chem. 2019, 77, 140–146. [Google Scholar] [CrossRef]
- QB/T 4257 2011; Ministry of Industry and Information Technology of the People’s Republic of China, General analysis method of brewing Daqu (QB/T4257 2011). China Light Industry Press: Beijing, China, 2011.
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Stackebrandt, E.; Goebel, B. Taxonomic note: A place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int. J. Syst. Evol. Micr. 1994, 44, 846–849. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian Classifier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [Green Version]
- Legendre, P.; Anderson, M.J. Distance-Based Redundancy Analysis: Testing Multispecies Responses in Multifactorial Ecological Experiments. Ecol. Monogr. 1999, 69, 512. [Google Scholar] [CrossRef]
- Jiao, S.; Chen, W.; Wang, J.; Du, N.; Li, Q.; Wei, G. Soil microbiomes with distinct assemblies through vertical soil profiles drive the cycling of multiple nutrients in reforested ecosystems. Microbiome 2018, 6, 146. [Google Scholar] [CrossRef] [Green Version]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Liu, X.; Gao, T.; Gu, S.; Wu, Y.; Zhao, L.; Ma, J.; Li, X.; Zhang, J. Unraveling the correlations between bacterial diversity, physicochemical properties and bacterial community succession during the fermentation of traditional Chinese strong-flavor Daqu. LWT Food Sci. Technol. 2021, 154, 112764. [Google Scholar] [CrossRef]
- He, M.; Jin, Y.; Zhou, R.; Zhao, D.; Zheng, J.; Wu, C. Dynamic succession of microbial community in Nongxiangxing daqu and microbial roles involved in flavor formation. Food Res. Int. 2022, 159, 111559. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Sun, B.; Fu, Z.; Xia, Y.; Huang, M.; Xu, C.; Li, X. Analysis of Physicochemical Indices, Volatile Flavor Components, and Microbial Community of a Light-Flavor Daqu. J. Am. Soc. Brew. Chem. 2018, 76, 209–218. [Google Scholar] [CrossRef]
- Yan, S.; Tong, Q.; Guang, J. Yeast dynamics and changes in volatile compounds during the fermentation of the traditional Chinese strong-flavor Daqu. LWT Food Sci. Technol. 2019, 106, 57–63. [Google Scholar] [CrossRef]
- Shi, W.; Chai, L.-J.; Fang, G.-Y.; Mei, J.-L.; Lu, Z.-M.; Zhang, X.-J.; Xiao, C.; Wang, S.-T.; Shen, C.-H.; Shi, J.-S.; et al. Spatial heterogeneity of the microbiome and metabolome profiles of high-temperature Daqu in the same workshop. Food Res. Int. 2022, 156, 111298. [Google Scholar] [CrossRef]
- Gan, S.-H.; Yang, F.; Sahu, S.K.; Luo, R.-Y.; Liao, S.-L.; Wang, H.-Y.; Jin, T.; Wang, L.; Zhang, P.-F.; Liu, X.; et al. Deciphering the Composition and Functional Profile of the Microbial Communities in Chinese Moutai Liquor Starters. Front. Microbiol. 2019, 10, 1540. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.; Yang, L.; Hui, L.; Yuan-Yuan, G.; Ming-Juan, Z.; Chun-Hui, X.; Ling, X.; Chi, C. Bacterial communities during the process of high-temperature Daqu production of roasted sesame-like flavour liquor. J. Inst. Brew. 2015, 121, 440–448. [Google Scholar] [CrossRef]
- Tang, J.; Rao, J.; Zou, Y.; Liao, L.; Huang, D.; Luo, H. The community assembly patterns determined differences between the surface and the core microbial communities of Nongxiangxing Daqu. LWT Food Sci. Technol. 2023, 183, 114936. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, S.-T.; Lu, Z.-M.; Zhang, X.-J.; Chai, L.-J.; Shen, C.-H.; Shi, J.-S.; Xu, Z.-H. Metagenomics unveils microbial roles involved in metabolic network of flavor development in medium-temperature daqu starter. Food Res. Int. 2021, 140, 110037. [Google Scholar] [CrossRef]
- Zhu, M.; Zheng, J.; Xie, J.; Zhao, D.; Qiao, Z.-W.; Huang, D.; Luo, H.-B. Effects of environmental factors on the microbial community changes during medium-high temperature Daqu manufacturing. Food Res. Int. 2022, 153, 110955. [Google Scholar] [CrossRef]
- Du, H.; Wang, X.; Zhang, Y.; Xu, Y. Exploring the impacts of raw materials and environments on the microbiota in Chinese Daqu starter. Int. J. Food Microbiol. 2019, 297, 32–40. [Google Scholar] [CrossRef]
- Hou, Q.; Wang, Y.; Cai, W.; Ni, H.; Zhao, H.; Zhang, Z.; Liu, Z.; Liu, J.; Zhong, J.; Guo, Z. Metagenomic and physicochemical analyses reveal microbial community and functional differences between three types of low-temperature Daqu. Food Res. Int. 2022, 156, 111167. [Google Scholar] [CrossRef]
- Kang, J.; Hu, Y.; Ding, Z.; Ye, L.; Li, H.; Cheng, J.; Fan, L.; Zhao, H.; Han, B.; Zheng, X. Deciphering the Shifts in Microbial Community Diversity from Material Pretreatment to Saccharification Process of Fuyu-Flavor Baijiu. Front. Microbiol. 2021, 12, 2354. [Google Scholar] [CrossRef]
- Yang, L.; Chen, R.; Liu, C.; Chen, L.; Yang, F.; Wang, L. Spatiotemporal accumulation differences of volatile compounds and bacteria metabolizing pickle like odor compounds during stacking fermentation of Maotai-flavor baijiu. Food Chem. 2023, 426, 136668. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, J.; Ding, F.; Deng, W.; Wang, X.; Xue, Y.; Chen, X.; Han, B.-Z. Multidimensional profiling indicates the shifts and functionality of wheat-origin microbiota during high-temperature Daqu incubation. Food Res. Int. 2022, 156, 111191. [Google Scholar] [CrossRef]
- Zhang, L.; Wu, C.; Ding, X.; Zheng, J.; Zhou, R. Characterisation of microbial communities in Chinese liquor fermentation starters Daqu using nested PCR-DGGE. World J. Microbiol. Biotechnol. 2014, 30, 3055–3063. [Google Scholar] [CrossRef]
- Deng, Y.; Huang, D.; Han, B.; Ning, X.; Yu, D.; Guo, H.; Zou, Y.; Jing, W.; Luo, H. Correlation: Between Autochthonous Microbial Diversity and Volatile Metabolites during the Fermentation of Nongxiang Daqu. Front. Microbiol. 2021, 12, 688891. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Cheng, Y.; Shi, Q.; Ge, X.; Yang, Y.; Huang, Y. Metagenomic analyses reveal microbial communities and functional differences between Daqu from seven provinces. Food Res. Int. 2023, 172, 113076. [Google Scholar] [CrossRef]
- He, M.; Jin, Y.; Liu, M.; Yang, G.; Zhou, R.; Zhao, J.; Wu, C. Metaproteomic investigation of enzyme profile in daqu used for the production of Nongxiangxing baijiu. Int. J. Food Microbiol. 2023, 400, 110250. [Google Scholar] [CrossRef]
- Zhao, G.; Liu, C.; Li, S.; Wang, X.; Yao, Y. Exploring the flavor formation mechanism under osmotic conditions during soy sauce fermentation in Aspergillus oryzae by proteomic analysis. Food Funct. 2020, 11, 640–648. [Google Scholar] [CrossRef]
- Xu, Y.; Wu, M.; Zhao, D.; Zheng, J.; Dai, M.; Li, X.; Li, W.; Zhang, C.; Sun, B. Simulated Fermentation of Strong-Flavor Baijiu through Functional Microbial Combination to Realize the Stable Synthesis of Important Flavor Chemicals. Foods 2023, 12, 644. [Google Scholar] [CrossRef]
- Yang, Y.; Hu, W.; Xia, Y.; Mu, Z.; Tao, L.; Song, X.; Zhang, H.; Ni, B.; Ai, L. Flavor Formation in Chinese Rice Wine (Huangjiu): Impacts of the Flavor-Active Microorganisms, Raw Materials, and Fermentation Technology. Front. Microbiol. 2020, 11, 580247. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yi, Z.; Jin, Y.; Zhao, Y.; He, K.; Liu, D.; Zhao, D.; He, H.; Luo, H.; Zhang, W.; et al. New microbial resource: Microbial diversity, function and dynamics in Chinese liquor starter. Sci. Rep. 2017, 7, 14577. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, P.; Jin, Y.; Liu, M.; Peng, L.; Yang, G.; Luo, Z.; Jiang, D.; Zhao, J.; Zhou, R.; Wu, C. Exploring the Successions in Microbial Community and Flavor of Daqu during Fermentation Produced by Different Pressing Patterns. Foods 2023, 12, 2603. https://doi.org/10.3390/foods12132603
Huang P, Jin Y, Liu M, Peng L, Yang G, Luo Z, Jiang D, Zhao J, Zhou R, Wu C. Exploring the Successions in Microbial Community and Flavor of Daqu during Fermentation Produced by Different Pressing Patterns. Foods. 2023; 12(13):2603. https://doi.org/10.3390/foods12132603
Chicago/Turabian StyleHuang, Ping, Yao Jin, Mingming Liu, Liqun Peng, Guanrong Yang, Zhi Luo, Dongcai Jiang, Jinsong Zhao, Rongqing Zhou, and Chongde Wu. 2023. "Exploring the Successions in Microbial Community and Flavor of Daqu during Fermentation Produced by Different Pressing Patterns" Foods 12, no. 13: 2603. https://doi.org/10.3390/foods12132603